The Influence of E1A C-Terminus on Adenovirus Replicative Cycle

Abstract
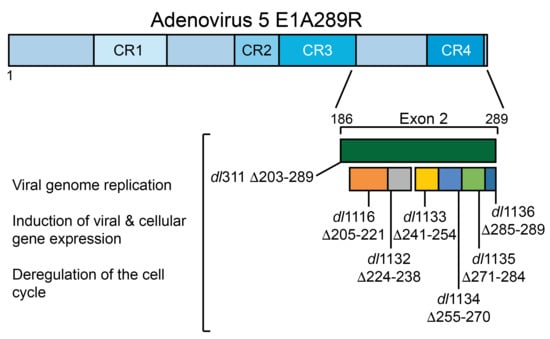
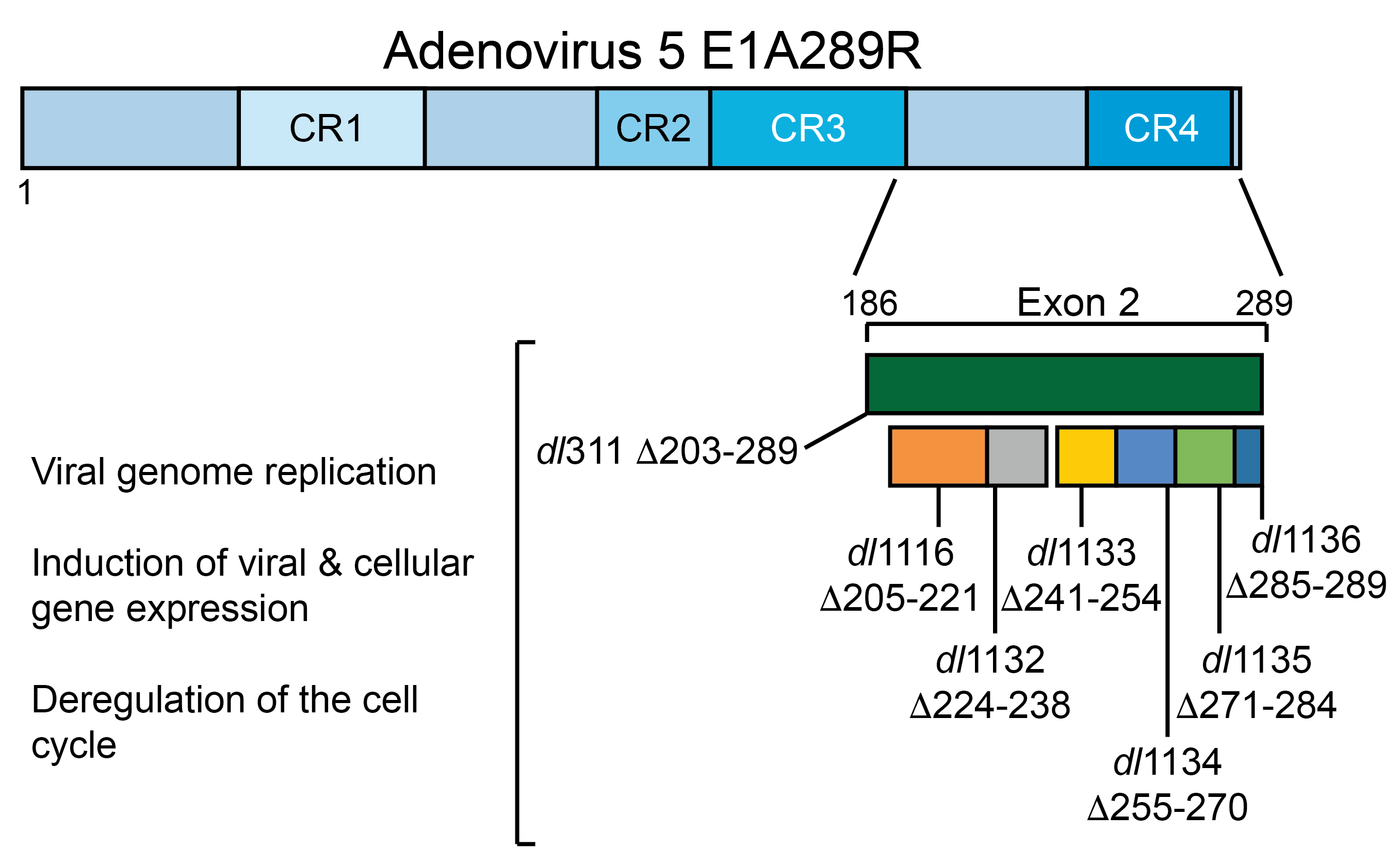
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
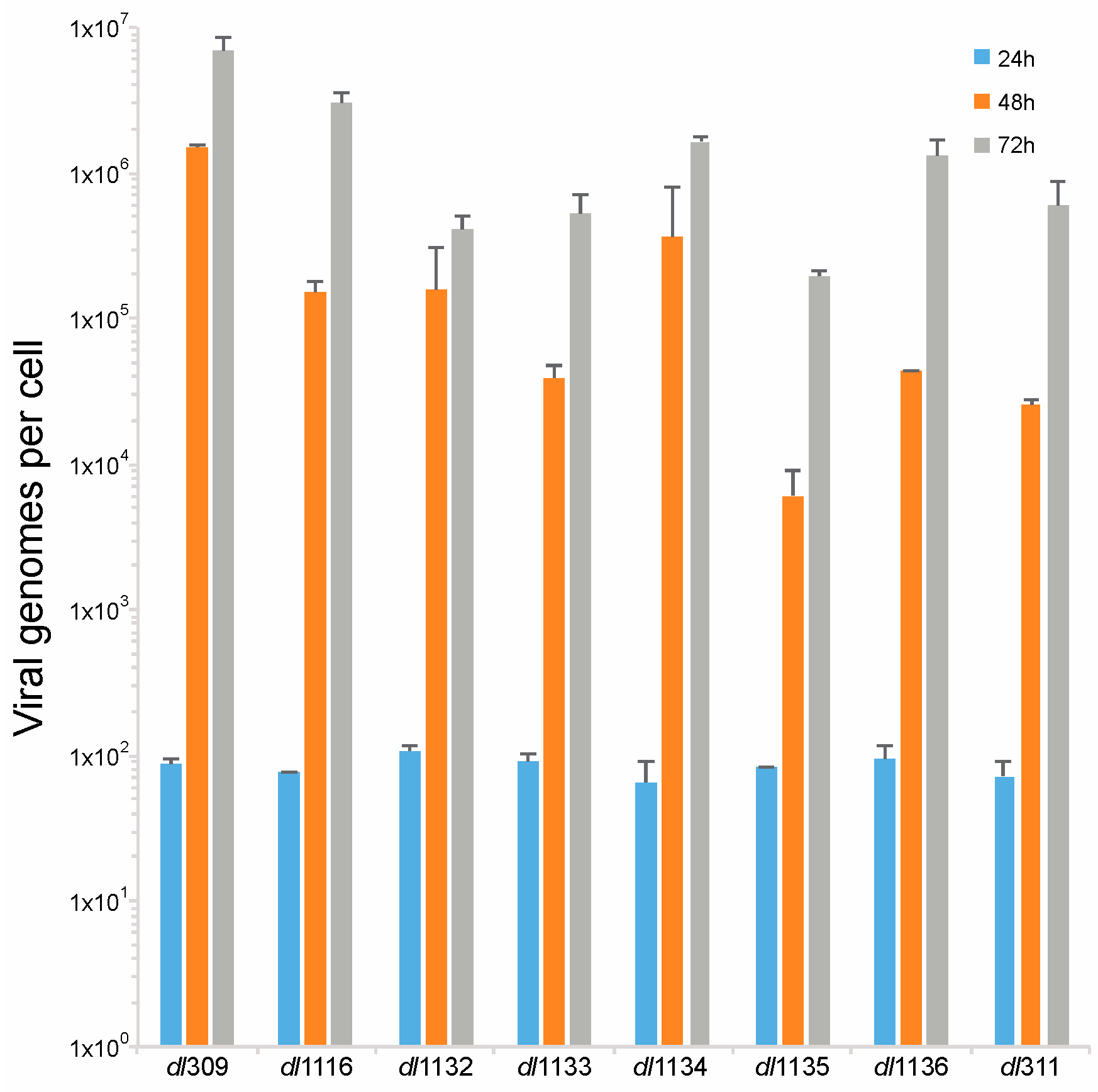{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Cell and Virus Culture
2.3. EdU Incorporation Assay
2.4. Immunofluorescence
2.5. PCR Primers
2.6. Real-Time Gene Expression Analysis
2.7. Statistical Analysis
2.8. Viral Genome Quantification
2.9. Virus Growth Assay
3. Results
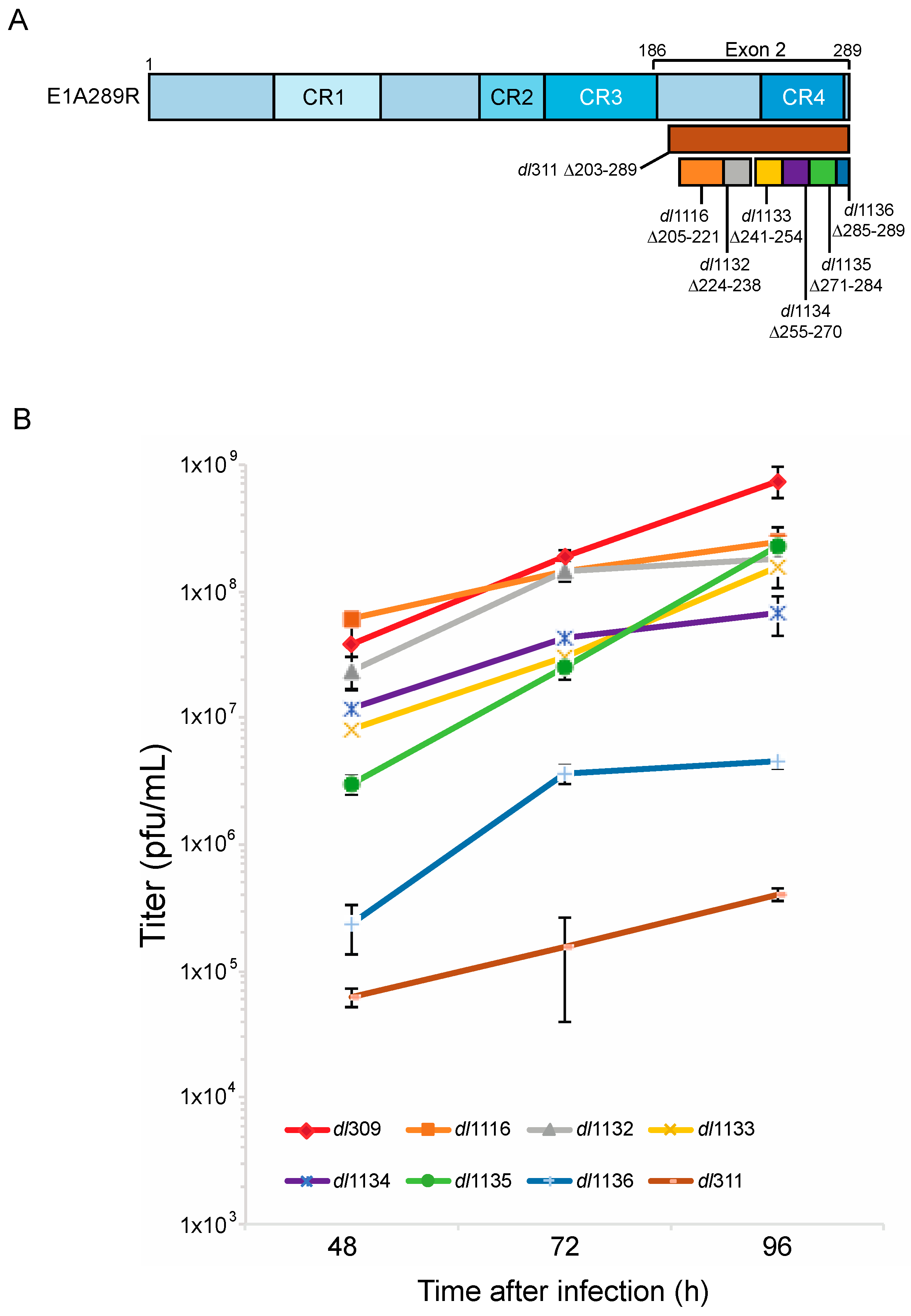
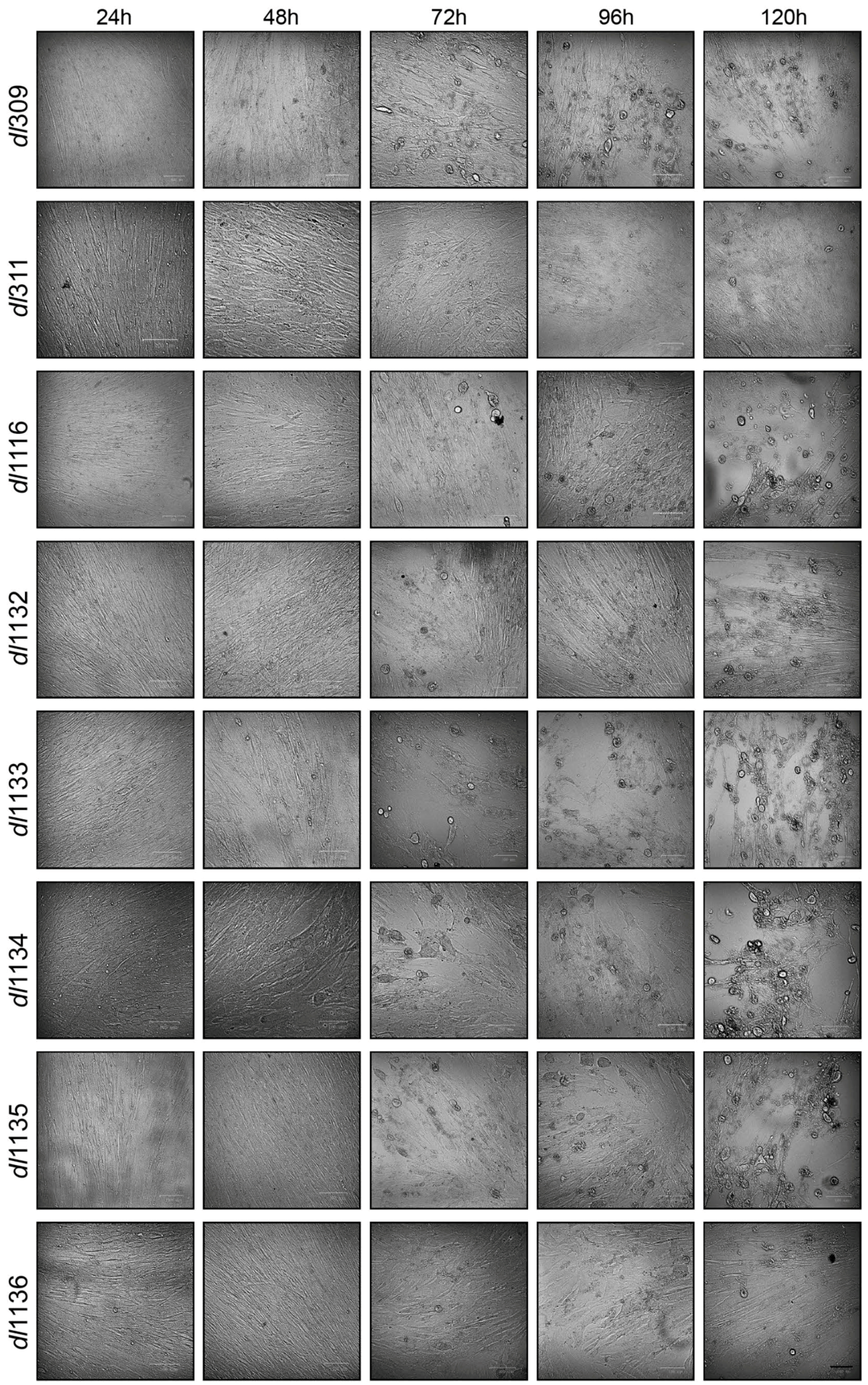
3.1. Growth of C-Terminus Deletion Mutants in Arrested Fibroblasts
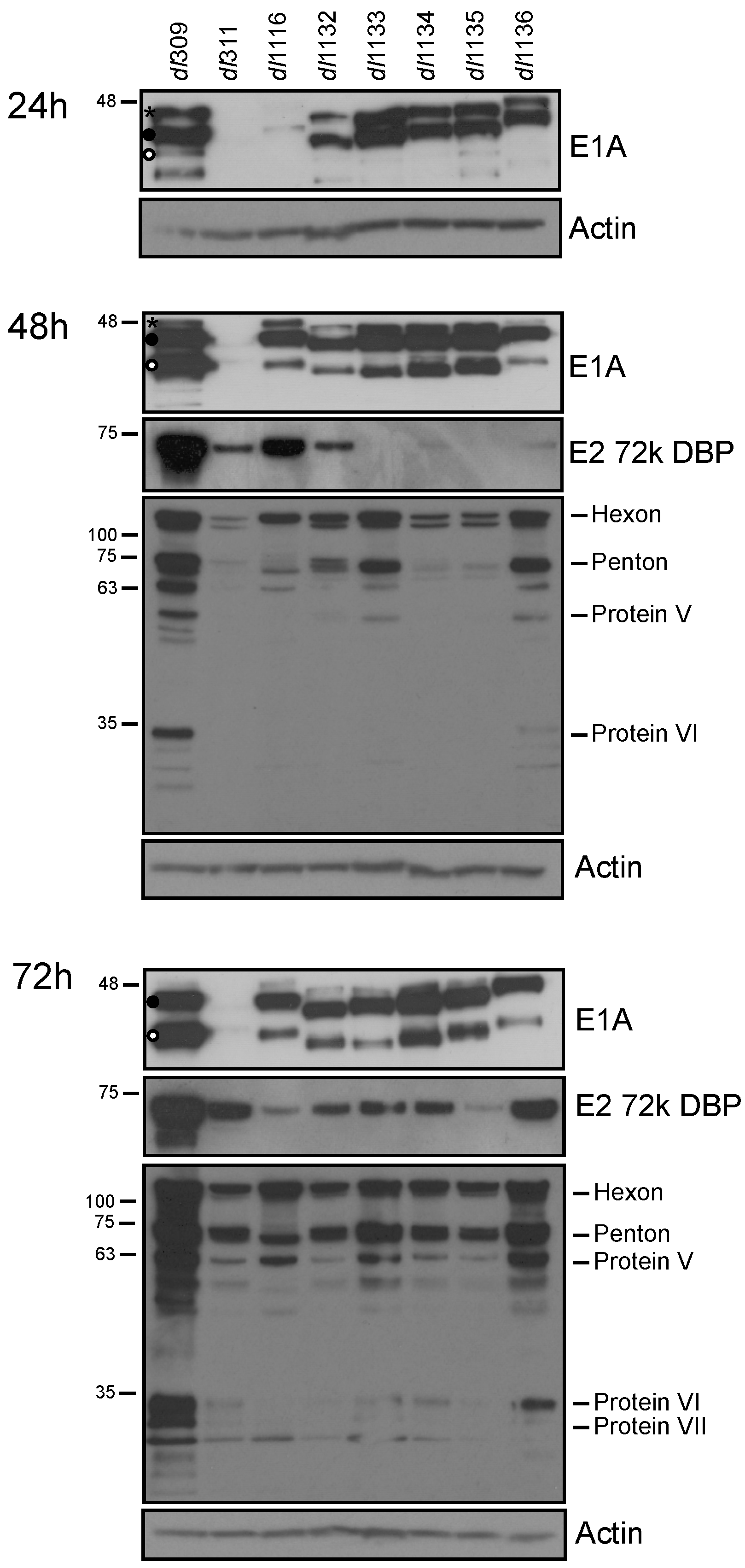
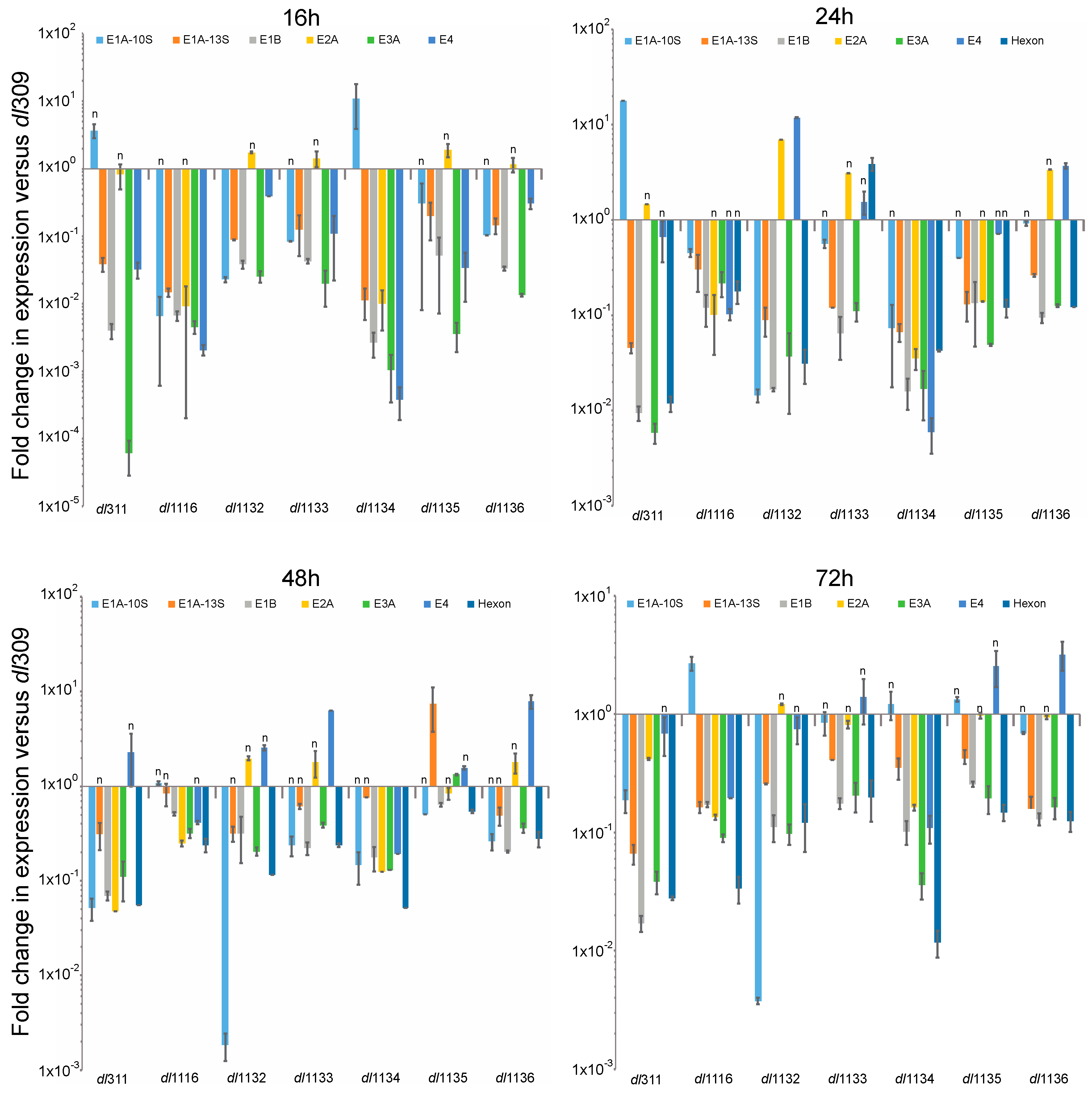
3.2. Viral Protein and Gene Expression
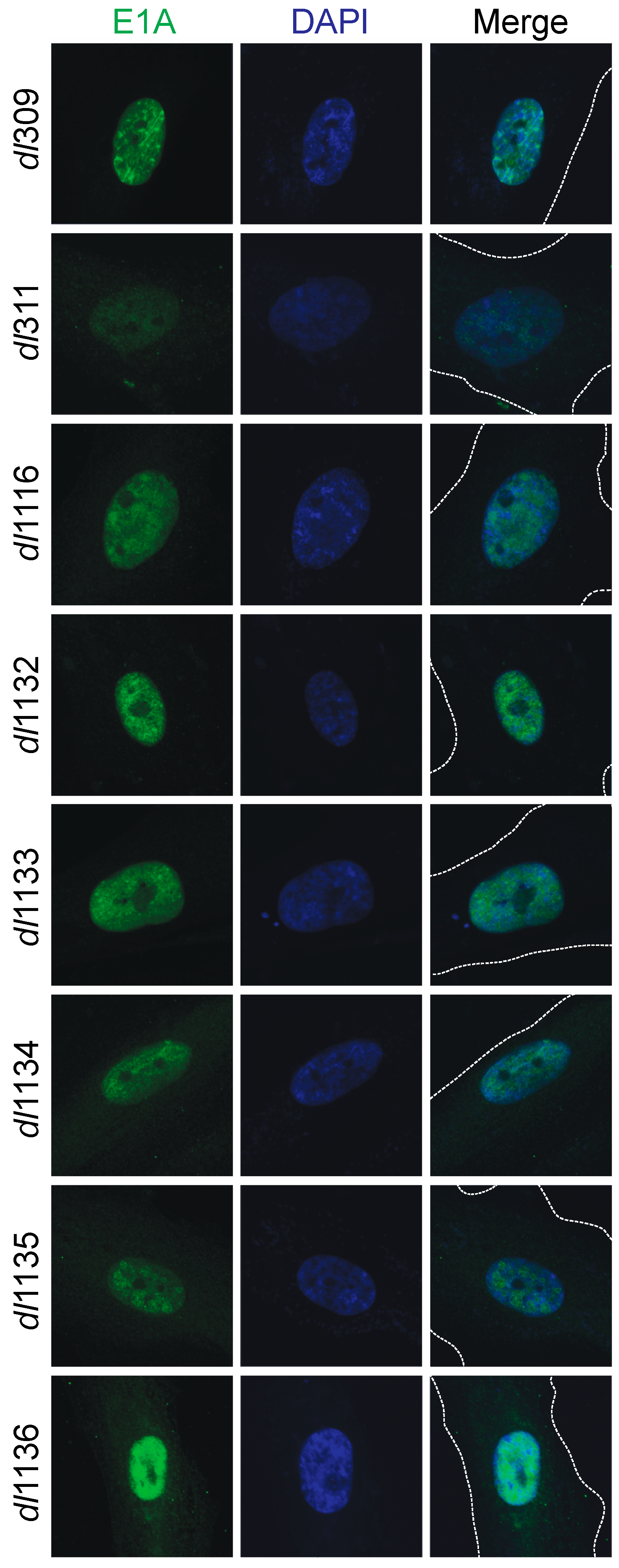
3.3. Sub-Cellular Localization of Mutant E1A Proteins
3.4. Viral Genome Replication
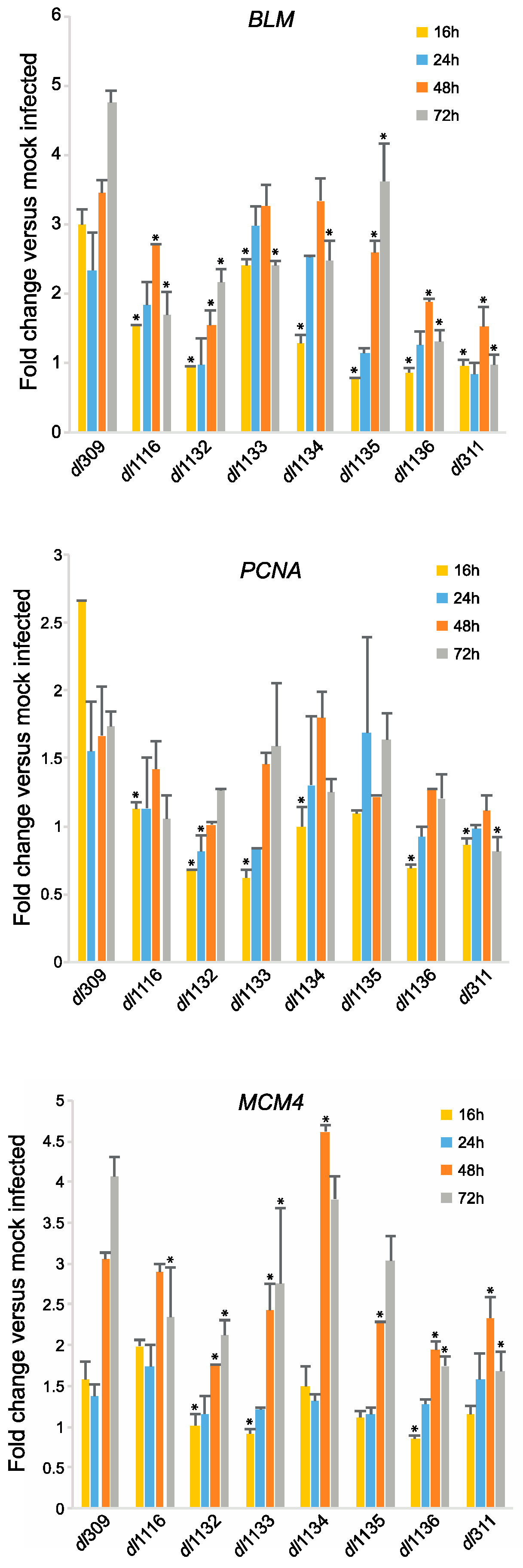
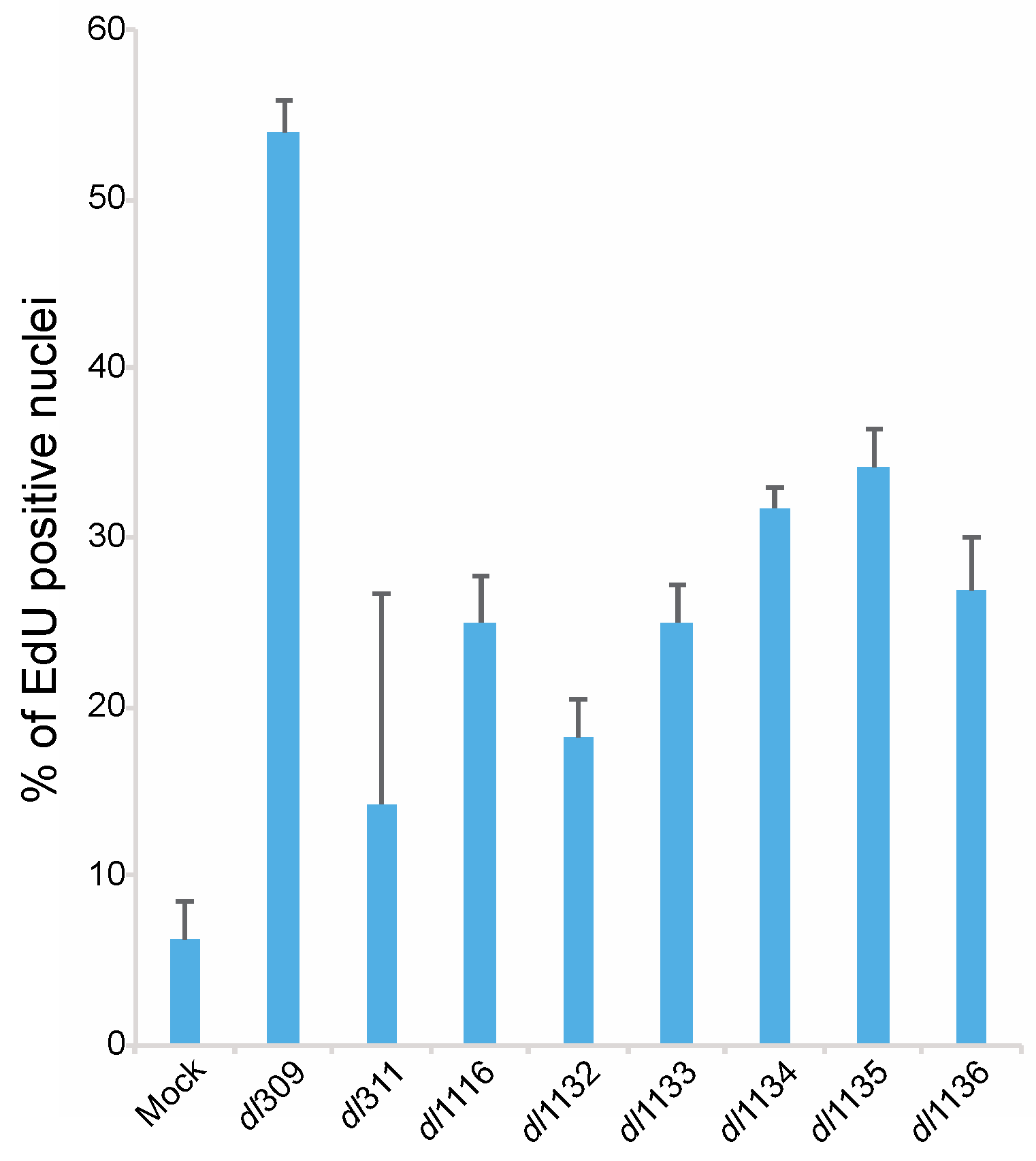
3.5. Effects of E1A C-Terminus Mutations on S-Phase Induction
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Berk, A.J. Chapter 55. Adenoviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Radko, S.; Jung, R.; Olanubi, O.; Pelka, P. Effects of adenovirus type 5 E1A isoforms on viral replication in arrested human cells. PLoS ONE 2015, 10, e0140124. [Google Scholar] [CrossRef] [PubMed]
