
A New Strategy to Reduce Influenza Escape: Detecting Therapeutic Targets Constituted of Invariance Groups

Abstract
:1. Introduction
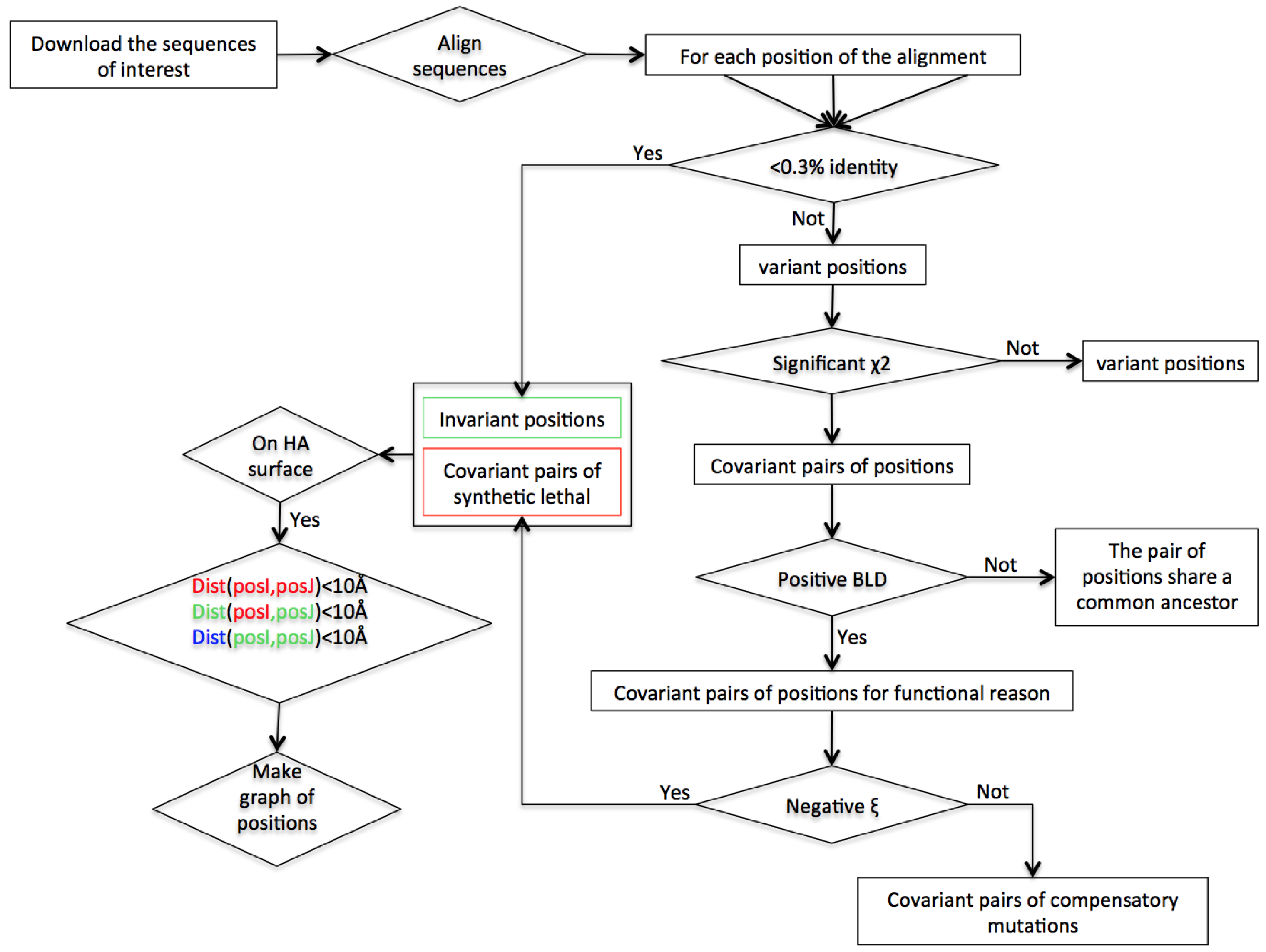
2. Materials and Methods
