Autophagy in Measles Virus Infection

Abstract
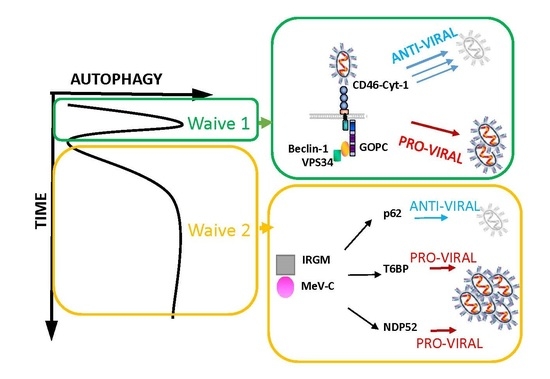
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
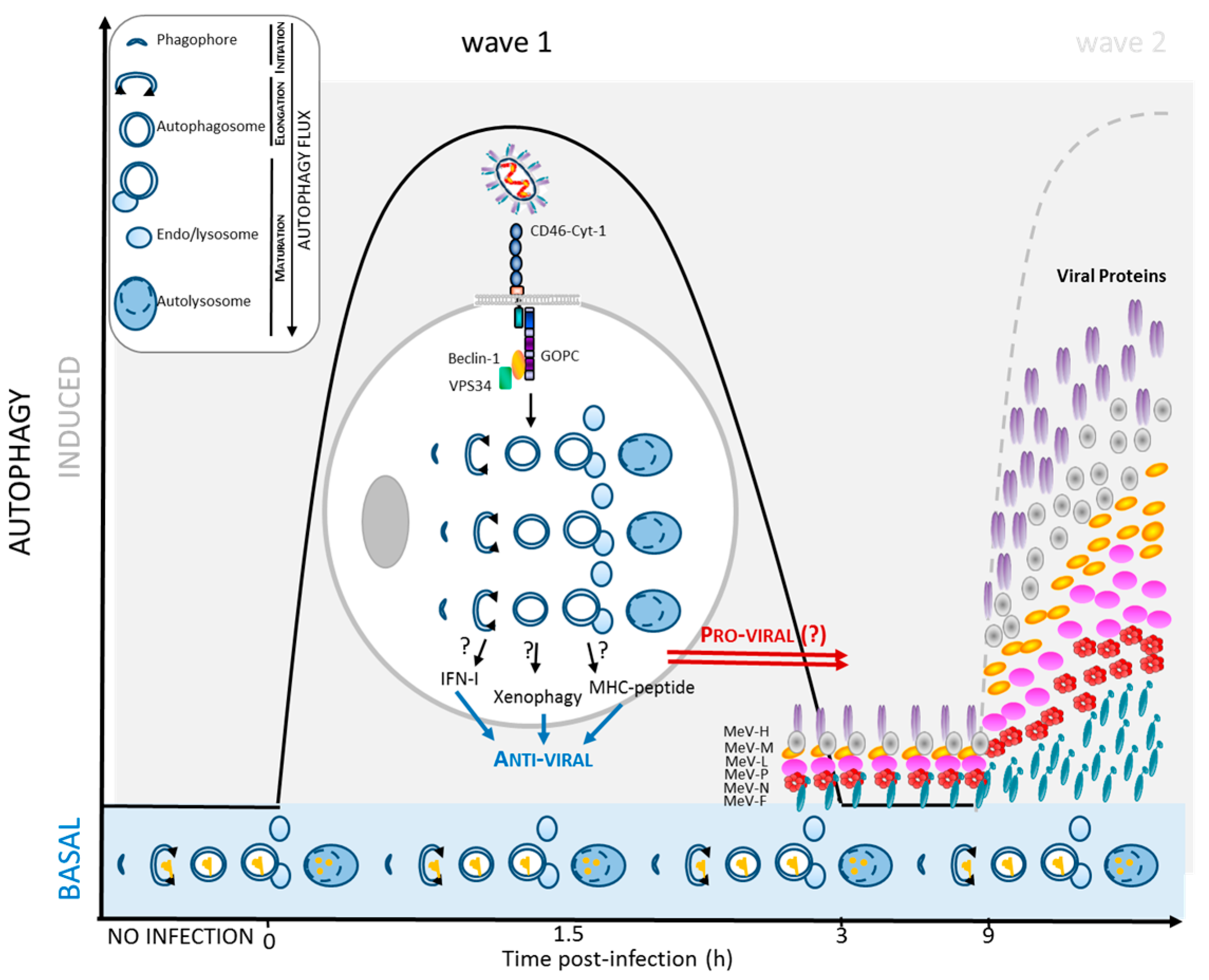
2. Autophagy Induction in MeV Infection
2.1. Autophagy Detection upon MeV Infection
2.2. MeV Entry and Early Autophagy Induction
2.3. MeV Replication and Late Autophagy Induction
2.4. MeV Replication and Very Late Autophagy Induction
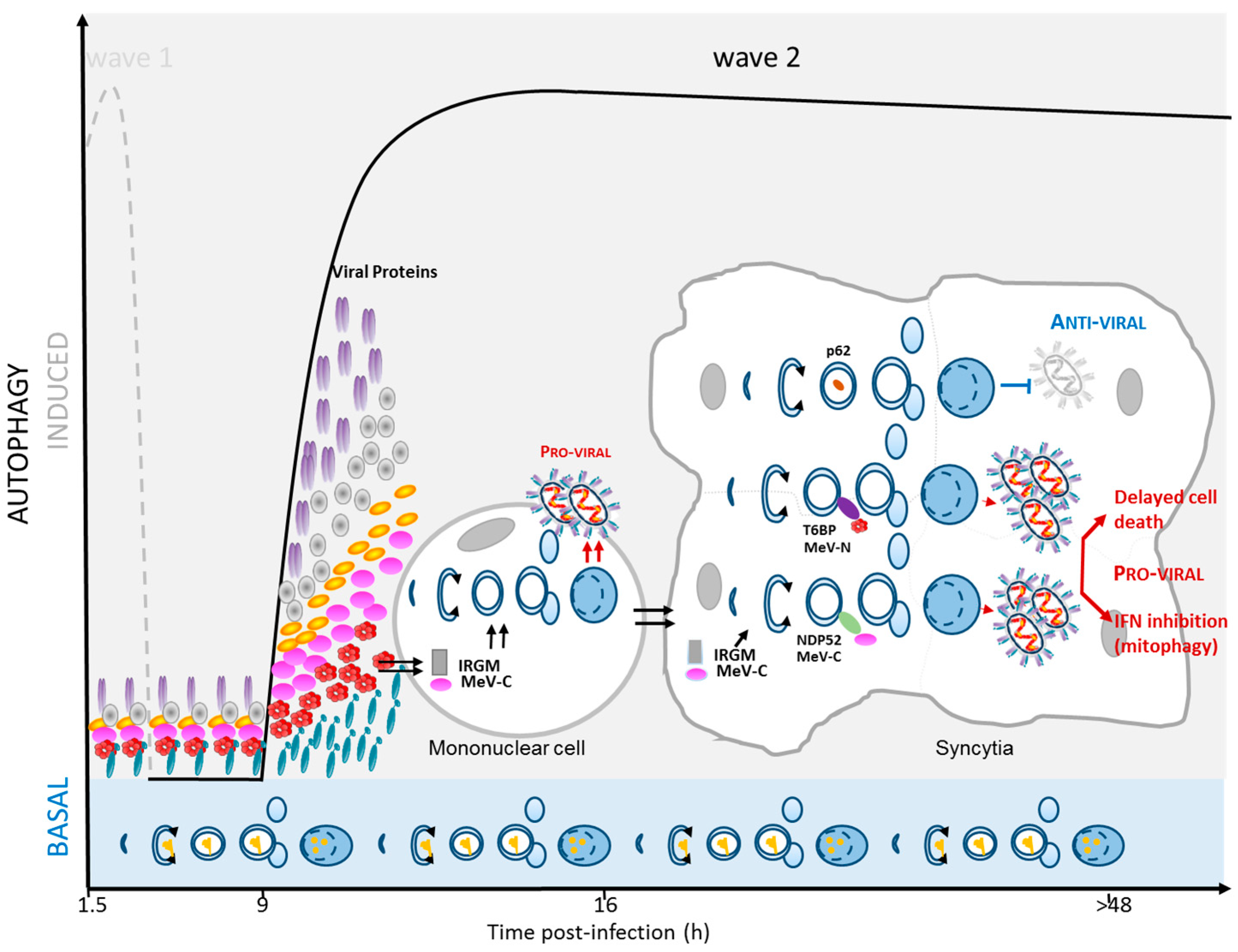
3. Autophagy as a Pro-MeV Mechanism
3.1. Autophagy Induction Promotes MeV Particle Production
3.2. Mitophagy Promotes MeV Replication
3.3. Autophagy Maturation Is Required for Efficient MeV Particle Production
4. Autophagy as an Anti-MeV Cellular Mechanism
4.1. Autophagy Contribution to Anti-MeV Immune Responses
4.2. Induction of Intrinsic and Innate Anti-Viral Responses
4.3. Induction of Anti-Viral Adaptive Immune Response
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Moss, W.J.; Griffin, D.E. Measles. Lancet 2012, 379, 153–164. [Google Scholar] [CrossRef]
- Griffin, D.E. Measles virus-induced suppression of immune responses. Immunol. Rev. 2010, 236, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Noyce, R.S.; Bondre, D.G.; Ha, M.N.; Lin, L.T.; Sisson, G.; Tsao, M.S.; Richardson, C.D. Tumor cell marker pvrl4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog. 2011, 7, e1002240. [Google Scholar] [CrossRef] [PubMed]
- Tatsuo, H.; Ono, N.; Tanaka, K.; Yanagi, Y. Slam (CDw150) is a cellular receptor for measles virus. Nature 2000, 406, 893–897. [Google Scholar] [PubMed]
- Muhlebach, M.D.; Mateo, M.; Sinn, P.L.; Prufer, S.; Uhlig, K.M.; Leonard, V.H.; Navaratnarajah, C.K.; Frenzke, M.; Wong, X.X.; Sawatsky, B.; et al. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 2011, 480, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Naniche, D.; Varior-Krishnan, G.; Cervoni, F.; Wild, T.F.; Rossi, B.; Rabourdin-Combe, C.; Gerlier, D. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J. Virol. 1993, 67, 6025–6032. [Google Scholar] [PubMed]
- Lamb, R.A.; Jardetzky, T.S. Structural basis of viral invasion: Lessons from paramyxovirus F. Curr. Opin. Struct. Biol. 2007, 17, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Faure, M.; Rabourdin-Combe, C. Innate immunity modulation in virus entry. Curr. Opin. Virol. 2011, 1, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Faure, M.; Lafont, F. Pathogen-induced autophagy signaling in innate immunity. J. Innate Immun. 2013, 5, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [PubMed]