- Pelka, P.; Ablack, J.N.; Fonseca, G.J.; Yousef, A.F.; Mymryk, J.S. Intrinsic structural disorder in adenovirus E1A: A viral molecular hub linking multiple diverse processes. J. Virol. 2008, 82, 7252–7263. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.J.; Yousef, A.F.; Massimi, P.; Fonseca, G.J.; Todorovic, B.; Pelka, P.; Turnell, A.S.; Banks, L.; Mymryk, J.S. Dissection of the C-terminal region of E1A re-defines the roles of CtBP and other cellular targets in oncogenic transformation. J. Virol. 2013, 87, 10348–10355. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.M.; Subramanian, T.; Schaeper, U.; La Regina, M.; Bayley, S.; Chinnadurai, G. A region in the C-terminus of adenovirus 2/5 E1A protein is required for association with a cellular phosphoprotein and important for the negative modulation of T24-ras mediated transformation, tumorigenesis and metastasis. EMBO J. 1993, 12, 469–478. [Google Scholar] [PubMed]
- Komorek, J.; Kuppuswamy, M.; Subramanian, T.; Vijayalingam, S.; Lomonosova, E.; Zhao, L.J.; Mymryk, J.S.; Schmitt, K.; Chinnadurai, G. Adenovirus type 5 E1A and E6 proteins of low-risk cutaneous beta-human papillomaviruses suppress cell transformation through interaction with FOXK1/K2 transcription factors. J. Virol. 2010, 84, 2719–2731. [Google Scholar] [CrossRef] [PubMed]
