Canonical and Non-Canonical Autophagy in HIV-1 Replication Cycle

Abstract
:1. Introduction
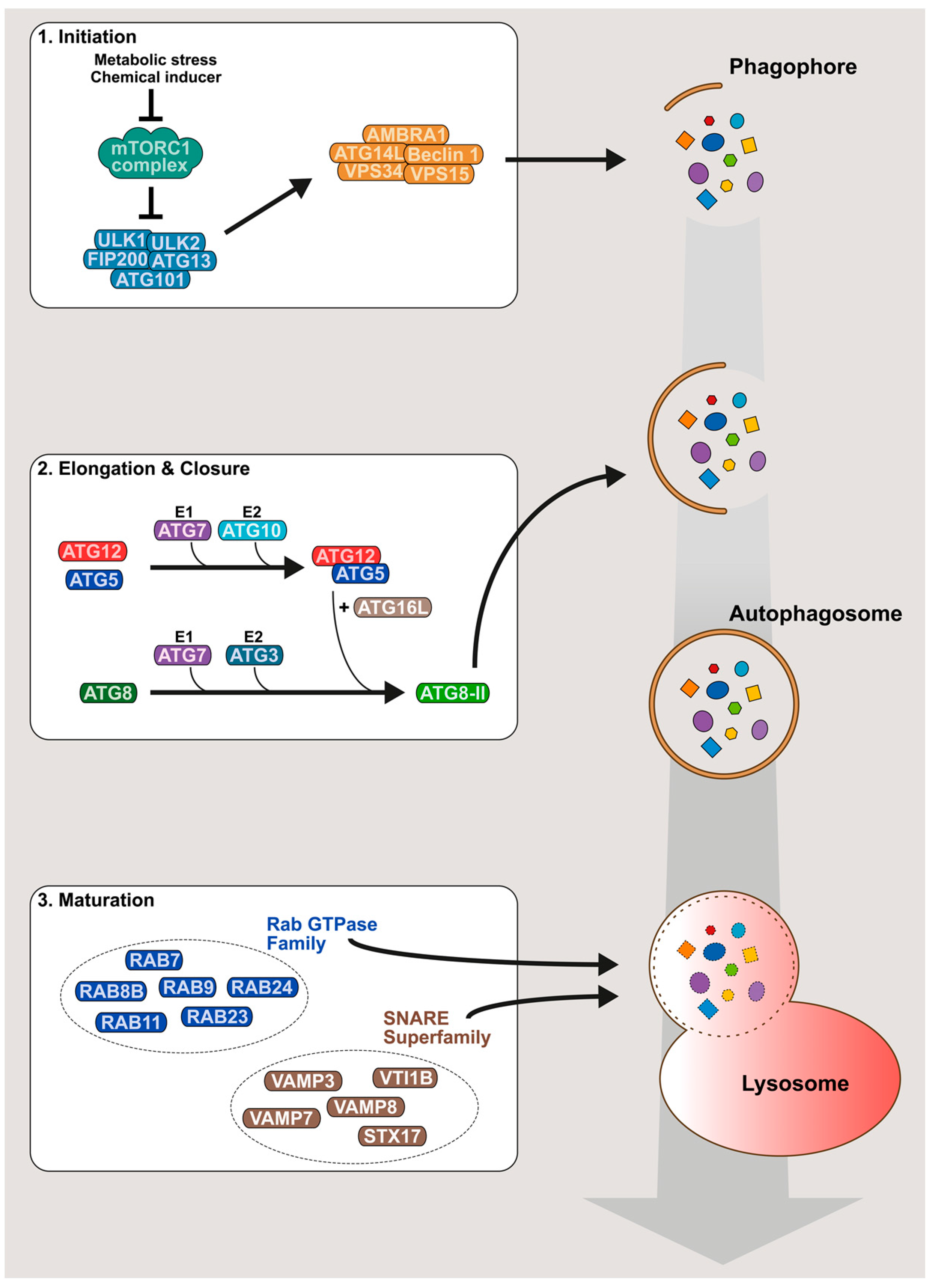
2. An Overview of the Autophagy Mechanism
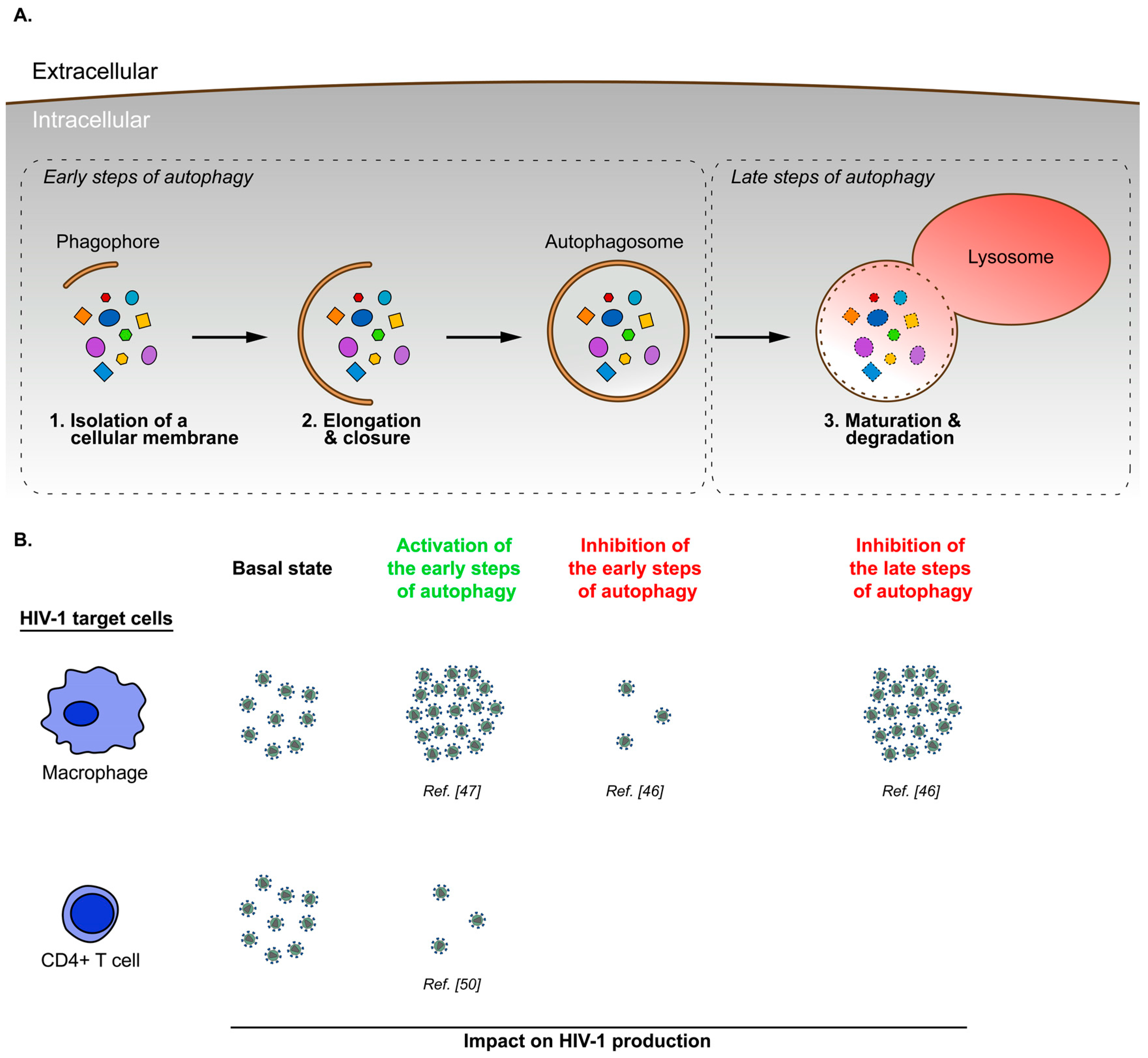
3. Cell Type Specific Modulation of Autophagy during HIV-1 Infection
4. Autophagy-Mediated Viral Restriction in HIV-1 Infected Cells
4.1. Degradation of the HIV-1 Tat Protein by the Selective Autophagy Mechanism
4.2. Contribution of Autophagy in the TRIM5α Restriction Factor Activities against HIV-1
4.3. Modulation of Vif-Mediated Countermeasure against APOBEC3G Restriction by Autophagy
4.4. Participation of Autophagy in the Induction of Innate Immunity to HIV-1 Infection
5. Disruption of Autophagy-Associated Adaptive Immune Response by HIV-1
6. Hijacking of Canonical Autophagy by HIV-1 Viral Proteins
6.1. Envelope Proteins
6.2. Tat Protein
6.3. Nef Protein
6.4. Vpr Protein
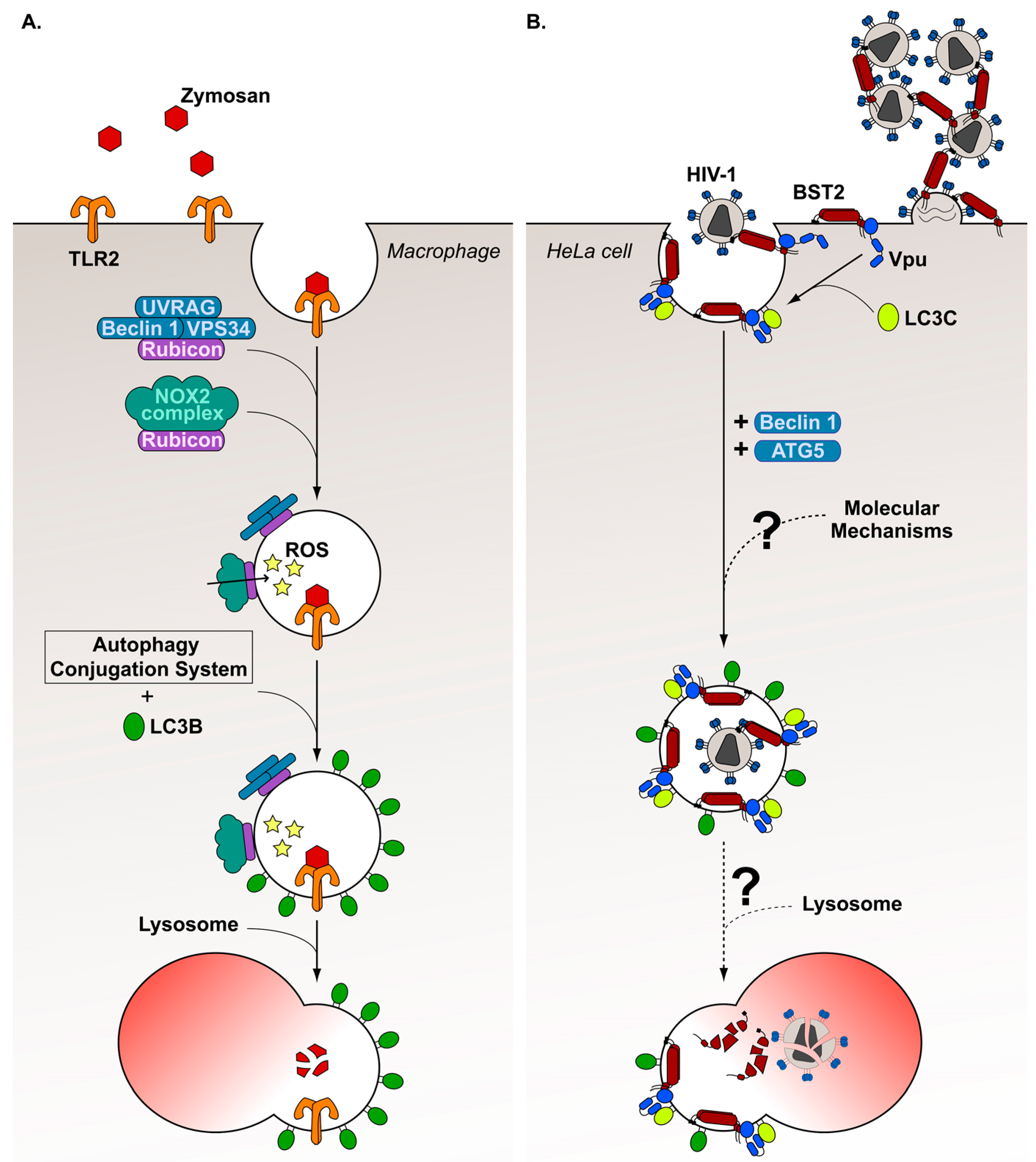
7. Diversion of a Non-Canonical Autophagy Pathway, the LC3-Associated Phagocytosis, by HIV-1 Viral Protein Vpu
8. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, I.; Amano, A.; Mizushima, N.; Yamamoto, A.; Yamaguchi, H.; Kamimoto, T.; Nara, A.; Funao, J.; Nakata, M.; Tsuda, K.; et al. Autophagy defends cells against invading group A Streptococcus. Science 2004, 306, 1037–1040. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Yoshimori, T.; Suzuki, T.; Sagara, H.; Mizushima, N.; Sasakawa, C. Escape of intracellular Shigella from autophagy. Science 2005, 307, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Münz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Wesselborg, S.; Stork, B. Autophagy signal transduction by ATG proteins: From hierarchies to networks. Cell. Mol. Life Sci. 2015, 72, 4721–4757. [Google Scholar] [CrossRef] [PubMed]
- Ylä-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.-L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Sasaki, T.; Iemura, S.I.; Natsume, T.; Hara, T.; Mizushima, N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009, 5, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Mercer, C.A.; Kaliappan, A.; Dennis, P.B. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 2009, 5, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 Complexes Mediate mTOR Signaling to the Autophagy Machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.-I.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 Association with the ULK1-Atg13-FIP200 Complex Required for Autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 Complex Mediates mTOR Signaling and Is Essential for Autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed]
- Shigemitsu, K.; Tsujishita, Y.; Hara, K.; Nanahoshi, M.; Avruch, J.; Yonezawa, K. Regulation of translational effectors by amino acid and mammalian target of rapamycin signaling pathways. Possible involvement of autophagy in cultured hepatoma cells. J. Biol. Chem. 1999, 274, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Karanasios, E.; Stapleton, E.; Manifava, M.; Kaizuka, T.; Mizushima, N.; Walker, S.A.; Ktistakis, N.T. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J. Cell Sci. 2013, 126, 5224–5238. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Petiot, A.; Ogier-Denis, E.; Blommaart, E.F.; Meijer, A.J.; Codogno, P. Distinct classes of phosphatidylinositol 3’-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 2000, 275, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Blommaart, E.F.; Krause, U.; Schellens, J.P.; Vreeling-Sindelárová, H.; Meijer, A.J. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 1997, 243, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER–mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Kirisako, T.; Baba, M.; Ishihara, N.; Miyazawa, K.; Ohsumi, M.; Yoshimori, T.; Noda, T.; Ohsumi, Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J. Cell Biol. 1999, 147, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Kuma, A.; Kobayashi, Y.; Yamamoto, A.; Matsubae, M.; Takao, T.; Natsume, T.; Ohsumi, Y.; Yoshimori, T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 2003, 116, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yamamoto, A.; Hatano, M.; Kobayashi, Y.; Kabeya, Y.; Suzuki, K.; Tokuhisa, T.; Ohsumi, Y.; Yoshimori, T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 2001, 152, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y.; Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Mizushima, N.; Ogawa, Y.; Matsuura, A.; Noda, T.; Ohsumi, Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J. 1999, 18, 5234–5241. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y.; Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Tanida-Miyake, E.; Ueno, T.; Kominami, E. The human homolog of Saccharomyces cerevisiae Apg7p is a Protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J. Biol. Chem. 2001, 276, 1701–1706. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in Mammalian Autophagy Research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Nair, U.; Klionsky, D.J. Atg8 Controls Phagophore Expansion during Autophagosome Formation. Mol. Biol. Cell 2008, 19, 3290–3298. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Hayashi-Nishino, M.; Fukumoto, H.; Omori, H.; Yamamoto, A.; Noda, T.; Yoshimori, T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell 2008, 19, 4651–4659. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, T.; Aikawa, C.; Goda, A.; Maruyama, F.; Hamada, S.; Nakagawa, I. The small GTPases Rab9A and Rab23 function at distinct steps in autophagy during Group A Streptococcus infection. Cell. Microbiol. 2012, 14, 1149–1165. [Google Scholar] [CrossRef] [PubMed]