- Richetta, C.; Gregoire, I.P.; Verlhac, P.; Azocar, O.; Baguet, J.; Flacher, M.; Tangy, F.; Rabourdin-Combe, C.; Faure, M. Sustained autophagy contributes to measles virus infectivity. PLoS Pathog. 2013, 9, e1003599. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar] [CrossRef] [PubMed]
- Joubert, P.E.; Meiffren, G.; Gregoire, I.P.; Pontini, G.; Richetta, C.; Flacher, M.; Azocar, O.; Vidalain, P.O.; Vidal, M.; Lotteau, V.; et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 2009, 6, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Gonzalez, P.; Li, C.; Meng, G.; Jiang, A.; Wang, H.; Gao, Q.; Debatin, K.M.; Beltinger, C.; Wei, J. Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. J. Virol. 2014, 88, 5152–5164. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Fara, A.F.; Dasgupta, P.; Kemper, C. CD46: The “multitasker” of complement proteins. Int. J. Biochem. Cell Biol. 2013, 45, 2808–2820. [Google Scholar] [CrossRef] [PubMed]
- Meiffren, G.; Flacher, M.; Azocar, O.; Rabourdin-Combe, C.; Faure, M. Cutting edge: Abortive proliferation of CD46-induced Tr1-like cells due to a defective Akt/Survivin signaling pathway. J. Immunol. 2006, 177, 4957–4961. [Google Scholar] [CrossRef] [PubMed]
- Kemper, C.; Chan, A.C.; Green, J.M.; Brett, K.A.; Murphy, K.M.; Atkinson, J.P. Activation of human CD4+ cells with CD3 and CD46 induces a T-regulatory cell 1 phenotype. Nature 2003, 421, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Purcell, D.F.; Russell, S.M.; Deacon, N.J.; Brown, M.A.; Hooker, D.J.; McKenzie, I.F. Alternatively spliced rnas encode several isoforms of CD46 (MCP), a regulator of complement activation. Immunogenetics 1991, 33, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Crimeen-Irwin, B.; Ellis, S.; Christiansen, D.; Ludford-Menting, M.J.; Milland, J.; Lanteri, M.; Loveland, B.E.; Gerlier, D.; Russell, S.M. Ligand binding determines whether CD46 is internalized by clathrin-coated pits or macropinocytosis. J. Biol. Chem. 2003, 278, 46927–46937. [Google Scholar] [CrossRef] [PubMed]