- Kohler, M.; Gorlich, D.; Hartmann, E.; Franke, J. Adenoviral E1A protein nuclear import is preferentially mediated by importin alpha3 in vitro. Virology 2001, 289, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Smith, M.M.; Mymryk, J.S. Interaction of the E1A oncoprotein with Yak1p, a novel regulator of yeast pseudohyphal differentiation, and related mammalian kinases. Mol. Biol. Cell 2001, 12, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Radko, S.; Koleva, M.; James, K.M.; Jung, R.; Mymryk, J.S.; Pelka, P. Adenovirus E1A targets the DREF nuclear factor to regulate virus gene expression, DNA replication, and growth. J. Virol. 2014, 88, 13469–13481. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.R.; Olanubi, O.; Cheng, S.K.; Soriano, A.; Crisostomo, L.; Lopez, A.; Pelka, P. The interaction of adenovirus E1A with the mammalian protein ku70/XRCC6. Virology 2016, 500, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Olanubi, O.; Frost, J.R.; Radko, S.; Pelka, P. Suppression of type I interferon signaling by E1A via RuvBL1/Pontin. J. Virol. 2017, 91, e02484-16. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.J.; King, C.R.; Dikeakos, J.D.; Mymryk, J.S. Functional analysis of the C-terminal region of human adenovirus E1A reveals a misidentified nuclear localization signal. Virology 2014, 468, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Harlow, E.; Franza, B.R., Jr.; Schley, C. Monoclonal antibodies specific for adenovirus early region 1A proteins: Extensive heterogeneity in early region 1A products. J. Virol. 1985, 55, 533–546. [Google Scholar] [PubMed]
- Stephens, C.; Harlow, E. Differential splicing yields novel adenovirus 5 E1A mrnas that encode 30 kd and 35 kd proteins. EMBO J. 1987, 6, 2027–2035. [Google Scholar] [PubMed]
- Reich, N.C.; Sarnow, P.; Duprey, E.; Levine, A.J. Monoclonal antibodies which recognize native and denatured forms of the adenovirus DNA-binding protein. Virology 1983, 128, 480–484. [Google Scholar] [CrossRef]
- Jones, N.; Shenk, T. Isolation of adenovirus type 5 host range deletion mutants defective for transformation of rat embryo cells. Cell 1979, 17, 683–689. [Google Scholar] [CrossRef]
- Jones, N.; Shenk, T. An adenovirus type 5 early gene function regulates expression of other early viral genes. Proc. Natl. Acad. Sci. USA 1979, 76, 3665–3669. [Google Scholar] [CrossRef] [PubMed]
