Subcellular Trafficking of the Papillomavirus Genome during Initial Infection: The Remarkable Abilities of Minor Capsid Protein L2

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
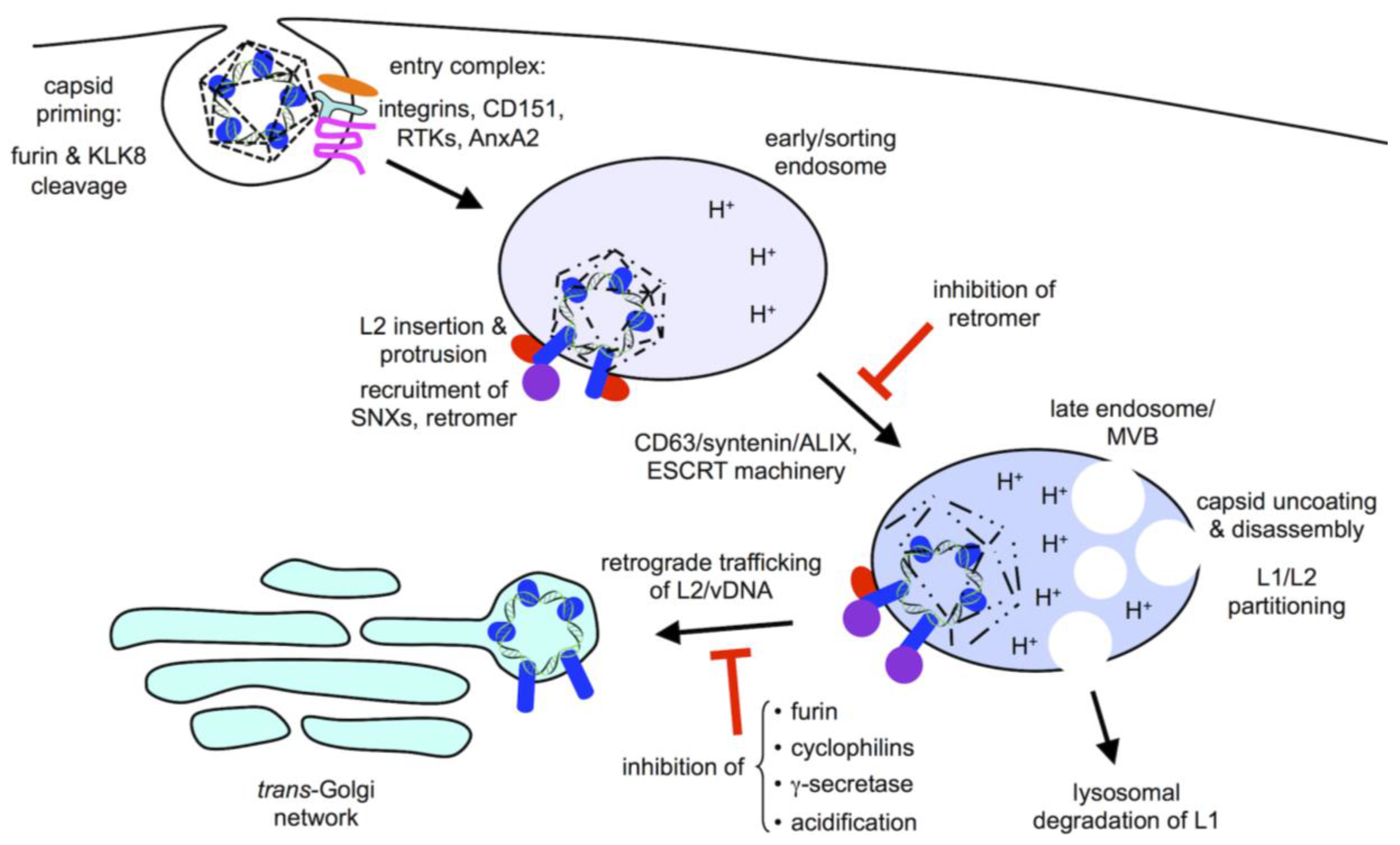
2. Viral Entry
3. Subcellular Trafficking
4. Retromer and Sorting Factors
5. γ-Secretase
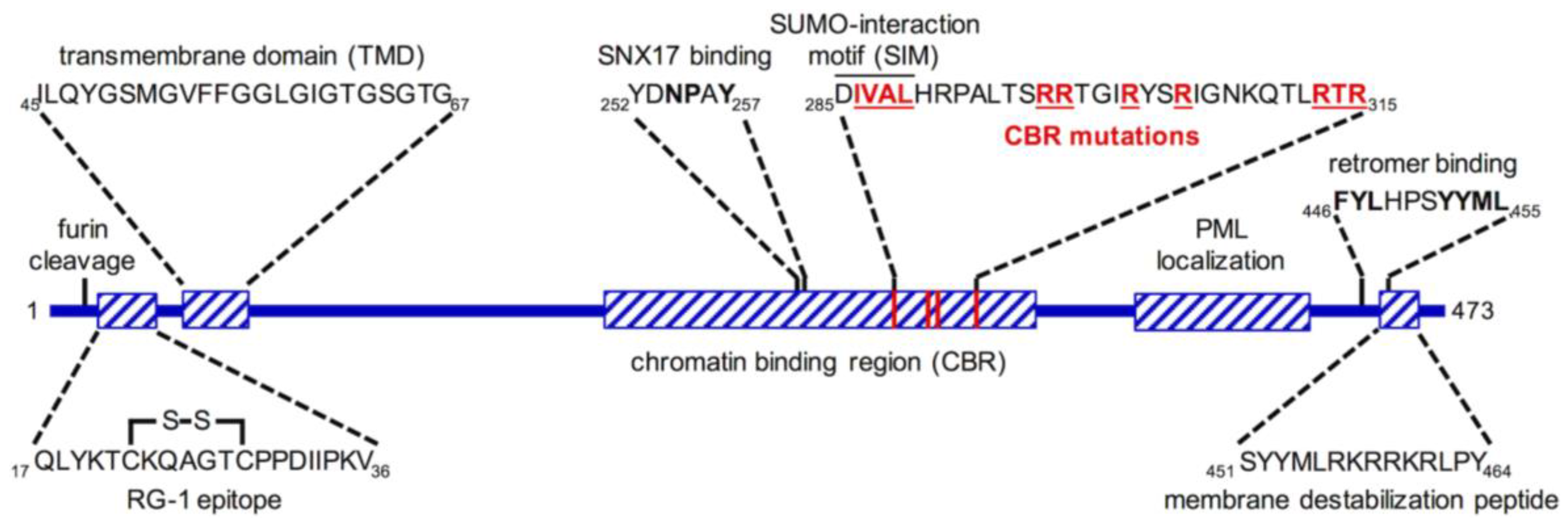
6. L2 Is an “Inducible Transmembrane” Protein
- (1)
- Insertion—Within the lumen, part(s) of L2 insert(s) into the local membrane.