- Munafó, D.B.; Colombo, M.I. Induction of autophagy causes dramatic changes in the subcellular distribution of GFP-Rab24. Traffic 2002, 3, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.M.; Sánchez, D.; Furlán, M.; Colombo, M.I. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 2008, 9, 230–250. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Furuta, N.; Fujita, N.; Noda, T.; Yoshimori, T.; Amano, A. Combinational Soluble N-Ethylmaleimide-sensitive Factor Attachment Protein Receptor Proteins VAMP8 and Vti1b Mediate Fusion of Antimicrobial and Canonical Autophagosomes with Lysosomes. Mol. Biol. Cell 2010, 21, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.M.; Sánchez, D.G.; Mestre, M.B.; Colombo, M.I. TI-VAMP/VAMP7 and VAMP3/cellubrevin: Two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim. Biophys. Acta-Mol. Cell Res. 2009, 1793, 1901–1916. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P. Long term non-progressor (LTNP) HIV infection. Indian J. Med. Res. 2013, 138, 291–293. [Google Scholar] [PubMed]
- Gaardbo, J.C.; Hartling, H.J.; Gerstoft, J.; Nielsen, S.D. Thirty Years with HIV Infection-Nonprogression Is Still Puzzling: Lessons to Be Learned from Controllers and Long-Term Nonprogressors. AIDS Res. Treat. 2012, 2012, 161584. [Google Scholar] [CrossRef] [PubMed]
- Okulicz, J.F.; Marconi, V.C.; Landrum, M.L.; Wegner, S.; Weintrob, A.; Ganesan, A.; Hale, B.; Crum-Cianflone, N.; Delmar, J.; Barthel, V.; et al. Infectious Disease Clinical Research Program (IDCRP) HIV Working Group Clinical outcomes of elite controllers, viremic controllers, and long-term nonprogressors in the US Department of Defense HIV natural history study. J. Infect. Dis. 2009, 200, 1714–1723. [Google Scholar] [CrossRef] [PubMed]
- Lambotte, O.; Boufassa, F.; Madec, Y.; Nguyen, A.; Goujard, C.; Meyer, L.; Rouzioux, C.; Venet, A.; Delfraissy, J.-F. SEROCO-HEMOCO Study Group HIV Controllers: A Homogeneous Group of HIV-1-Infected Patients with Spontaneous Control of Viral Replication. Clin. Infect. Dis. 2005, 41, 1053–1056. [Google Scholar] [CrossRef] [PubMed]
- Grabar, S.; Selinger-Leneman, H.; Abgrall, S.; Pialoux, G.; Weiss, L.; Costagliola, D. Prevalence and comparative characteristics of long-term nonprogressors and HIV controller patients in the French Hospital Database on HIV. AIDS 2009, 23, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Nardacci, R.; Amendola, A.; Ciccosanti, F.; Corazzari, M.; Esposito, V.; Vlassi, C.; Taibi, C.; Fimia, G.M.; del Nonno, F.; Ippolito, G.; et al. Autophagy plays an important role in the containment of HIV-1 in nonprogressor-infected patients. Autophagy 2014, 10, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Varbanov, M.; Robert-Hebmann, V.; Sagnier, S.; Robbins, I.; Sanchez, F.; Lafont, V.; Biard-Piechaczyk, M. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS ONE 2009, 4, e5787. [Google Scholar] [CrossRef] [PubMed]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Spector, S.A. Human immunodeficiency virus type-1 infection inhibits autophagy. AIDS 2008, 22, 695. [Google Scholar] [CrossRef] [PubMed]
- Laforge, M.; Limou, S.; Harper, F.; Casartelli, N.; Rodrigues, V.; Silvestre, R.; Haloui, H.; Zagury, J.F.; Senik, A.; Estaquier, J. DRAM Triggers Lysosomal Membrane Permeabilization and Cell Death in CD4+ T Cells Infected with HIV. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Sagnier, S.; Daussy, C.F.; Borel, S.; Robert-Hebmann, V.; Faure, M.; Blanchet, F.P.; Beaumelle, B.; Biard-Piechaczyk, M.; Espert, L. Autophagy Restricts HIV-1 Infection by Selectively Degrading Tat in CD4 + T Lymphocytes. J. Virol. 2015, 89, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-Induced Modulator of Autophagy, Is Critical for Apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Bieniasz, P.D. Intrinsic immunity: A front-line defense against viral attack. Nat. Immunol. 2004, 5, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.; Nava, B.; Caputi, M. Tat is a multifunctional viral protein that modulates cellular gene expression and functions. Oncotarget 2017, 8, 27569–27581. [Google Scholar] [CrossRef] [PubMed]
- Pertel, T.; Hausmann, S.; Morger, D.; Züger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Ganser-Pornillos, B.K.; Chandrasekaran, V.; Pornillos, O.; Sodroski, J.G.; Sundquist, W.I.; Yeager, M. Hexagonal assembly of a restricting TRIM5α protein. Proc. Natl. Acad. Sci. USA 2011, 108, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5α restriction factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef] [PubMed]