- Jelsma, T.N.; Howe, J.A.; Evelegh, C.M.; Cunniff, N.F.; Skiadopoulos, M.H.; Floroff, M.R.; Denman, J.E.; Bayley, S.T. Use of deletion and point mutants spanning the coding region of the adenovirus 5 E1A gene to define a domain that is essential for transcriptional activation. Virology 1988, 163, 494–502. [Google Scholar] [CrossRef]
- Douglas, J.L.; Quinlan, M.P. Efficient nuclear localization of the Ad5 E1A 12S protein is necessary for immortalization but not cotransformation of primary epithelial cells. Cell Growth Differ. 1994, 5, 475–483. [Google Scholar] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Reza Etemadi, M.; Ling, K.H.; Zainal Abidin, S.; Chee, H.Y.; Sekawi, Z. Gene expression patterns induced at different stages of rhinovirus infection in human alveolar epithelial cells. PLoS ONE 2017, 12, e0176947. [Google Scholar] [CrossRef] [PubMed]
- Spindler, K.R.; Eng, C.Y.; Berk, A.J. An adenovirus early region 1A protein is required for maximal viral DNA replication in growth-arrested human cells. J. Virol. 1985, 53, 742–750. [Google Scholar] [PubMed]
- Braithwaite, A.W.; Cheetham, B.F.; Li, P.; Parish, C.R.; Waldron-Stevens, L.K.; Bellett, A.J. Adenovirus-induced alterations of the cell growth cycle: A requirement for expression of E1A but not of E1B. J. Virol. 1983, 45, 192–199. [Google Scholar] [PubMed]