- (2)
- Protrusion—L2 becomes a transmembrane protein. Part of L2 remains lumenal and is complexed with the vDNA, while other parts of L2 stick through the membrane and are accessible to cytosolic proteins.
- (3)
- Translocation—L2/vDNA exits vesicular compartments, passing across the limiting membrane to establish infection within the cell nucleus.
7. Insertion and Protrusion of L2
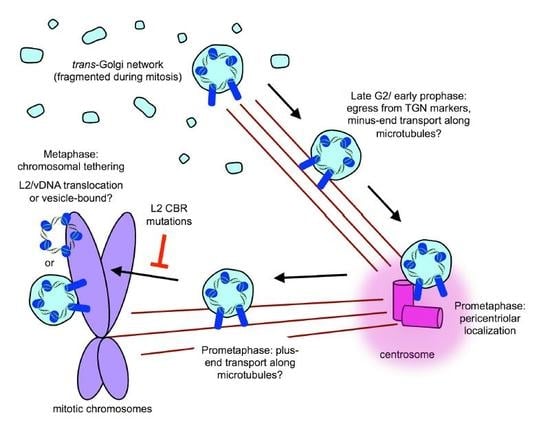
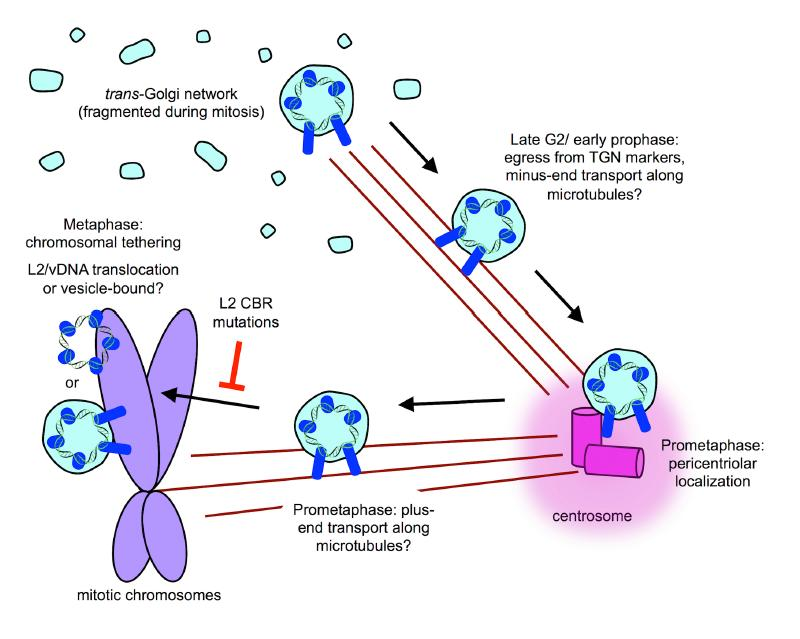
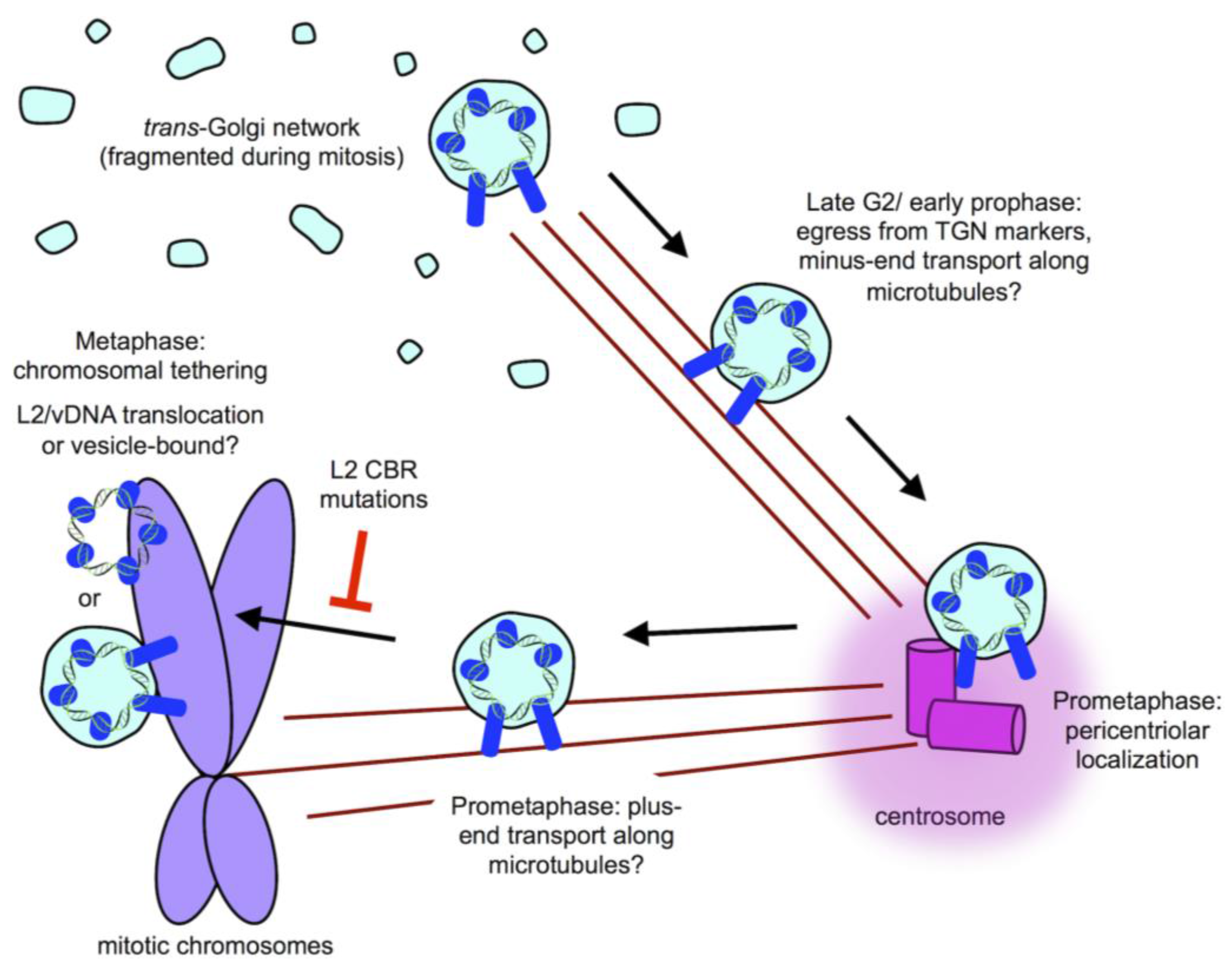
8. Post-TGN Transport- Mitosis, Translocation, and Chromatin Binding
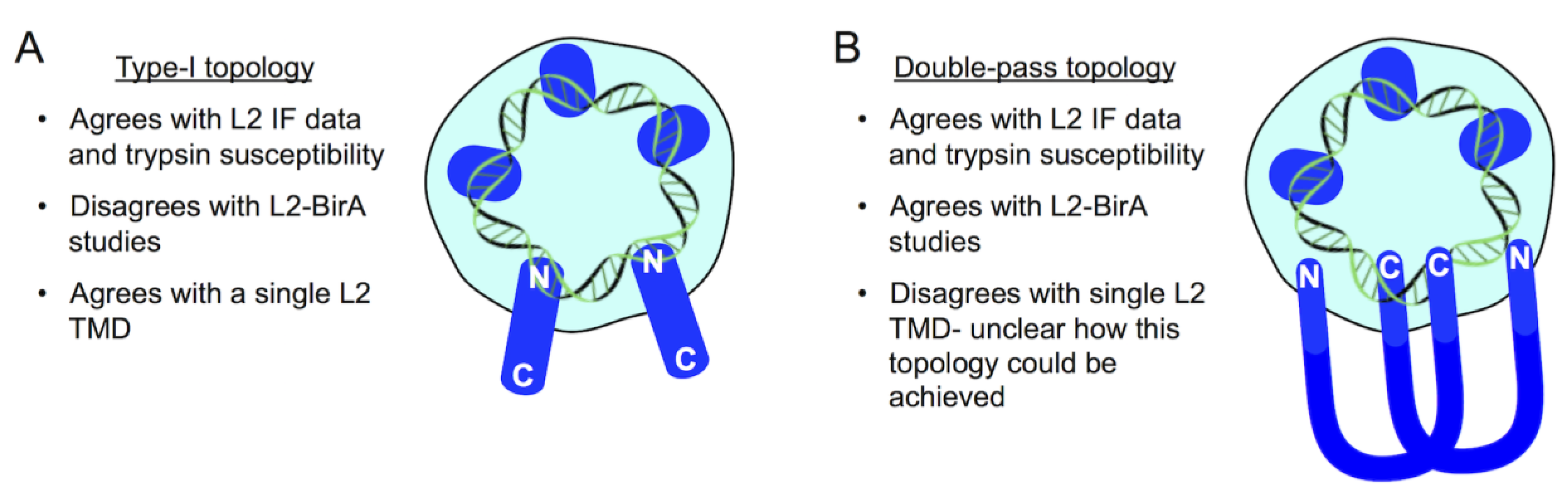
9. A Topology Conundrum
10. PML Bodies and Beyond
11. Conclusions & Future Directions
Acknowledgments
Conflicts of Interest
References
- Bzhalava, D.; Muhr, L.S.; Lagheden, C.; Ekstrom, J.; Forslund, O.; Dillner, J.; Hultin, E. Deep sequencing extends the diversity of human papillomaviruses in human skin. Sci. Rep. 2014, 4, 5807. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K. Evolution of the papillomaviridae. Virology 2013, 445, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K.; Li, Z.; Xirasagar, S.; Maes, P.; Kaminsky, D.; Liou, D.; Sun, Q.; Kaur, R.; Huyen, Y.; McBride, A.A. The papillomavirus episteme: A major update to the papillomavirus sequence database. Nucleic Acids Res. 2017, 45, D499–D506. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Forman, D.; de Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M.; et al. Global burden of human papillomavirus and related diseases. Vaccine 2012, 30 (Suppl. 5), F12–F23. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; de Sanjose, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Prim. 2016, 2, 16086. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Cheng, N.; Thompson, C.D.; Lowy, D.R.; Steven, A.C.; Schiller, J.T.; Trus, B.L. Arrangement of L2 within the papillomavirus capsid. J. Virol. 2008, 82, 5190–5197. [Google Scholar] [CrossRef] [PubMed]
- Bywaters, S.M.; Brendle, S.A.; Tossi, K.P.; Biryukov, J.; Meyers, C.; Christensen, N.D. Antibody competition reveals surface location of HPV L2 minor capsid protein residues 17–36. Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Pastrana, D.V.; Gambhira, R.; Buck, C.B.; Pang, Y.Y.; Thompson, C.D.; Culp, T.D.; Christensen, N.D.; Lowy, D.R.; Schiller, J.T.; Roden, R.B. Cross-neutralization of cutaneous and mucosal papillomavirus types with anti-sera to the amino terminus of L2. Virology 2005, 337, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Ishii, Y.; Ochi, H.; Matsumoto, T.; Yoshikawa, H.; Kanda, T. Neutralization of HPV16, 18, 31, and 58 pseudovirions with antisera induced by immunizing rabbits with synthetic peptides representing segments of the HPV16 minor capsid protein L2 surface region. Virology 2007, 358, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Roden, R.B. L2, the minor capsid protein of papillomavirus. Virology 2013, 445, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Bordeaux, J.; Forte, S.; Harding, E.; Darshan, M.S.; Klucevsek, K.; Moroianu, J. The L2 minor capsid protein of low-risk human papillomavirus type 11 interacts with host nuclear import receptors and viral DNA. J. Virol. 2006, 80, 8259–8262. [Google Scholar] [CrossRef] [PubMed]
- Fay, A.; Yutzy, W.H., IV; Roden, R.B.S.; Moroianu, J. The positively charged termini of L2 minor capsid protein required for bovine papillomavirus infection function separately in nuclear import and DNA binding. J. Virol. 2004, 78, 13447–13454. [Google Scholar] [CrossRef] [PubMed]
- Klucevsek, K.; Daley, J.; Darshan, M.S.; Bordeaux, J.; Moroianu, J. Nuclear import strategies of high-risk HPV18 L2 minor capsid protein. Virology 2006, 352, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Sun, X.Y.; Louis, K.; Frazer, I.H. Interaction of human papillomavirus (HPV) type 16 capsid proteins with HPV DNA requires an intact L2 N-terminal sequence. J. Virol. 1994, 68, 619–625. [Google Scholar] [PubMed]
- Buck, C.B.; Thompson, C.D. Production of papillomavirus-based gene transfer vectors. Curr. Protoc. Cell Biol. 2007. [Google Scholar] [CrossRef]