- Meiffren, G.; Joubert, P.E.; Gregoire, I.P.; Codogno, P.; Rabourdin-Combe, C.; Faure, M. Pathogen recognition by the cell surface receptor CD46 induces autophagy. Autophagy 2010, 6, 299–300. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Singh, S.; de Haro, S.; Master, S.; Ponpuak, M.; Dinkins, C.; Ornatowski, W.; Vergne, I.; Deretic, V. Autophagy and pattern recognition receptors in innate immunity. Immunol. Rev. 2009, 227, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Plumet, S.; Herschke, F.; Bourhis, J.M.; Valentin, H.; Longhi, S.; Gerlier, D. Cytosolic 5′-triphosphate ended viral leader transcript of measles virus as activator of the RIG I-mediated interferon response. PLoS ONE 2007, 2, e279. [Google Scholar] [CrossRef] [PubMed]
- Guirimand, T.; Delmotte, S.; Navratil, V. Virhostnet 2.0: Surfing on the web of virus/host molecular interactions data. Nucleic Acids Res. 2015, 43, D583–587. [Google Scholar] [CrossRef] [PubMed]
- Petkova, D.S.; Verlhac, P.; Rozieres, A.; Baguet, J.; Claviere, M.; Kretz-Remy, C.; Mahieux, R.; Viret, C.; Faure, M. Distinct contributions of autophagy receptors in measles virus replication. Viruses 2017, 9, 123. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, I.P.; Rabourdin-Combe, C.; Faure, M. Autophagy and RNA virus interactomes reveal IRGM as a common target. Autophagy 2012, 8, 1136–1137. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Davis, A.S.; Taylor, G.A.; Deretic, V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 2006, 313, 1438–1441. [Google Scholar] [CrossRef] [PubMed]
- Lapaquette, P.; Glasser, A.L.; Huett, A.; Xavier, R.J.; Darfeuille-Michaud, A. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell. Microbiol. 2010, 12, 99–113. [Google Scholar] [CrossRef] [PubMed]
- McCarroll, S.A.; Huett, A.; Kuballa, P.; Chilewski, S.D.; Landry, A.; Goyette, P.; Zody, M.C.; Hall, J.L.; Brant, S.R.; Cho, J.H.; et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat. Genet. 2008, 40, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Brest, P.; Lapaquette, P.; Souidi, M.; Lebrigand, K.; Cesaro, A.; Vouret-Craviari, V.; Mari, B.; Barbry, P.; Mosnier, J.F.; Hebuterne, X.; et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat. Genet. 2011, 43, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.; Mandell, M.A.; Deretic, V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol. Cell 2015, 58, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Petkova, D.S.; Viret, C.; Faure, M. IRGM in autophagy and viral infections. Front. Immunol. 