- Pelka, P.; Miller, M.S.; Cecchini, M.; Yousef, A.F.; Bowdish, D.M.; Dick, F.; Whyte, P.; Mymryk, J.S. Adenovirus E1A directly targets the E2F/DP-1 complex. J. Virol. 2011, 85, 8841–8851. [Google Scholar] [CrossRef] [PubMed]
- Yousef, A.F.; Fonseca, G.J.; Cohen, M.J.; Mymryk, J.S. The C-terminal region of E1A: A molecular tool for cellular cartography. Biochem. Cell Biol. 2012, 90, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Bruton, R.K.; Pelka, P.; Mapp, K.L.; Fonseca, G.J.; Torchia, J.; Turnell, A.S.; Mymryk, J.S.; Grand, R.J. Identification of a second CtBP binding site in adenovirus type 5 E1A conserved region 3. J. Virol. 2008, 82, 8476–8486. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.S.; Quinlan, M.P. The C terminus of E1A regulates tumor progression and epithelial cell differentiation. Virology 1998, 249, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.S.; Quinlan, M.P. While E1A can facilitate epithelial cell transformation by several dominant oncogenes, the C-terminus seems only to regulate RAC and CDC42 function, but in both epithelial and fibroblastic cells. Virology 2000, 269, 404–419. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.L.; Gopalakrishnan, S.; Quinlan, M.P. Modulation of transformation of primary epithelial cells by the second exon of the Ad5 E1A12S gene. Oncogene 1991, 6, 2093–2103. [Google Scholar] [PubMed]