- Biryukov, J.; Meyers, C. Papillomavirus infectious pathways: A comparison of systems. Viruses 2015, 7, 4303–4325. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, P.; Gottschalk, E.Y.; Meneses, P.I. HPV entry into cells. Mutat. Res. Rev. Mutat. Res. 2017, 772, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Schelhaas, M. Concepts of papillomavirus entry into host cells. Curr. Opin. Virol. 2014, 4, 24–31. [Google Scholar] [CrossRef] [PubMed]
- DiGiuseppe, S.; Bienkowska-Haba, M.; Guion, L.G.; Sapp, M. Cruising the cellular highways: How human papillomavirus travels from the surface to the nucleus. Virus Res. 2017, 231, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.M.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, C.; Samperio Ventayol, P.; Vogeley, C.; Schelhaas, M. Kallikrein-8 proteolytically processes human papillomaviruses in the extracellular space to facilitate entry into host cells. J. Virol. 2015, 89, 7038–7052. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Thompson, C.D.; Schowalter, R.M.; Lowy, D.R.; Schiller, J.T. Identification of a role for the trans-Golgi network in human papillomavirus 16 pseudovirus infection. J. Virol. 2013, 87, 3862–3870. [Google Scholar] [CrossRef] [PubMed]
- Schelhaas, M.; Shah, B.; Holzer, M.; Blattmann, P.; Kuhling, L.; Day, P.M.; Schiller, J.T.; Helenius, A. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog. 2012, 8, e1002657. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, P.; Abban, C.Y.; Kiyashka, E.; Qiang, W.; Meneses, P.I. HPV16 infection of HaCaTs is dependent on β4 integrin, and α6 integrin processing. Virology 2014, 449, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Dziduszko, A.; Ozbun, M.A. Annexin A2 and S100A10 regulate human papillomavirus type 16 entry and intracellular trafficking in human keratinocytes. J. Virol. 2013, 87, 7502–7515. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, K.D.; Gawlitza, A.; Spoden, G.A.; Zhang, X.A.; Lambert, C.; Berditchevski, F.; Florin, L. Tetraspanin CD151 mediates papillomavirus type 16 endocytosis. J. Virol. 2013, 87, 3435–3446. [Google Scholar] [CrossRef] [PubMed]
- Spoden, G.; Freitag, K.; Husmann, M.; Boller, K.; Sapp, M.; Lambert, C.; Florin, L. Clathrin- and caveolin-independent entry of human papillomavirus type 16—Involvement of tetraspanin-enriched microdomains (tems). PLoS ONE 2008, 3, e3313. [Google Scholar] [CrossRef] [PubMed]
- Spoden, G.; Kuhling, L.; Cordes, N.; Frenzel, B.; Sapp, M.; Boller, K.; Florin, L.; Schelhaas, M. Human papillomavirus types 16, 18, and 31 share similar endocytic requirements for entry. J. Virol. 2013, 87, 7765–7773. [Google Scholar] [CrossRef] [PubMed]
- Surviladze, Z.; Dziduszko, A.; Ozbun, M.A. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog. 2012, 8, e1002519. [Google Scholar] [CrossRef] [PubMed]
- Woodham, A.W.; da Silva, D.M.; Skeate, J.G.; Raff, A.B.; Ambroso, M.R.; Brand, H.E.; Isas, J.M.; Langen, R.; Kast, W.M. The S100A10 subunit of the annexin A2 heterotetramer facilitates L2-mediated human papillomavirus infection. PLoS ONE 2012, 7, e43519. [Google Scholar] [CrossRef] [PubMed]
- Wustenhagen, E.; Hampe, L.; Boukhallouk, F.; Schneider, M.A.; Spoden, G.A.; Negwer, I.; Koynov, K.; Kast, W.M.; Florin, L. The cytoskeletal adaptor obscurin-like 1 interacts with the human papillomavirus 16 (HPV16) capsid protein L2 and is required for HPV16 endocytosis. J. Virol. 2016, 90, 10629–10641. [Google Scholar] [CrossRef] [PubMed]
- Earnest, J.T.; Hantak, M.P.; Li, K.; McCray, P.B., Jr.; Perlman, S.; Gallagher, T. The tetraspanin CD9 facilitates mers-coronavirus entry by scaffolding host cell receptors and proteases. PLoS Pathog. 2017, 13, e1006546. [Google Scholar] [CrossRef] [PubMed]
- Feneant, L.; Levy, S.; Cocquerel, L. CD81 and hepatitis c virus (HCV) infection. Viruses 2014, 6, 535–572. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Schiller, J.T. The role of furin in papillomavirus infection. Future Microbiol. 2009, 4, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Bronnimann, M.P.; Calton, C.M.; Chiquette, S.F.; Li, S.; Lu, M.; Chapman, J.A.; Bratton, K.N.; Schlegel, A.M.; Campos, S.K. Furin cleavage of L2 during papillomavirus infection: Minimal dependence on cyclophilins. J. Virol. 2016, 90, 6224–6234. [Google Scholar] [CrossRef] [PubMed]