- Mandell, M.A.; Jain, A.; Arko-Mensah, J.; Chauhan, S.; Kimura, T.; Dinkins, C.; Silvestri, G.; Münch, J.; Kirchhoff, F.; Simonsen, A.; et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev. Cell 2014, 30, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.M.S.; Sarrami-Forooshani, R.; Setiawan, L.C.; Zijlstra-Willems, E.M.; van Hamme, J.L.; Tigchelaar, W.; van der Wel, N.N.; Kootstra, N.A.; Gringhuis, S.I.; Geijtenbeek, T.B.H. Receptor usage dictates HIV-1 restriction by human TRIM5α in dendritic cell subsets. Nature 2016, 1, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Yap, M.W.; Nisole, S.; Stoye, J.P. A single amino acid change in the SPRY domain of human TRIM5α leads to HIV-1 restriction. Curr. Biol. 2005, 15, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Wu, L.I.; Emerman, M.; Malik, H.S. Positive selection of primate TRIM5α identifies a critical species-specific retroviral restriction domain. Proc. Natl. Acad. Sci. USA 2005, 102, 2832–2837. [Google Scholar] [CrossRef] [PubMed]
- de Witte, L.; Nabatov, A.; Pion, M.; Fluitsma, D.; de Jong, M.A.W.P.; de Gruijl, T.; Piguet, V.; van Kooyk, Y.; Geijtenbeek, T.B.H. Langerin is a natural barrier to HIV-1 transmission by Langerhans cells. Nat. Med. 2007, 13, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Gringhuis, S.I.; van der Vlist, M.; van den Berg, L.M.; den Dunnen, J.; Litjens, M.; Geijtenbeek, T.B.H. HIV-1 exploits innate signaling by TLR8 and DC-SIGN for productive infection of dendritic cells. Nat. Immunol. 2010, 11, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Mc Dermott, R.; Ziylan, U.; Spehner, D.; Bausinger, H.; Lipsker, D.; Mommaas, M.; Cazenave, J.-P.; Raposo, G.; Goud, B.; de la Salle, H.; et al. Birbeck granules are subdomains of endosomal recycling compartment in human epidermal Langerhans cells, which form where Langerin accumulates. Mol. Biol. Cell 2002, 13, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Mehle, A.; Strack, B.; Ancuta, P.; Zhang, C.; McPike, M.; Gabuzda, D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem. 2004, 279, 7792–7798. [Google Scholar] [CrossRef] [PubMed]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Iwatani, Y.; Chan, D.S.B.; Wang, F.; Maynard, K.S.; Sugiura, W.; Gronenborn, A.M.; Rouzina, I.; Williams, M.C.; Musier-Forsyth, K.; et al. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007, 35, 7096–7108. [Google Scholar] [CrossRef] [PubMed]
- Mbisa, J.L.; Barr, R.; Thomas, J.A.; Vandegraaff, N.; Dorweiler, I.J.; Svarovskaia, E.S.; Brown, W.L.; Mansky, L.M.; Gorelick, R.J.; Harris, R.S.; et al. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 2007, 81, 7099–7110. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Cen, S.; Niu, M.; Saadatmand, J.; Kleiman, L. Inhibition of tRNA₃(Lys)-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J. Virol. 2006, 80, 11710–11722. [Google Scholar] [CrossRef] [PubMed]
- Mbisa, J.L.; Bu, W.; Pathak, V.K. APOBEC3F and APOBEC3G inhibit HIV-1 DNA integration by different mechanisms. J. Virol. 2010, 84, 5250–5259. [Google Scholar] [CrossRef] [PubMed]
- Valera, M.-S.; de Armas-Rillo, L.; Barroso-González, J.; Ziglio, S.; Batisse, J.; Dubois, N.; Marrero-Hernández, S.; Borel, S.; García-Expósito, L.; Biard-Piechaczyk, M.; et al. The HDAC6/APOBEC3G complex regulates HIV-1 infectiveness by inducing Vif autophagic degradation. Retrovirology 2015, 12, 53. [Google Scholar] [CrossRef] [PubMed]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.-S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Bruckman, R.S.; Chu, Y.L.; Spector, S.A. Autophagy induction by histone deacetylase inhibitors inhibits HIV type 1. J. Biol. Chem. 2015, 290, 5028–5040. [Google Scholar] [CrossRef] [PubMed]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin. Epigenet. 2012, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Spector, S.A. Toll-Like Receptor 8 Ligands Activate a Vitamin D Mediated Autophagic Response that Inhibits Human Immunodeficiency Virus Type 1. PLoS Pathog. 2012, 8, 1003017. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.-M.; Shin, D.-M.; Lee, H.-M.; Yang, C.-S.; Jin, H.S.; Kim, K.-K.; Lee, Z.-W.; Lee, S.-H.; Kim, J.-M.; Jo, E.-K. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 2009, 6, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Kang, K.H.; Spector, S.A. Production of Interferon by Human Immunodeficiency Virus Type 1 in Human Plasmacytoid Dendritic Cells Is Dependent on Induction of Autophagy. J. Infect. Dis. 2012, 205, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.; Manches, O.; Bhardwaj, N. Plasmacytoid dendritic cells in HIV infection. Adv. Exp. Med. Biol. 2013, 762, 71–107. [Google Scholar] [PubMed]
- Coulon, P.-G.; Richetta, C.; Rouers, A.; Blanchet, F.P.; Urrutia, A.; Guerbois, M.; Piguet, V.; Theodorou, I.; Bet, A.; Schwartz, O.; et al. HIV-Infected Dendritic Cells Present Endogenous MHC Class II–Restricted Antigens to HIV-Specific CD4+ T Cells. J. Immunol. 2016, 197, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Granelli-Piperno, A.; Golebiowska, A.; Trumpfheller, C.; Siegal, F.P.; Steinman, R.M. HIV-1-infected monocyte-derived dendritic cells do not undergo maturation but can elicit IL-10 production and T cell regulation. Proc. Natl. Acad. Sci. USA 2004, 101, 7669–7674. [Google Scholar] [CrossRef] [PubMed]