2.1. Primary Sequence and Quaternary Structure References
2.2. Construction of Sequence Dataset
2.3. Identification of Accessible Variant Positions
2.4. Identification of Interdependent Positions: χ2 Test
2.5. Identification of the Background Linkage Disequilibrium (BLD)
2.6. Characterization of Interdependent Pairs into Compensatory Mutations (CM) or Synthetic Lethals (SL)
2.7. Determination of Binding Sites Generating Little Resistance to a Small Drug Molecule
2.8. Accession Numbers
3. Results
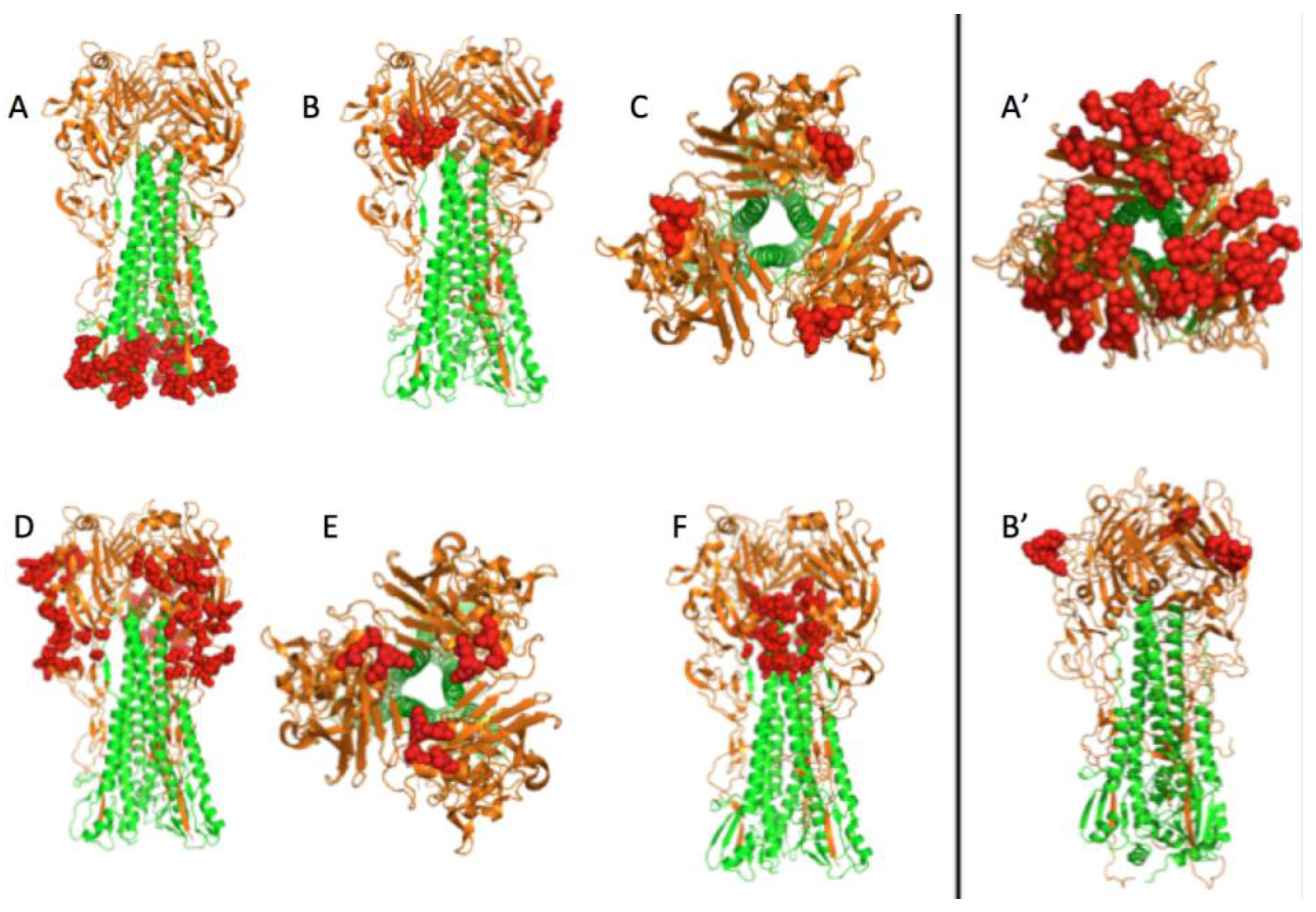
3.1. Prediction of Therapeutic Targets In Silico