- Gambhira, R.; Karanam, B.; Jagu, S.; Roberts, J.N.; Buck, C.B.; Bossis, I.; Alphs, H.H.; Culp, T.; Christensen, N.D.; Roden, R.B.S. A protective and broadly cross-neutralizing epitope of human papillomavirus L2. J. Virol. 2007, 81, 13927–13931. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Gambhira, R.; Roden, R.B.; Lowy, D.R.; Schiller, J.T. Mechanisms of human papillomavirus type 16 neutralization by L2 cross-neutralizing and L1 type-specific antibodies. J. Virol. 2008, 82, 4638–4646. [Google Scholar] [CrossRef] [PubMed]
- Bienkowska-Haba, M.; Patel, H.D.; Sapp, M. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog. 2009, 5, e1000524. [Google Scholar] [CrossRef] [PubMed]
- Gräßel, L.; Fast, L.A.; Scheffer, K.D.; Boukhallouk, F.; Spoden, G.A.; Tenzer, S.; Boller, K.; Bago, R.; Rajesh, S.; Overduin, M.; et al. The CD63-syntenin-1 complex controls post-endocytic trafficking of oncogenic human papillomaviruses. Sci. Rep. 2016, 6, 32337. [Google Scholar] [CrossRef] [PubMed]
- Broniarczyk, J.; Bergant, M.; Gozdzicka-Jozefiak, A.; Banks, L. Human papillomavirus infection requires the TSG101 component of the ESCRT machinery. Virology 2014, 460–461, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Broniarczyk, J.; Pim, D.; Massimi, P.; Bergant, M.; Gozdzicka-Jozefiak, A.; Crump, C.; Banks, L. The VPS4 component of the ESCRT machinery plays an essential role in hpv infectious entry and capsid disassembly. Sci. Rep. 2017, 7, 45159. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.H.; Spoden, G.A.; Scheffer, K.D.; Brunnhofer, R.; de Brabander, J.K.; Maier, M.E.; Florin, L.; Muller, C.P. Inhibition by cellular vacuolar ATPase impairs human papillomavirus uncoating and infection. Antimicrob. Agents Chemother. 2014, 58, 2905–2911. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Campos, S.K.; Wandinger-Ness, A.; Ozbun, M.A. Caveolin-1-dependent infectious entry of human papillomavirus type 31 in human keratinocytes proceeds to the endosomal pathway for pH-dependent uncoating. J. Virol. 2008, 82, 9505–9512. [Google Scholar] [CrossRef] [PubMed]
- Sapp, M.; Kraus, U.; Volpers, C.; Snijders, P.J.; Walboomers, J.M.; Streeck, R.E. Analysis of type-restricted and cross-reactive epitopes on virus-like particles of human papillomavirus type 33 and in infected tissues using monoclonal antibodies to the major capsid protein. J. Gen. Virol. 1994, 75 Pt 12, 3375–3383. [Google Scholar] [CrossRef] [PubMed]
- Bienkowska-Haba, M.; Williams, C.; Kim, S.M.; Garcea, R.L.; Sapp, M. Cyclophilins facilitate dissociation of the human papillomavirus type 16 capsid protein L1 from the L2/DNA complex following virus entry. J. Virol. 2012, 86, 9875–9887. [Google Scholar] [CrossRef] [PubMed]
- Lipovsky, A.; Popa, A.; Pimienta, G.; Wyler, M.; Bhan, A.; Kuruvilla, L.; Guie, M.A.; Poffenberger, A.C.; Nelson, C.D.; Atwood, W.J.; et al. Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc. Natl. Acad. Sci. USA 2013, 110, 7452–7457. [Google Scholar] [CrossRef] [PubMed]
- Popa, A.; Zhang, W.; Harrison, M.S.; Goodner, K.; Kazakov, T.; Goodwin, E.C.; Lipovsky, A.; Burd, C.G.; DiMaio, D. Direct binding of retromer to human papillomavirus type 16 minor capsid protein L2 mediates endosome exit during viral infection. PLoS Pathog. 2015, 11, e1004699. [Google Scholar] [CrossRef] [PubMed]
- Aydin, I.; Villalonga-Planells, R.; Greune, L.; Bronnimann, M.P.; Calton, C.M.; Becker, M.; Lai, K.Y.; Campos, S.K.; Schmidt, M.A.; Schelhaas, M. A central region in the minor capsid protein of papillomaviruses facilitates viral genome tethering and membrane penetration for mitotic nuclear entry. PLoS Pathog. 2017, 13, e1006308. [Google Scholar] [CrossRef] [PubMed]
- Aydin, I.; Weber, S.; Snijder, B.; Samperio Ventayol, P.; Kuhbacher, A.; Becker, M.; Day, P.M.; Schiller, J.T.; Kann, M.; Pelkmans, L.; et al. Large scale RNAi reveals the requirement of nuclear envelope breakdown for nuclear import of human papillomaviruses. PLoS Pathog. 2014, 10, e1004162. [Google Scholar] [CrossRef] [PubMed]