- Macatonia, S.E.; Lau, R.; Patterson, S.; Pinching, A.J.; Knight, S.C. Dendritic cell infection, depletion and dysfunction in HIV-infected individuals. Immunology 1990, 71, 38–45. [Google Scholar] [PubMed]
- Münz, C. Autophagy Beyond Intracellular MHC Class II Antigen Presentation. Trends Immunol. 2016, 37, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Mattei, L.M.; Steinberg, B.E.; Alberts, P.; Lee, Y.H.; Chervonsky, A.; Mizushima, N.; Grinstein, S.; Iwasaki, A. In Vivo Requirement for Atg5 in Antigen Presentation by Dendritic Cells. Immunity 2010, 32, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Pypaert, M.; Münz, C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Leib, D.A.; Alexander, D.E.; Cox, D.; Yin, J.; Ferguson, T.A. Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T-cell responses. J. Virol. 2009, 83, 12164–12171. [Google Scholar] [CrossRef] [PubMed]
- Jagannath, C.; Lindsey, D.R.; Dhandayuthapani, S.; Xu, Y.; Hunter, R.L.; Eissa, N.T. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat. Med. 2009, 15, 267–276. [Google Scholar] [CrossRef] [PubMed]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippé, R.; et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, F.P.; Moris, A.; Nikolic, D.S.; Lehmann, M.; Cardinaud, S.; Stalder, R.; Garcia, E.; Dinkins, C.; Leuba, F.; Wu, L.; et al. Human Immunodeficiency Virus-1 Inhibition of Immunoamphisomes in Dendritic Cells Impairs Early Innate and Adaptive Immune Responses. Immunity 2010, 32, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Sun, C.; Feng, L.; Li, P.; Xiao, L.; Ren, Y.; Wang, D.; Li, C.; Chen, L. Regulation of SIV Antigen-Specific CD4+ T Cellular Immunity via Autophagosome-Mediated MHC II Molecule-Targeting Antigen Presentation in Mice. PLoS ONE 2014, 9, e93143. [Google Scholar] [CrossRef] [PubMed]
- Andersen, A.N.; Landsverk, O.J.; Simonsen, A.; Bogen, B.; Corthay, A.; Øynebråten, I. Coupling of HIV-1 Antigen to the Selective Autophagy Receptor SQSTM1/p62 Promotes T-Cell-Mediated Immunity. Front. Immunol. 2016, 7, 167. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Investig. 2006, 116, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Denizot, M.; Varbanov, M.; Espert, L.; Robert-Hebmann, V.; Sagnier, S.; Garcia, E.; Curriu, M.; Mamoun, R.; Blanco, J.; Biard-Piechaczyk, M. HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy 2008, 4, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Masliah, E.; Spector, S.A. Autophagy is increased in postmortem brains of persons with HIV-1-associated encephalitis. J. Infect. Dis. 2011, 203, 1647–1657. [Google Scholar] [CrossRef] [PubMed]
- Pandhare, J.; Dash, S.; Jones, B.; Villalta, F.; Dash, C. A novel role of proline oxidase in HIV-1 envelope glycoprotein-induced neuronal autophagy. J. Biol. Chem. 2015, 290, 25439–25451. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Qiao, L.; Zhang, Y.; Zang, Y.; Shi, Y.; Liu, K.; Zhang, X.; Lu, X.; Yuan, L.; Su, B.; et al. ASPP2 plays a dual role in gp120-induced autophagy and apoptosis of neuroblastoma cells. Front. Neurosci. 2017, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Fu, M.; Kumar, S.; Kumar, A. Methamphetamine potentiates HIV-1 gp120-mediated autophagy via Beclin-1 and Atg5/7 as a pro-survival response in astrocytes. Cell Death Dis. 2016, 7, e2425. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.; Dumaop, W.; Eleuteri, S.; Elueteri, S.; Campos, S.; Serger, E.; Trejo, M.; Kosberg, K.; Adame, A.; Spencer, B.; et al. HIV-1 Tat alters neuronal autophagy by modulating autophagosome fusion to the lysosome: implications for HIV-associated neurocognitive disorders. J. Neurosci. 2015, 35, 1921–1938. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.A.; Metcalf, J.; Overk, C.; Adame, A.; Spencer, B.; Wrasidlo, W.; Florio, J.; Rockenstein, E.; He, J.J.; Masliah, E. The anticancer drug sunitinib promotes autophagyand protects from neurotoxicity in an HIV-1 Tat model of neurodegeneration. J. Neurovirol. 2017, 23, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.P.; de Simone, F.I.; Iorio, V.; De Marco, M.; Khalili, K.; Sariyer, I.K.; Capunzo, M.; Nori, S.L.; Rosati, A. HIV-1 Tat protein induces glial cell autophagy through enhancement of BAG3 protein levels. Cell Cycle 2014, 13, 3640–3644. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Chen, X.; Haughey, N.J.; Geiger, J.D. Role of endolysosomes in HIV-1 Tat-induced neurotoxicity. ASN Neuro 2012, 4, 243–252. [Google Scholar] [CrossRef] [PubMed]
- De Simone, F.I.; Darbinian, N.; Amini, S.; Muniswamy, M.; White, M.K.; Elrod, J.W.; Datta, P.K.; Langford, D.; Khalili, K. HIV-1 Tat and Cocaine Impair Survival of Cultured Primary Neuronal Cells via a Mitochondrial Pathway. J. Neuroimmune Pharmacol. 2016, 11, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Li, J.C.; Au, K.; Fang, J.; Yim, H.C.; Chow, K.; Ho, P.; Lau, A.S. HIV-1 trans-activator protein dysregulates IFN-γ signaling and contributes to the suppression of autophagy induction. AIDS 2011, 25, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Van Grol, J.; Subauste, C.; Andrade, R.M.; Fujinaga, K.; Nelson, J.; Subauste, C.S. HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PLoS ONE 2010, 5, e11733. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Rawat, P.; Bruckman, R.S.; Spector, S.A. Human Immunodeficiency Virus Type 1 Nef Inhibits Autophagy through Transcription Factor EB Sequestration. PLoS Pathog. 2015, 11, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Saribas, A.S.; Khalili, K.; Sariyer, I.K. Dysregulation of autophagy by HIV-1 Nef in human astrocytes. Cell Cycle 2015, 14, 2899–2904. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zheng, Y.; He, Y.; Chen, Z.; He, B. The role of autophagy in THP-1 macrophages resistance to HIV- Vpr-induced apoptosis. Exp. Cell Res. 2017, 351, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Madjo, U.; Leymarie, O.; Frémont, S.; Kuster, A.; Nehlich, M.; Gallois-Montbrun, S.; Janvier, K.; Berlioz-Torrent, C. LC3C Contributes to Vpu-Mediated Antagonism of BST2/Tetherin Restriction on HIV-1 Release through a Non-canonical Autophagy Pathway. Cell Rep. 2016, 17, 2221–2233. [Google Scholar] [CrossRef] [PubMed]
- Nath, A. Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J. Infect. Dis. 2002, 186, S193–S198. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.R.; Ruiz, A.P.; Prasad, V.R. Viral and cellular factors underlying neuropathogenesis in HIV associated neurocognitive disorders (HAND). AIDS Res. Ther. 2014, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Cherra, S.J.; Chu, C.T. Autophagy in neuroprotection and neurodegeneration: A question of balance. Future Neurol. 2008, 3, 309–323. [Google Scholar] [PubMed]
- Pérez, A.; Probert, A.W.; Wang, K.K.; Sharmeen, L. Evaluation of HIV-1 Tat induced neurotoxicity in rat cortical cell culture. J. Neurovirol. 2001, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- King, J.E.; Eugenin, E.A.; Buckner, C.M.; Berman, J.W. HIV tat and neurotoxicity. Microbes Infect. 2006, 8, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Buscemi, L.; Ramonet, D.; Geiger, J.D. Human immunodeficiency virus type-1 protein Tat induces tumor necrosis factor-α-mediated neurotoxicity. Neurobiol. Dis. 2007, 26, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, L.; Louboutin, J.-P.; Reyes, B.A.S.; Van Bockstaele, E.J.; Strayer, D.S. HIV-1 Tat neurotoxicity: A model of acute and chronic exposure, and neuroprotection by gene delivery of antioxidant enzymes. Neurobiol. Dis. 2012, 45, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Robinson, S.M.; Nath, A. Permeability of the blood–brain barrier to HIV-1 Tat. Exp. Neurol. 2005, 193, 218–227. [Google Scholar] [CrossRef] [PubMed]
- He, J.J.; Liu, Y.; Jones, M.; Hingtgen, C.M.; Bu, G.; Laribee, N.; Tanzi, R.E.; Moir, R.D.; Nath, A. Uptake of HIV-1 Tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat. Med. 2000, 6, 1380–1387. [Google Scholar] [CrossRef] [PubMed]
- Gutheil, W.G.; Subramanyam, M.; Flentke, G.R.; Sanford, D.G.; Munoz, E.; Huber, B.T.; Bachovchin, W.W. Human immunodeficiency virus 1 Tat binds to dipeptidyl aminopeptidase IV (CD26): A possible mechanism for Tat’s immunosuppressive activity. Proc. Natl. Acad. Sci. USA 1994, 91, 6594–6598. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Neuveut, C.; Tiffany, H.L.; Benkirane, M.; Rich, E.A.; Murphy, P.M.; Jeang, K.T. Selective CXCR4 antagonism by Tat: Implications for in vivo expansion of coreceptor use by HIV-1. Proc. Natl. Acad. Sci. USA 2000, 97, 11466–11471. [Google Scholar] [CrossRef] [PubMed]
- Vendeville, A.; Rayne, F.; Bonhoure, A.; Bettache, N.; Montcourrier, P.; Beaumelle, B. HIV-1 Tat enters T cells using coated pits before translocating from acidified endosomes and eliciting biological responses. Mol. Biol. Cell 2004, 15, 2347–2360. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, M.; Rusnati, M.; Presta, M.; Giacca, M. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J. Biol. Chem. 2001, 276, 3254–3261. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Tanaka, Y.; Guhde, G.; Suter, A.; Eskelinen, E.-L.; Hartmann, D.; Lüllmann-Rauch, R.; Janssen, P.M.L.; Blanz, J.; von Figura, K. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000, 406, 902–906. [Google Scholar] [CrossRef] [PubMed]
- Farrow, J.M.; Yang, J.C.; Evans, C.P. Autophagy as a modulator and target in prostate cancer. Nat. Rev. Urol. 2014, 11, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Palmieri, M.; Loehr, J.A.; Li, S.; Abo-Zahrah, R.; Monroe, T.O.; Thakur, P.B.; Sardiello, M.; Rodney, G.G. Src-dependent impairment of autophagy by oxidative stress in a mouse model of Duchenne muscular dystrophy. Nat. Commun. 2014, 5, 4425. [Google Scholar] [CrossRef] [PubMed]
- Lum, J.J.; DeBerardinis, R.J.; Thompson, C.B. Autophagy in metazoans: Cell survival in the land of plenty. Nat. Rev. Mol. Cell Biol. 2005, 6, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Basmaciogullari, S.; Pizzato, M. The activity of Nef on HIV-1 infectivity. Front. Microbiol. 2014, 5, 232. [Google Scholar] [CrossRef] [PubMed]
- Schindler, M.; Würfl, S.; Benaroch, P.; Greenough, T.C.; Daniels, R.; Easterbrook, P.; Brenner, M.; Münch, J.; Kirchhoff, F. Down-modulation of mature major histocompatibility complex class II and up-regulation of invariant chain cell surface expression are well-conserved functions of human and simian immunodeficiency virus nef alleles. J. Virol. 2003, 77, 10548–10556. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, O.; Maréchal, V.; Le Gall, S.; Lemonnier, F.; Heard, J.M. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 1996, 2, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Aiken, C.; Konner, J.; Landau, N.R.; Lenburg, M.E.; Trono, D. Nef induces CD4 endocytosis: requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell 1994, 76, 853–864. [Google Scholar] [CrossRef]