- Quinlan, M.P.; Douglas, J.L. Immortalization of primary epithelial cells requires first- and second-exon functions of adenovirus type 5 12S. J. Virol. 1992, 66, 2020–2030. [Google Scholar] [PubMed]
- Mymryk, J.S.; Bayley, S.T. Induction of gene expression by exon 2 of the major E1A proteins of adenovirus type 5. J. Virol. 1993, 67, 6922–6928. [Google Scholar] [PubMed]
- Jung, R.; Radko, S.; Pelka, P. The dual nature of Nek9 in adenovirus replication. J. Virol. 2015, 90, 1931–1943. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.A.; Nevins, J.R. Identification of distinct roles for separate E1A domains in disruption of E2F complexes. Mol. Cell. Biol. 1993, 13, 7029–7035. [Google Scholar] [CrossRef] [PubMed]
- Marshall, K.S.; Cohen, M.J.; Fonseca, G.J.; Todorovic, B.; King, C.R.; Yousef, A.F.; Zhang, Z.; Mymryk, J.S. Identification and characterization of multiple conserved nuclear localization signals within adenovirus E1A. Virology 2014, 454, 206–214. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, R.J.; Hearing, P. The E4-6/7 protein functionally compensates for the loss of E1A expression in adenovirus infection. J. Virol. 