- Calton, C.M.; Bronnimann, M.P.; Manson, A.R.; Li, S.; Chapman, J.A.; Suarez-Berumen, M.; Williamson, T.R.; Molugu, S.K.; Bernal, R.A.; Campos, S.K. Translocation of the papillomavirus L2/vDNA complex across the limiting membrane requires the onset of mitosis. PLoS Pathog. 2017, 13, e1006200. [Google Scholar] [CrossRef] [PubMed]
- DiGiuseppe, S.; Bienkowska-Haba, M.; Guion, L.G.M.; Keiffer, T.R.; Sapp, M. Human papillomavirus major capsid protein L1 remains associated with the incoming viral genome throughout the entry process. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Nakahara, T.; Kataoka, M.; Kusumoto-Matsuo, R.; Mori, S.; Takeuchi, T.; Kukimoto, I. Identification of TrappC8 as a host factor required for human papillomavirus cell entry. PLoS ONE 2013, 8, e80297. [Google Scholar] [CrossRef] [PubMed]
- Lipovsky, A.; Zhang, W.; Iwasaki, A.; DiMaio, D. Application of the proximity-dependent assay and fluorescence imaging approaches to study viral entry pathways. Methods Mol. Biol. 2015, 1270, 437–451. [Google Scholar] [PubMed]
- Zhang, W.; Kazakov, T.; Popa, A.; DiMaio, D. Vesicular trafficking of incoming human papillomavirus 16 to the Golgi apparatus and endoplasmic reticulum requires γ-secretase activity. mBio 2014, 5, e01777-14. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Navarro, N.; Miller, E. Protein sorting at the ER-Golgi interface. J. Cell Biol. 2016, 215, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, J.; Duran, J.; Scarpa, M.; Bassaganyas, L.; van Galen, J.; Malhotra, V. Golgi enzymes do not cycle through the endoplasmic reticulum during protein secretion or mitosis. Mol. Biol. Cell 2017, 28, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Hurley, J.H. Retromer. Curr. Opin. Cell Biol. 2008, 20, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.; Cullen, P.J. Retromer: A master conductor of endosome sorting. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Bergant Marusic, M.; Ozbun, M.A.; Campos, S.K.; Myers, M.P.; Banks, L. Human papillomavirus L2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic 2012, 13, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Broniarczyk, J.; Bergant, M.; Playford, M.P.; Banks, L. A novel PDZ domain interaction mediates the binding between human papillomavirus 16 L2 and sorting nexin 27 and modulates virion trafficking. J. Virol. 2015, 89, 10145–10155. [Google Scholar] [CrossRef] [PubMed]
- Bergant, M.; Peternel, S.; Pim, D.; Broniarczyk, J.; Banks, L. Characterizing the spatio-temporal role of sorting nexin 17 in human papillomavirus trafficking. J. Gen. Virol. 2017, 98, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Liu, D.; Liu, N.; Xu, L.; Li, S.; Lin, S.; Shu, X.; Pei, D. SNX17 regulates notch pathway and pancreas development through the retromer-dependent recycling of jag1. Cell Regen. 2012, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- McNally, K.E.; Faulkner, R.; Steinberg, F.; Gallon, M.; Ghai, R.; Pim, D.; Langton, P.; Pearson, N.; Danson, C.M.; Nagele, H.; et al. Retriever is a multiprotein complex for retromer-independent endosomal cargo recycling. Nat. Cell Biol. 2017, 19, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and γ-secretase: Structure, function, and role in alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006304. [Google Scholar] [CrossRef] [PubMed]
- Beel, A.J.; Sanders, C.R. Substrate specificity of γ-secretase and other intramembrane proteases. Cell. Mol. Life Sci. 2008, 65, 1311–1334. [Google Scholar] [CrossRef] [PubMed]