- Garcia, J.V.; Miller, A.D. Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature 1991, 350, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Matheson, N.J.; Sumner, J.; Wals, K.; Rapiteanu, R.; Weekes, M.P.; Vigan, R.; Weinelt, J.; Schindler, M.; Antrobus, R.; Costa, A.S.H.; et al. Cell Surface Proteomic Map of HIV Infection Reveals Antagonism of Amino Acid Metabolism by Vpu and Nef. Cell Host Microbe 2015, 18, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Chande, A.; Ziglio, S.; de Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [PubMed]
- Busca, A.; Saxena, M.; Kumar, A. Critical role for antiapoptotic Bcl-xL and Mcl-1 in human macrophage survival and cellular IAP1/2 (cIAP1/2) in resistance to HIV-Vpr-induced apoptosis. J. Biol. Chem. 2012, 287, 15118–15133. [Google Scholar] [CrossRef] [PubMed]
- Godet, A.N.; Guergnon, J.; Croset, A.; Cayla, X.; Falanga, P.B.; Colle, J.-H.; Garcia, A. PP2A1 binding, cell transducing and apoptotic properties of Vpr(77–92): A new functional domain of HIV-1 Vpr proteins. PLoS ONE 2010, 5, e13760. [Google Scholar] [CrossRef] [PubMed]
- Mukerjee, R.; Chang, J.R.; del Valle, L.; Bagashev, A.; Gayed, M.M.; Lyde, R.B.; Hawkins, B.J.; Brailoiu, E.; Cohen, E.; Power, C.; et al. Deregulation of microRNAs by HIV-1 Vpr protein leads to the development of neurocognitive disorders. J. Biol. Chem. 2011, 286, 34976–34985. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.A.; Mukhtar, M.; Pomerantz, R.J. Human immunodeficiency virus type 1 Vpr induces apoptosis in human neuronal cells. J. Virol. 2000, 74, 9717–9726. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Busca, A.; Pandey, S.; Kryworuchko, M.; Kumar, A. CpG protects human monocytic cells against HIV-Vpr-induced apoptosis by cellular inhibitor of apoptosis-2 through the calcium-activated JNK pathway in a TLR9-independent manner. J. Immunol. 2011, 187, 5865–5878. [Google Scholar] [CrossRef] [PubMed]
- Arokium, H.; Kamata, M.; Chen, I. Virion-associated Vpr of human immunodeficiency virus type 1 triggers activation of apoptotic events and enhances fas-induced apoptosis in human T cells. J. Virol. 2009, 83, 11283–11297. [Google Scholar] [CrossRef] [PubMed]
- Muthumani, K.; Hwang, D.S.; Desai, B.M.; Zhang, D.; Dayes, N.; Green, D.R.; Weiner, D.B. HIV-1 Vpr induces apoptosis through caspase 9 in T cells and peripheral blood mononuclear cells. J. Biol. Chem. 2002, 277, 37820–37831. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, A.; Nonnemacher, M.; Wigdahl, B. Cellular phenotype impacts human immunodeficiency virus type 1 viral protein R subcellular localization. Virol. J. 2011, 8, 397. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.A.; Ehrlich, L.S. Cell Biology of HIV-1 Infection of Macrophages. Annu. Rev. Microbiol. 2008, 62, 425–443. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.D.; Rota, T.R.; Hirsch, M.S. Infection of monocyte/macrophages by human T lymphotropic virus type III. J. Clin. Investig. 1986, 77, 1712–1715. [Google Scholar] [CrossRef] [PubMed]
- Coleman, C.M.; Wu, L. HIV interactions with monocytes and dendritic cells: Viral latency and reservoirs. Retrovirology 2009, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Reynoso, R.; Wieser, M.; Ojeda, D.; Bönisch, M.; Kühnel, H.; Bolcic, F.; Quendler, H.; Grillari, J.; Grillari-Voglauer, R.; Quarleri, J. HIV-1 induces telomerase activity in monocyte-derived macrophages, possibly safeguarding one of its reservoirs. J. Virol. 2012, 86, 10327–10337. [Google Scholar] [CrossRef] [PubMed]
- Swingler, S.; Mann, A.M.; Zhou, J.; Swingler, C.; Stevenson, M. Apoptotic killing of HIV-1-infected macrophages is subverted by the viral envelope glycoprotein. PLoS Pathog. 2007, 3, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Codogno, P.; Mehrpour, M.; Proikas-Cezanne, T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat. Rev. Mol. Cell Biol. 2012, 13, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.G.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Florey, O.; Kim, S.E.; Sandoval, C.P.; Haynes, C.M.; Overholtzer, M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat. Cell Biol. 2011, 13, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Almendinger, J.; Oberst, A.; Ness, R.; Dillon, C.P.; Fitzgerald, P.; Hengartner, M.O.; Green, D.R. Microtubule-associates protein 1 light chain 3 α (LC3)-associated phagocytosis is required for the ef fi cient clearance of dead cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17396–17401. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Malireddi, R.K.S.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.-L.; Tan, H.; Peng, J.; et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 2015, 17, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Cullinane, M.; Treerat, P.; Ramm, G.; Prescott, M.; Adler, B.; Boyce, J.D.; Devenish, R.J. The Burkholderia pseudomallei Type III Secretion System and BopA Are Required for Evasion of LC3-Associated Phagocytosis. PLoS ONE 2011, 6, e17852. [Google Scholar] [CrossRef] [PubMed]