2012, 3, 426. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; He, X.; Zhu, G.; Tu, H.; Liu, Z.; Li, W.; Han, S.; Yin, J.; Peng, B.; Liu, W. Coxsackievirus A16 elicits incomplete autophagy involving the mTOR and ERK pathways. PLoS ONE 2015, 10, e0122109. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.D.; Johnsen, I.B.; Stiberg, K.A.; Sherstova, T.; Wakita, T.; Richard, G.M.; Kandasamy, R.K.; Meurs, E.F.; Anthonsen, M.W. Hepatitis C virus triggers Golgi fragmentation and autophagy through the immunity-related GTPase M. Proc. Natl. Acad. Sci. USA 2017, 114, E3462–E3471. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hou, L.; Du, J.; Zhou, L.; Ge, X.; Guo, X.; Yang, H. Capsid, membrane and NS3 are the major viral proteins involved in autophagy induced by japanese encephalitis virus. Vet. Microbiol. 2015, 178, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Jung, C.H.; Seo, M.; Kim, E.K.; Park, J.M.; Bae, S.S.; Kim, D.H. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol. Cell 2015, 57, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Delpeut, S.; Rudd, P.A.; Labonte, P.; von Messling, V. Membrane fusion-mediated autophagy induction enhances morbillivirus cell-to-cell spread. J. Virol. 2012, 86, 8527–8535. [Google Scholar] [CrossRef] [PubMed]
- Herschke, F.; Plumet, S.; Duhen, T.; Azocar, O.; Druelle, J.; Laine, D.; Wild, T.F.; Rabourdin-Combe, C.; Gerlier, D.; Valentin, H. Cell-cell fusion induced by measles virus amplifies the type I interferon response. J. Virol. 2007, 81, 12859–12871. [Google Scholar] [CrossRef] [PubMed]
- Baertsch, M.A.; Leber, M.F.; Bossow, S.; Singh, M.; Engeland, C.E.; Albert, J.; Grossardt, C.; Jager, D.; von Kalle, C.; Ungerechts, G. MicroRNA-mediated multi-tissue detargeting of oncolytic measles virus. Cancer Gene Ther. 2014, 21, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.; Fesler, A.; Ju, J. MicroRNA: A third dimension in autophagy. Cell Cycle 2013, 12, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Frankel, L.B.; Lubas, M.; Lund, A.H. Emerging connections between RNA and autophagy. Autophagy 2017, 13, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, Y.; Tan, X.; Jing, H. MicroRNAs in autophagy and their emerging