- Andrew, R.J.; Kellett, K.A.; Thinakaran, G.; Hooper, N.M. A greek tragedy: The growing complexity of alzheimer amyloid precursor protein proteolysis. J. Biol. Chem. 2016, 291, 19235–19244. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J.; et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of notch intracellular domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Herreman, A.; Serneels, L.; Annaert, W.; Collen, D.; Schoonjans, L.; De Strooper, B. Total inactivation of gamma-secretase activity in presenilin-deficient embryonic stem cells. Nat. Cell Biol. 2000, 2, 461–462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Nadeau, P.; Song, W.; Donoviel, D.; Yuan, M.; Bernstein, A.; Yankner, B.A. Presenilins are required for γ-secretase cleavage of β-APP and transmembrane cleavage of Notch-1. Nat. Cell Biol. 2000, 2, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Karanam, B.; Peng, S.; Li, T.; Buck, C.; Day, P.M.; Roden, R.B. Papillomavirus infection requires gamma secretase. J. Virol. 2010, 84, 10661–10670. [Google Scholar] [CrossRef] [PubMed]
- Kwak, K.; Jiang, R.; Wang, J.W.; Jagu, S.; Kirnbauer, R.; Roden, R.B. Impact of inhibitors and L2 antibodies upon the infectivity of diverse alpha and beta human papillomavirus types. PLoS ONE 2014, 9, e97232. [Google Scholar] [CrossRef] [PubMed]
- Duggan, S.P.; McCarthy, J.V. Beyond γ-secretase activity: The multifunctional nature of presenilins in cell signalling pathways. Cell. Signal. 2016, 28, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Choy, R.W.; Cheng, Z.; Schekman, R. Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid β (Aβ) production in the trans-Golgi network. Proc. Natl. Acad. Sci. USA 2012, 109, E2077–E2082. [Google Scholar] [CrossRef] [PubMed]
- Small, S.A.; Gandy, S. Sorting through the cell biology of alzheimer’s disease: Intracellular pathways to pathogenesis. Neuron 2006, 52, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Auvinen, E.; Kujari, H.; Arstila, P.; Hukkanen, V. Expression of the L2 and E7 genes of the human papillomavirus type 16 in female genital dysplasias. Am. J. Pathol. 1992, 141, 1217–1224. [Google Scholar] [PubMed]
- Hagensee, M.E.; Yaegashi, N.; Galloway, D.A. Self-assembly of human papillomavirus type 1 capsids by expression of the L1 protein alone or by coexpression of the L1 and L2 capsid proteins. J. Virol. 1993, 67, 315–322. [Google Scholar] [PubMed]
- Kämper, N.; Day, P.M.; Nowak, T.; Selinka, H.C.; Florin, L.; Bolscher, J.; Hilbig, L.; Schiller, J.T.; Sapp, M. A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes. J. Virol. 2006, 80, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Bronnimann, M.P.; Chapman, J.A.; Park, C.K.; Campos, S.K. A transmembrane domain and GxxxG motifs within L2 are essential for papillomavirus infection. J. Virol. 2013, 87, 464–473. [Google Scholar] [CrossRef] [PubMed]
- DiGiuseppe, S.; Keiffer, T.R.; Bienkowska-Haba, M.; Luszczek, W.; Guion, L.G.; Muller, M.; Sapp, M. Topography of the human papillomavirus minor capsid protein L2 during vesicular trafficking of infectious entry. J. Virol. 2015, 89, 10442–10452. [Google Scholar] [CrossRef] [PubMed]
- DiGiuseppe, S.; Luszczek, W.; Keiffer, T.R.; Bienkowska-Haba, M.; Guion, L.G.; Sapp, M.J. Incoming human papillomavirus type 16 genome resides in a vesicular compartment throughout mitosis. Proc. Natl. Acad. Sci. USA 2016, 113, 6289–6294. [Google Scholar] [CrossRef] [PubMed]
- Garred, O.; van Deurs, B.; Sandvig, K. Furin-induced cleavage and activation of shiga toxin. J. Biol. Chem. 1995, 270, 10817–10821. [Google Scholar] [CrossRef] [PubMed]
- Gordon, V.M.; Klimpel, K.R.; Arora, N.; Henderson, M.A.; Leppla, S.H. Proteolytic activation of bacterial toxins by eukaryotic cells is performed by furin and by additional cellular proteases. Infect. Immun. 1995, 63, 82–87. [Google Scholar] [PubMed]
- Epand, R.M. Fusion peptides and the mechanism of viral fusion. Biochim. Biophys. Acta 2003, 1614, 116–121. [Google Scholar] [CrossRef]