- Lam, G.Y.; Cemma, M.; Muise, A.M.; Higgins, D.E.; Brumell, J.H. Host and bacterial factors that regulate LC3 recruitment to Listeria monocytogenes during the early stages of macrophage infection. Autophagy 2013, 9, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Baxt, L.A.; Goldberg, M.B. Host and bacterial proteins that repress recruitment of LC3 to Shigella early during infection. PLoS ONE 2014, 9, e94653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligeon, L.-A.; Moreau, K.; Barois, N.; Bongiovanni, A.; Lacorre, D.-A.; Werkmeister, E.; Proux-Gillardeaux, V.; Galli, T.; Lafont, F. Role of VAMP3 and VAMP7 in the commitment of Yersinia pseudotuberculosis to LC3-associated pathways involving single- or double-membrane vacuoles. Autophagy 2014, 10, 1588–1602. [Google Scholar] [CrossRef] [PubMed]
- Abnave, P.; Mottola, G.; Gimenez, G.; Boucherit, N.; Trouplin, V.; Torre, C.; Conti, F.; Ben Amara, A.; Lepolard, C.; Djian, B.; et al. Screening in planarians identifies MORN2 as a key component in LC3-associated phagocytosis and resistance to bacterial infection. Cell Host Microbe 2014, 16, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Mimouna, S.; Bazin, M.; Mograbi, B.; Darfeuille-Michaud, A.; Brest, P.; Hofman, P.; Vouret-Craviari, V. HIF1A regulates xenophagic degradation of adherent and invasive Escherichia coli (AIEC). Autophagy 2014, 10, 2333–2345. [Google Scholar] [CrossRef] [PubMed]
- Hubber, A.; Kubori, T.; Coban, C.; Matsuzawa, T.; Ogawa, M.; Kawabata, T.; Yoshimori, T.; Nagai, H. Bacterial secretion system skews the fate of Legionella-containing vacuoles towards LC3-associated phagocytosis. Sci. Rep. 2017, 7, 44795. [Google Scholar] [CrossRef] [PubMed]
- Deen, N.S.; Gong, L.; Naderer, T.; Devenish, R.J.; Kwok, T. Analysis of the Relative Contribution of Phagocytosis, LC3-Associated Phagocytosis, and Canonical Autophagy During Helicobacter pylori Infection of Macrophages. Helicobacter 2015, 20, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.M.; Mansour, M.K.; Khan, N.S.; Seward, M.; Puranam, S.; Tanne, A.; Sokolovska, A.; Becker, C.E.; Acharya, M.; Baird, M.A.; et al. Dectin-1–Dependent LC3 Recruitment to Phagosomes Enhances Fungicidal Activity in Macrophages. J. Infect. Dis. 2014, 210, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, V.; Moretti, S.; Renga, G.; Galosi, C.; Borghi, M.; Pariano, M.; Puccetti, M.; Palmerini, C.A.; Amico, L.; Carotti, A.; et al. Noncanonical Fungal Autophagy Inhibits Inflammation in Response to IFN-γ via DAPK1. Cell Host Microbe 2016, 20, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Akoumianaki, T.; Kyrmizi, I.; Valsecchi, I.; Gresnigt, M.S.; Samonis, G.; Drakos, E.; Boumpas, D.; Muszkieta, L.; Prevost, M.-C.; Kontoyiannis, D.P.; et al. Aspergillus Cell Wall Melanin Blocks LC3-Associated Phagocytosis to Promote Pathogenicity. Cell Host Microbe 2016, 19, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Boonhok, R.; Rachaphaew, N.; Duangmanee, A.; Chobson, P.; Pattaradilokrat, S.; Utaisincharoen, P.; Sattabongkot, J.; Ponpuak, M. LAP-like process as an immune mechanism downstream of IFN-γ in control of the human malaria Plasmodium vivax liver stage. Proc. Natl. Acad. Sci. USA 2016, 113, e3519–e3528. [Google Scholar] [CrossRef] [PubMed]
- Crauwels, P.; Bohn, R.; Thomas, M.; Gottwalt, S.; Jäckel, F.; Krämer, S.; Bank, E.; Tenzer, S.; Walther, P.; Bastian, M.; et al. Apoptotic-like Leishmania exploit the host’s autophagy machinery to reduce T-cell-mediated parasite elimination. Autophagy 2015, 11, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Matte, C.; Casgrain, P.-A.; Séguin, O.; Moradin, N.; Hong, W.J.; Descoteaux, A. Leishmania major Promastigotes Evade LC3-Associated Phagocytosis through the Action of GP63. PLoS Pathog. 2016, 12, e1005690. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.M.; Crowe, S.M.; Mak, J. Lipid rafts and HIV-1: From viral entry to assembly of progeny virions. J. Clin. Virol. 2001, 22, 217–227. [Google Scholar] [CrossRef]
- Rollason, R.; Korolchuk, V.; Hamilton, C.; Jepson, M.; Banting, G. A CD317/tetherin-RICH2 complex plays a critical role in the organization of the subapical actin cytoskeleton in polarized epithelial cells. J. Cell Biol. 2009, 184, 721–736. [Google Scholar] [CrossRef] [PubMed]
- Billcliff, P.G.; Rollason, R.; Prior, I.; Owen, D.M.; Gaus, K.; Banting, G. CD317/tetherin is an organiser of membrane microdomains. J. Cell Sci. 2013, 126, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Rollason, R.; Dunstan, K.; Billcliff, P.G.; Bishop, P.; Gleeson, P.; Wise, H.; Digard, P.; Banting, G. Expression of HIV-1 Vpu Leads to Loss of the Viral Restriction Factor CD317/Tetherin from Lipid Rafts and Its Enhanced Lysosomal Degradation. PLoS ONE 2013, 8, e75680. [Google Scholar] [CrossRef] [PubMed]
- Dubé, M.; Bhusan Roy, B.; Guiot-Guillain, P.; Binette, J.; Mercier, J.; Chiasson, A.; Cohen, É.A. Antagonism of Tetherin Restriction of HIV-1 Release by Vpu Involves Binding and Sequestration of the Restriction Factor in a Perinuclear Compartment. PLoS Pathog. 2010, 6, e1000856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 2009, 5, e1000450. [Google Scholar] [CrossRef] [PubMed]