roles in crosstalk with apoptosis. Autophagy 2012, 8, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Kissova, I.; Salin, B.; Schaeffer, J.; Bhatia, S.; Manon, S.; Camougrand, N. Selective and non-selective autophagic degradation of mitochondria in yeast. Autophagy 2007, 3, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Zhang, L.; Li, Z.; Zhong, Y.; Tang, Q.; Qin, Y.; Chen, M. The matrix protein of human parainfluenza virus type 3 induces mitophagy that suppresses interferon responses. Cell Host Microbe 2017, 21, 538–547.e4. [Google Scholar] [CrossRef] [PubMed]
- Faure, M. The p value of HPIV3-mediated autophagy inhibition. Cell Host Microbe 2014, 15, 519–521. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Meng, G.; Jiang, A.; Chen, A.; Dahlhaus, M.; Gonzalez, P.; Beltinger, C.; Wei, J. Mitophagy switches cell death from apoptosis to necrosis in NSCLC cells treated with oncolytic measles virus. Oncotarget 2014, 5, 3907–3918. [Google Scholar] [CrossRef] [PubMed]
- Verlhac, P.; Gregoire, I.P.; Azocar, O.; Petkova, D.S.; Baguet, J.; Viret, C.; Faure, M. Autophagy receptor NDP52 regulates pathogen-containing autophagosome maturation. Cell Host Microbe 2015, 17, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Tumbarello, D.A.; Manna, P.T.; Allen, M.; Bycroft, M.; Arden, S.D.; Kendrick-Jones, J.; Buss, F. Correction: The autophagy receptor TAX1BP1 and the molecular motor myosin VI are required for clearance of salmonella typhimurium by autophagy. PLoS Pathog. 2016, 12, e1005433. [Google Scholar] [CrossRef] [PubMed]
- Verlhac, P.; Viret, C.; Faure, M. Dual function of CALCOCO2/NDP52 during xenophagy. Autophagy 2015, 11, 965–966. [Google Scholar] [CrossRef] [PubMed]
- Verlhac, P.; Viret, C.; Faure, M. Handcuffs for bacteria—NDP52 orchestrates xenophagy of intracellular salmonella. Microb. Cell 2015, 2, 214–215. [Google Scholar] [CrossRef] [PubMed]
- Tumbarello, D.A.; Waxse, B.J.; Arden, S.D.; Bright, N.A.; Kendrick-Jones, J.; Buss, F. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat. Cell Biol. 2012, 14, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Munz, C. The autophagic machinery in viral exocytosis. Front. Microbiol. 2017, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Munz, C. Autophagy beyond intracellular MHC CLASS II antigen presentation. Trends Immunol. 