- Tamm, L.K.; Han, X.; Li, Y.; Lai, A.L. Structure and function of membrane fusion peptides. Biopolymers 2002, 66, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Lorieau, J.L.; Louis, J.M.; Bax, A. The complete influenza hemagglutinin fusion domain adopts a tight helical hairpin arrangement at the lipid:Water interface. Proc. Natl. Acad. Sci. USA 2010, 107, 11341–11346. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.W.; Weise, K.; Ollesch, J.; Agrawal, P.; Stalz, H.; Stelzer, W.; Hulsbergen, F.; de Groot, H.; Gerwert, K.; Reed, J.; et al. De novo design of conformationally flexible transmembrane peptides driving membrane fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 14776–14781. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, T.; Craessaerts, K.; Bammens, L.; Bentahir, M.; Borgions, F.; Herdewijn, P.; Staes, A.; Timmerman, E.; Vandekerckhove, J.; Rubinstein, E.; et al. Analysis of the γ-secretase interactome and validation of its association with tetraspanin-enriched microdomains. Nat. Cell Biol. 2009, 11, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. Structure, mechanism and inhibition of gamma-secretase and presenilin-like proteases. Biol. Chem. 2010, 391, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Kornilova, A.Y.; Das, C.; Wolfe, M.S. Differential effects of inhibitors on the γ-secretase complex. Mechanistic implications. J. Biol. Chem. 2003, 278, 16470–16473. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bohm, C.; Dodd, R.; Chen, F.; Qamar, S.; Schmitt-Ulms, G.; Fraser, P.E.; St George-Hyslop, P.H. Structural biology of presenilin 1 complexes. Mol. Neurodegener. 2014, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Lipovsky, A.; Erden, A.; Kanaya, E.; Zhang, W.; Crite, M.; Bradfield, C.; MacMicking, J.; DiMaio, D.; Schoggins, J.W.; Iwasaki, A. The cellular endosomal protein stannin inhibits intracellular trafficking of human papillomavirus during virus entry. J. Gen. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wiens, M.E.; Smith, J.G. α-defensin HD5 inhibits human papillomavirus 16 infection via capsid stabilization and redirection to the lysosome. mBio 2017, 8, e02304-16. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T. Interferon gamma prevents infectious entry of human papillomavirus 16 via an L2-dependent mechanism. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Calton, C.M.; Schlegel, A.M.; Chapman, J.A.; Campos, S.K. Human papillomavirus type 16 does not require cathepsin L or B for infection. J. Gen. Virol. 2013, 94, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Akache, B.; Grimm, D.; Shen, X.; Fuess, S.; Yant, S.R.; Glazer, D.S.; Park, J.; Kay, M.A. A two-hybrid screen identifies cathepsins B and L as uncoating factors for adeno-associated virus 2 and 8. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Ebert, D.H.; Deussing, J.; Peters, C.; Dermody, T.S. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J. Biol. Chem. 2002, 277, 24609–24617. [Google Scholar] [CrossRef] [PubMed]
- Schafer, G.; Graham, L.M.; Lang, D.M.; Blumenthal, M.J.; Bergant Marusic, M.; Katz, A.A. Vimentin modulates infectious internalization of human papillomavirus 16 pseudovirions. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.T.; Day, P.M.; Kines, R.C. Current understanding of the mechanism of HPV infection. Gynecol. Oncol. 2010, 118, S12–S17. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.E.; Helenius, A. How viruses enter animal cells. Science 2004, 304, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Tsai, B. Penetration of nonenveloped viruses into the cytoplasm. Annu. Rev. Cell Dev. Biol. 2007, 23, 23–43. [Google Scholar] [CrossRef] [PubMed]
- Pyeon, D.; Pearce, S.M.; Lank, S.M.; Ahlquist, P.; Lambert, P.F. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009, 5, e1000318. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.F.; Emerman, M. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J. Virol. 1994, 68, 510–516. [Google Scholar] [PubMed]
- Schatz, P.J. Use of peptide libraries to map the substrate specificity of a peptide-modifying enzyme: A 13 residue consensus peptide specifies biotinylation in Escherichia coli. Nat. Biotechnol. 1993, 11, 1138–1143. [Google Scholar] [CrossRef]
- Murphy, J.R. Mechanism of diphtheria toxin catalytic domain delivery to the eukaryotic cell cytosol and the cellular factors that directly participate in the process. Toxins 2011, 3, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Pirazzini, M.; Azarnia Tehran, D.; Leka, O.; Zanetti, G.; Rossetto, O.; Montecucco, C. On the translocation of botulinum and tetanus neurotoxins across the membrane of acidic intracellular compartments. Biochim. Biophys. Acta 2016, 1858, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Baker, C.C.; Lowy, D.R.; Schiller, J.T. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc. Natl. Acad. Sci. USA 2004, 101, 14252–14257. [Google Scholar] [CrossRef] [PubMed]