2000, 74, 5819–5824. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.L.; Quinlan, M.P. Efficient nuclear localization and immortalizing ability, two functions dependent on the adenovirus type 5 (Ad5) E1A second exon, are necessary for cotransformation with Ad5 E1B but not with T24ras. J. Virol. 1995, 69, 8061–8065. [Google Scholar] [PubMed]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crisostomo, L.; Soriano, A.M.; Frost, J.R.; Olanubi, O.; Mendez, M.; Pelka, P. The Influence of E1A C-Terminus on Adenovirus Replicative Cycle. Viruses 2017, 9, 387. https://doi.org/10.3390/v9120387
Crisostomo L, Soriano AM, Frost JR, Olanubi O, Mendez M, Pelka P. The Influence of E1A C-Terminus on Adenovirus Replicative Cycle. Viruses. 2017; 9(12):387. https://doi.org/10.3390/v9120387
Chicago/Turabian StyleCrisostomo, Leandro, Andrea Michelle Soriano, Jasmine Rae Frost, Oladunni Olanubi, Megan Mendez, and Peter Pelka. 2017. "The Influence of E1A C-Terminus on Adenovirus Replicative Cycle" Viruses 9, no. 12: 387. https://doi.org/10.3390/v9120387
APA StyleCrisostomo, L., Soriano, A. M., Frost, J. R., Olanubi, O., Mendez, M., & Pelka, P. (2017). The Influence of E1A C-Terminus on Adenovirus Replicative Cycle. Viruses, 9(12), 387. https://doi.org/10.3390/v9120387