2016, 37, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Richetta, C.; Faure, M. Autophagy in antiviral innate immunity. Cell. Microbiol. 2013, 15, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Talloczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Judith, D.; Mostowy, S.; Bourai, M.; Gangneux, N.; Lelek, M.; Lucas-Hourani, M.; Cayet, N.; Jacob, Y.; Prevost, M.C.; Pierre, P.; et al. Species-specific impact of the autophagy machinery on Chikungunya virus infection. EMBO Rep. 2013, 14, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef] [PubMed]
- Okabayashi, T.; Kojima, T.; Masaki, T.; Yokota, S.; Imaizumi, T.; Tsutsumi, H.; Himi, T.; Fujii, N.; Sawada, N. Type-III interferon, not type-I, is the predominant interferon induced by respiratory viruses in nasal epithelial cells. Virus Res. 2011, 160, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Pypaert, M.; Munz, C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Munz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Gerlier, D.; Trescol-Biemont, M.C.; Varior-Krishnan, G.; Naniche, D.; Fugier-Vivier, I.; Rabourdin-Combe, C. Efficient major histocompatibility complex class II-restricted presentation of measles virus relies on hemagglutinin-mediated targeting to its cellular receptor human CD46 expressed by murine B cells. J. Exp. Med. 1994, 179, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Gerlier, D.; Trescol-Biemont, M.C.; Varior-Krishnan, G.; Naniche, D.; Fugier-Vivier, I.; Rabourdin-Combe, C. Efficient mhc class II-restricted presentation of measles virus to T cells relies on its targeting to its cellular receptor human CD46 and involves an endosomal pathway. Cell Biol. Int. 1994, 18, 315–320. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rozières, A.; Viret, C.; Faure, M. Autophagy in Measles Virus Infection. Viruses 2017, 9, 359. https://doi.org/10.3390/v9120359
Rozières A, Viret C, Faure M. Autophagy in Measles Virus Infection. Viruses. 2017; 9(12):359. https://doi.org/10.3390/v9120359
Chicago/Turabian StyleRozières, Aurore, Christophe Viret, and Mathias Faure. 2017. "Autophagy in Measles Virus Infection" Viruses 9, no. 12: 359. https://doi.org/10.3390/v9120359
APA StyleRozières, A., Viret, C., & Faure, M. (2017). Autophagy in Measles Virus Infection. Viruses, 9(12), 359. https://doi.org/10.3390/v9120359