- Lallemand-Breitenbach, V.; de The, H. PML nuclear bodies. Cold Spring Harb. Perspect. Biol. 2010, 2, a000661. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Lallemand-Breitenbach, V.; de The, H. PML nuclear bodies: Regulation, function and therapeutic perspectives. J. Pathol. 2014, 234, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Scherer, M.; Stamminger, T. Emerging role of PML nuclear bodies in innate immune signaling. J. Virol. 2016, 90, 5850–5854. [Google Scholar] [CrossRef] [PubMed]
- Weidtkamp-Peters, S.; Lenser, T.; Negorev, D.; Gerstner, N.; Hofmann, T.G.; Schwanitz, G.; Hoischen, C.; Maul, G.; Dittrich, P.; Hemmerich, P. Dynamics of component exchange at PML nuclear bodies. J. Cell Sci. 2008, 121, 2731–2743. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Ferhi, O.; Jeanne, M.; Benhenda, S.; Berthier, C.; Jollivet, F.; Niwa-Kawakita, M.; Faklaris, O.; Setterblad, N.; de The, H.; et al. Oxidative stress-induced assembly of pml nuclear bodies controls sumoylation of partner proteins. J. Cell Biol. 2014, 204, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D. DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 2001, 20, 7266–7273. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Chelbi-Alix, M.K. PML and PML nuclear bodies: Implications in antiviral defence. Biochimie 2007, 89, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Florin, L.; Schafer, F.; Sotlar, K.; Streeck, R.E.; Sapp, M. Reorganization of nuclear domain 10 induced by papillomavirus capsid protein L2. Virology 2002, 295, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.A.; Florin, L.; Sapp, C.; Sapp, M. Dissection of human papillomavirus type 33 L2 domains involved in nuclear domains (ND) 10 homing and reorganization. Virology 2003, 314, 161–167. [Google Scholar] [CrossRef]
- Bund, T.; Spoden, G.A.; Koynov, K.; Hellmann, N.; Boukhallouk, F.; Arnold, P.; Hinderberger, D.; Florin, L. An L2 sumo interacting motif is important for PML localization and infection of human papillomavirus type 16. Cell. Microbiol. 2014, 16, 1179–1200. [Google Scholar] [CrossRef] [PubMed]
- Marusic, M.B.; Mencin, N.; Licen, M.; Banks, L.; Grm, H.S. Modification of human papillomavirus minor capsid protein L2 by sumoylation. J. Virol. 2010, 84, 11585–11589. [Google Scholar] [CrossRef] [PubMed]
- Kieback, E.; Muller, M. Factors influencing subcellular localization of the human papillomavirus L2 minor structural protein. Virology 2006, 345, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Stepp, W.H.; Meyers, J.M.; McBride, A.A. Sp100 provides intrinsic immunity against human papillomavirus infection. mBio 2013, 4, e00845-13. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Kappel, C.; Beaudouin, J.; Eils, R.; Spector, D.L. Live cell dynamics of promyelocytic leukemia nuclear bodies upon entry into and exit from mitosis. Mol. Biol. Cell 2008, 19, 3147–3162. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos, S.K. Subcellular Trafficking of the Papillomavirus Genome during Initial Infection: The Remarkable Abilities of Minor Capsid Protein L2. Viruses 2017, 9, 370. https://doi.org/10.3390/v9120370
Campos SK. Subcellular Trafficking of the Papillomavirus Genome during Initial Infection: The Remarkable Abilities of Minor Capsid Protein L2. Viruses. 2017; 9(12):370. https://doi.org/10.3390/v9120370
Chicago/Turabian StyleCampos, Samuel K. 2017. "Subcellular Trafficking of the Papillomavirus Genome during Initial Infection: The Remarkable Abilities of Minor Capsid Protein L2" Viruses 9, no. 12: 370. https://doi.org/10.3390/v9120370
APA StyleCampos, S. K. (2017). Subcellular Trafficking of the Papillomavirus Genome during Initial Infection: The Remarkable Abilities of Minor Capsid Protein L2. Viruses, 9(12), 370. https://doi.org/10.3390/v9120370