- Janvier, K.; Pelchen-Matthews, A.; Renaud, J.-B.; Caillet, M.; Marsh, M.; Berlioz-Torrent, C. The ESCRT-0 component HRS is required for HIV-1 Vpu-mediated BST-2/tetherin down-regulation. PLoS Pathog. 2011, 7, e1001265. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Pacini, G.; Berlioz-Torrent, C.; Janvier, K. Characterization of E3 ligases involved in lysosomal sorting of the HIV-1 restriction factor BST2. J. Cell Sci. 2017, 130, 1596–1611. [Google Scholar] [CrossRef] [PubMed]
- Kueck, T.; Neil, S.J.D. A cytoplasmic tail determinant in HIV-1 Vpu mediates targeting of tetherin for endosomal degradation and counteracts interferon-induced restriction. PLoS Pathog. 2012, 8, e1002609. [Google Scholar] [CrossRef] [PubMed]
- Kueck, T.; Foster, T.L.; Weinelt, J.; Sumner, J.C.; Pickering, S.; Neil, S.J.D. Serine Phosphorylation of HIV-1 Vpu and Its Binding to Tetherin Regulates Interaction with Clathrin Adaptors. PLoS Pathog. 2015, 11, e1005141. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Weber, E.; Tokarev, A.; Lewinski, M.; Rizk, M.; Suarez, M.; Guatelli, J.; Xiong, Y. Structural basis of HIV-1 Vpu-mediated BST2 antagonism via hijacking of the clathrin adaptor protein complex 1. eLife 2014, 3, e02362. [Google Scholar] [CrossRef] [PubMed]
- McNatt, M.W.; Zang, T.; Bieniasz, P.D. Vpu Binds Directly to Tetherin and Displaces It from Nascent Virions. PLoS Pathog. 2013, 9, e1003299. [Google Scholar] [CrossRef] [PubMed]
- Lewinski, M.K.; Jafari, M.; Zhang, H.; Opella, S.J.; Guatelli, J. Membrane anchoring by a C-terminal tryptophan enables HIV-1 Vpu to displace bone marrow stromal antigen 2 (BST2) from sites of viral assembly. J. Biol. Chem. 2015, 290, 10919–10933. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Guatelli, J.; Lewinski, M.K. Activities of transmitted/founder and chronic clade B HIV-1 Vpu and a C-terminal polymorphism specifically affecting virion release. J. Virol. 2014, 88, 5062–5078. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| HIV-1 Proteins | Non-Autophagic Functions | Autophagic Functions | References (Associated with Autophagy) |
|---|---|---|---|
| Env (gp120 and gp41) | Binds to HIV-1 receptor and coreceptors; mediates fusion between virus envelope and plasma membrane. | Induces apoptosis in CD4+ T cell; impairs dendritic cells-mediated CD4+ T cell response; increases HIV-1 spreading from dendritic cells to CD4+ T cells; reduces TLR signaling response; upregulates autophagy in neurons. | [92,95,96,97,98,99,100] |
| Tat | Regulates HIV-1 and cellular genes transcription; upregulates cell surface CXCR4 expression in uninfected CD4+ T cells; mediates T cell activation; impedes CD4+ and CD8+ maturation; alters mature CD8+ activity; modulates apoptosis. | Blocks IFNγ-mediated autophagy in macrophages/monocytes; inhibits autophagyin uninfected macrophages/monocytes by stimulating Src-Akt signaling pathway; alters autophagosome-lysosomes fusion in neurons. | [101,102,103,104,105,106,107] |
| Nef | Downregulates cell surface expression of CD4, MHC-I, MHC-II, CD28, T cell receptor/CD3 complex; prevents incorporation of restriction factors SERINC3/5 in budding viruses; modulates CD4+ T cell activation. | Prevents autophagy-mediated HIV-1 degradation by blocking autophagosome maturation in macrophages and astrocytes; blocks nuclear translocation of the pro-autophagy transcription factor TFEB in macrophages. | [47,108,109,110] |
| Vpr | Induces apoptosis in infected and/or uninfected T cells, DCs, monocytes and neurons; blocks the cell cycle at G2 phase; reactivates viral expression in latently infected cells; influences reverse transcription accuracy; regulates nuclear import of the HIV-1 pre-integration complex. | Prevents apoptosis in macrophages by diverting early steps of autophagy. | [111] |
| Vpu | Induces CD4 degradation; downregulates cell surface expression of MHC-I, CD1d, CCR7, CD40, CD81, SLAMF6; induces degradation of the restriction factor BST2; induces mistrafficking of BST2; drives displacing of BST2 from virus budding sites; depolarizes membrane potential by forming ion channel. | Drives displacing of BST2 from virus budding sites via a non-canonical autophagy mechanism (LC3-associated phagocytosis). | [112] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leymarie, O.; Lepont, L.; Berlioz-Torrent, C. Canonical and Non-Canonical Autophagy in HIV-1 Replication Cycle. Viruses 2017, 9, 270. https://doi.org/10.3390/v9100270
Leymarie O, Lepont L, Berlioz-Torrent C. Canonical and Non-Canonical Autophagy in HIV-1 Replication Cycle. Viruses. 2017; 9(10):270. https://doi.org/10.3390/v9100270
Chicago/Turabian StyleLeymarie, Olivier, Leslie Lepont, and Clarisse Berlioz-Torrent. 2017. "Canonical and Non-Canonical Autophagy in HIV-1 Replication Cycle" Viruses 9, no. 10: 270. https://doi.org/10.3390/v9100270
APA StyleLeymarie, O., Lepont, L., & Berlioz-Torrent, C. (2017). Canonical and Non-Canonical Autophagy in HIV-1 Replication Cycle. Viruses, 9(10), 270. https://doi.org/10.3390/v9100270