3.2. The H1N1 Case
3.2.1. Can These Pockets Tie Up Small Drug Molecules?
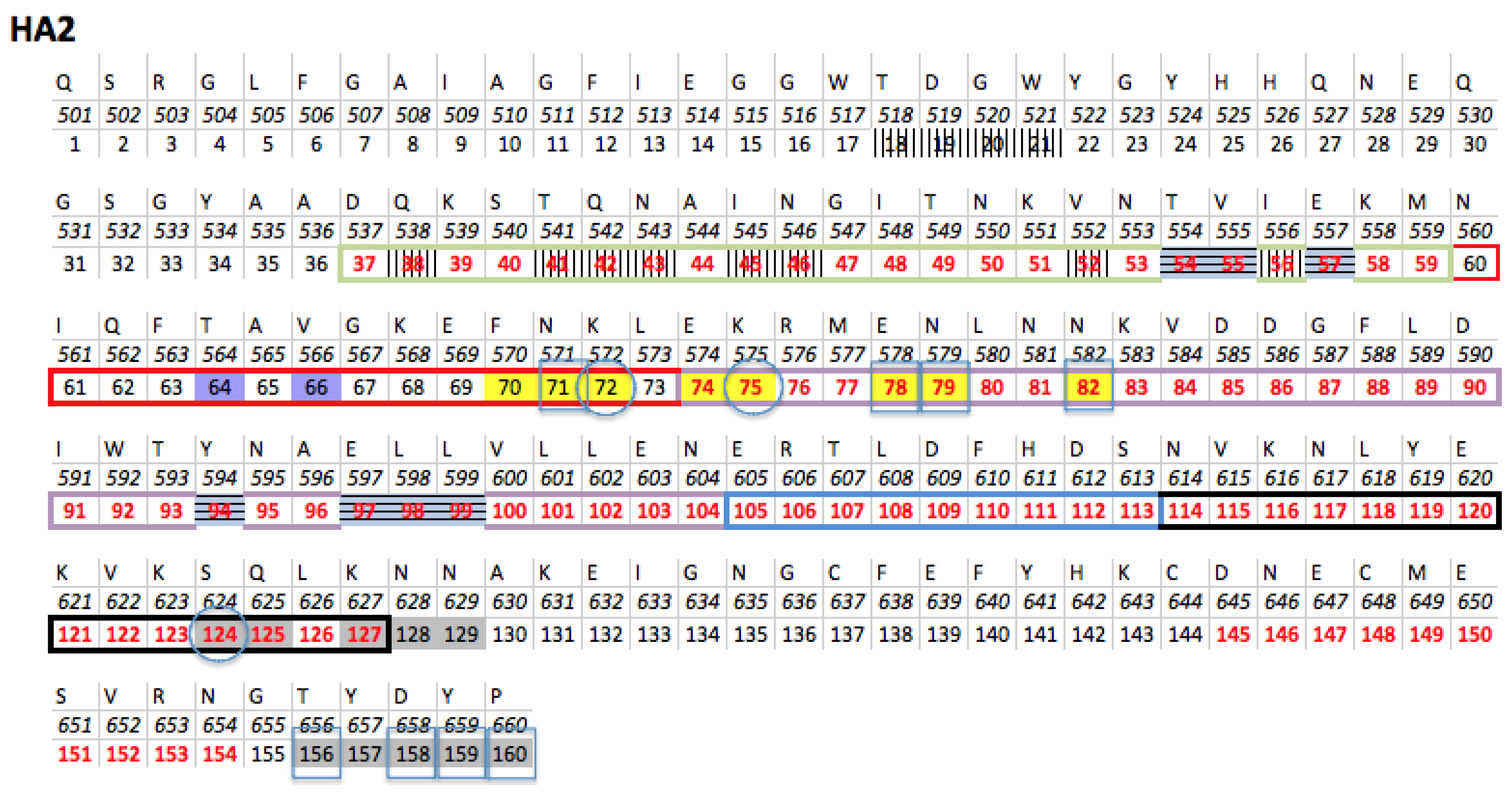
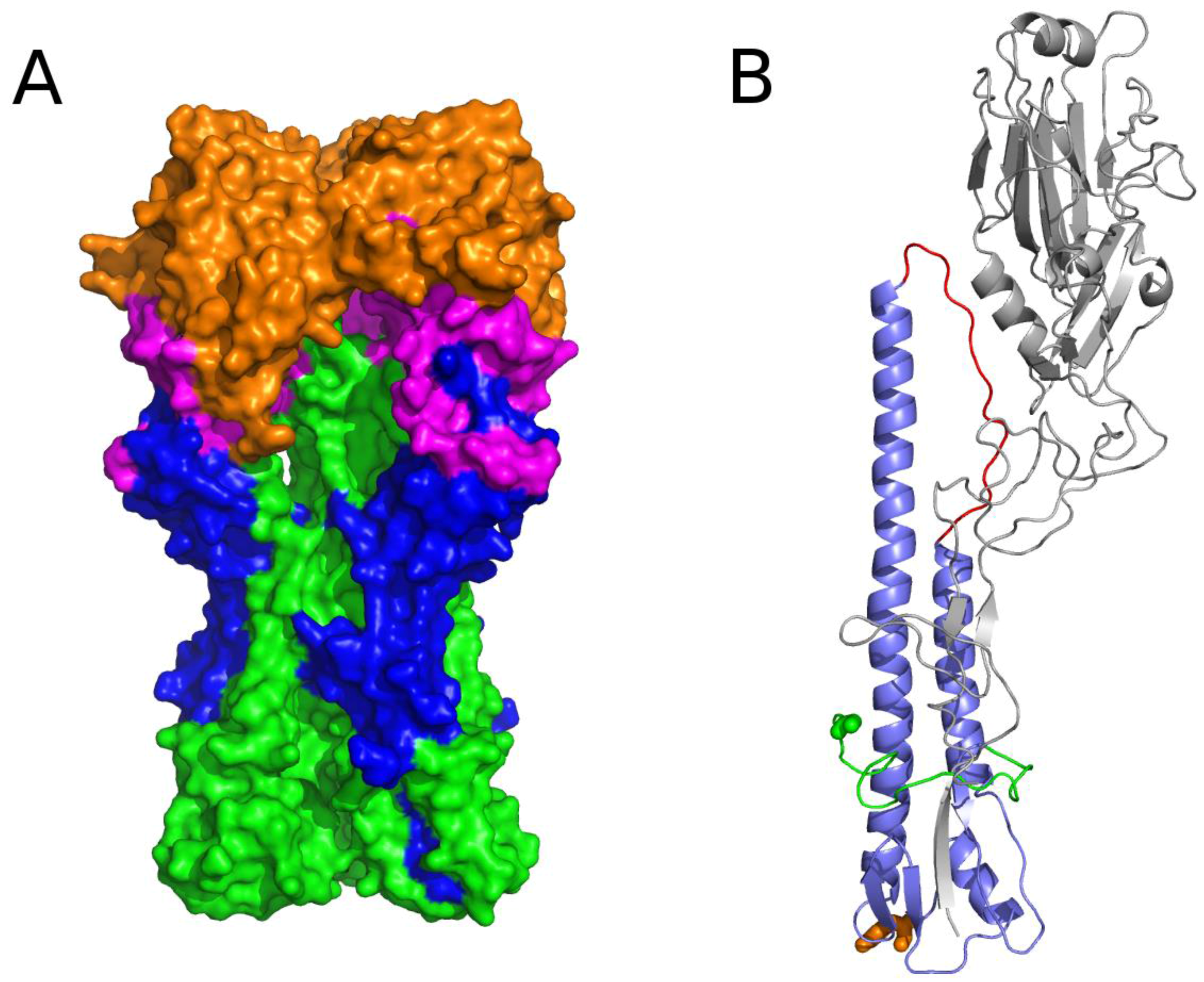
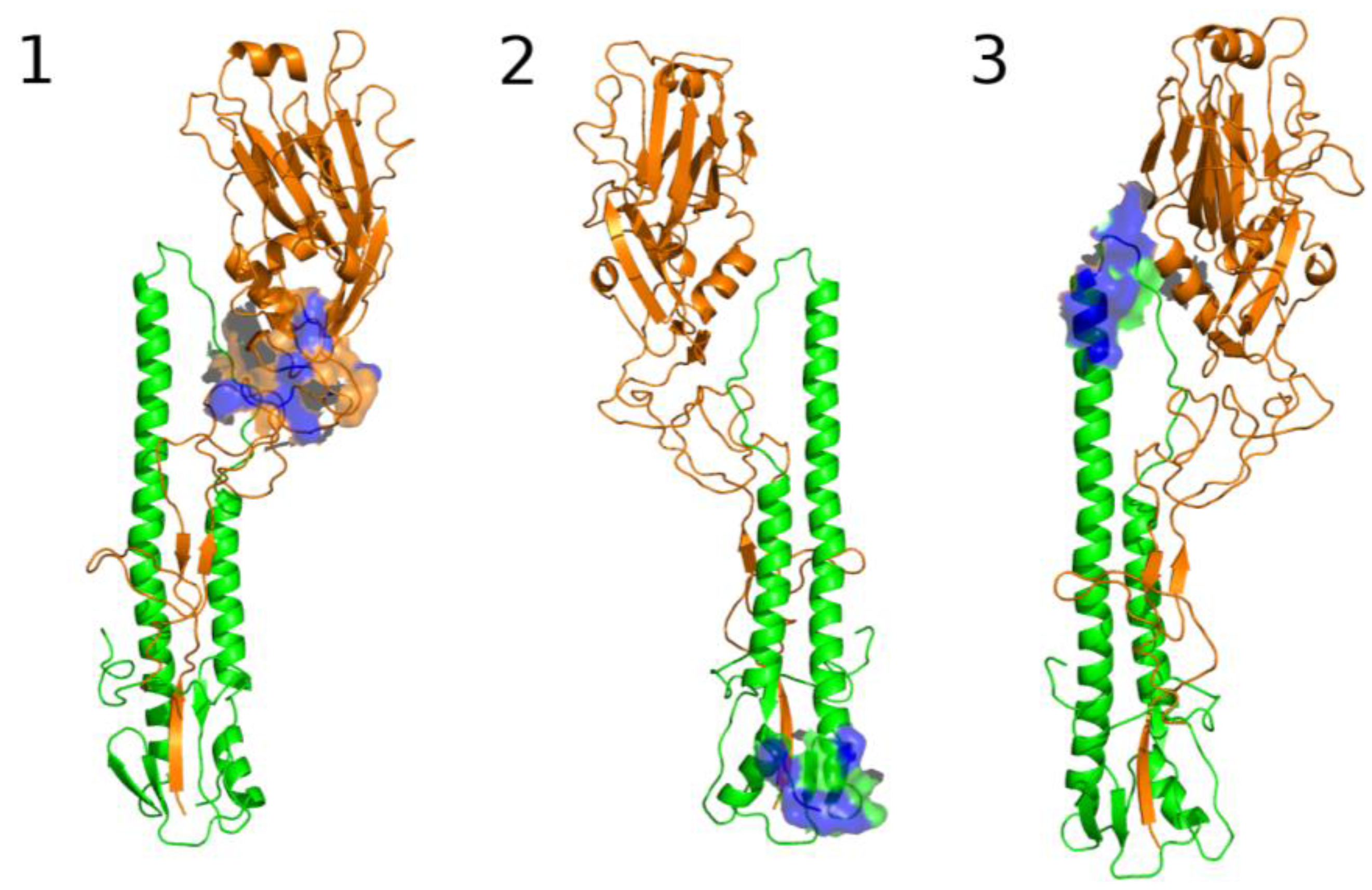
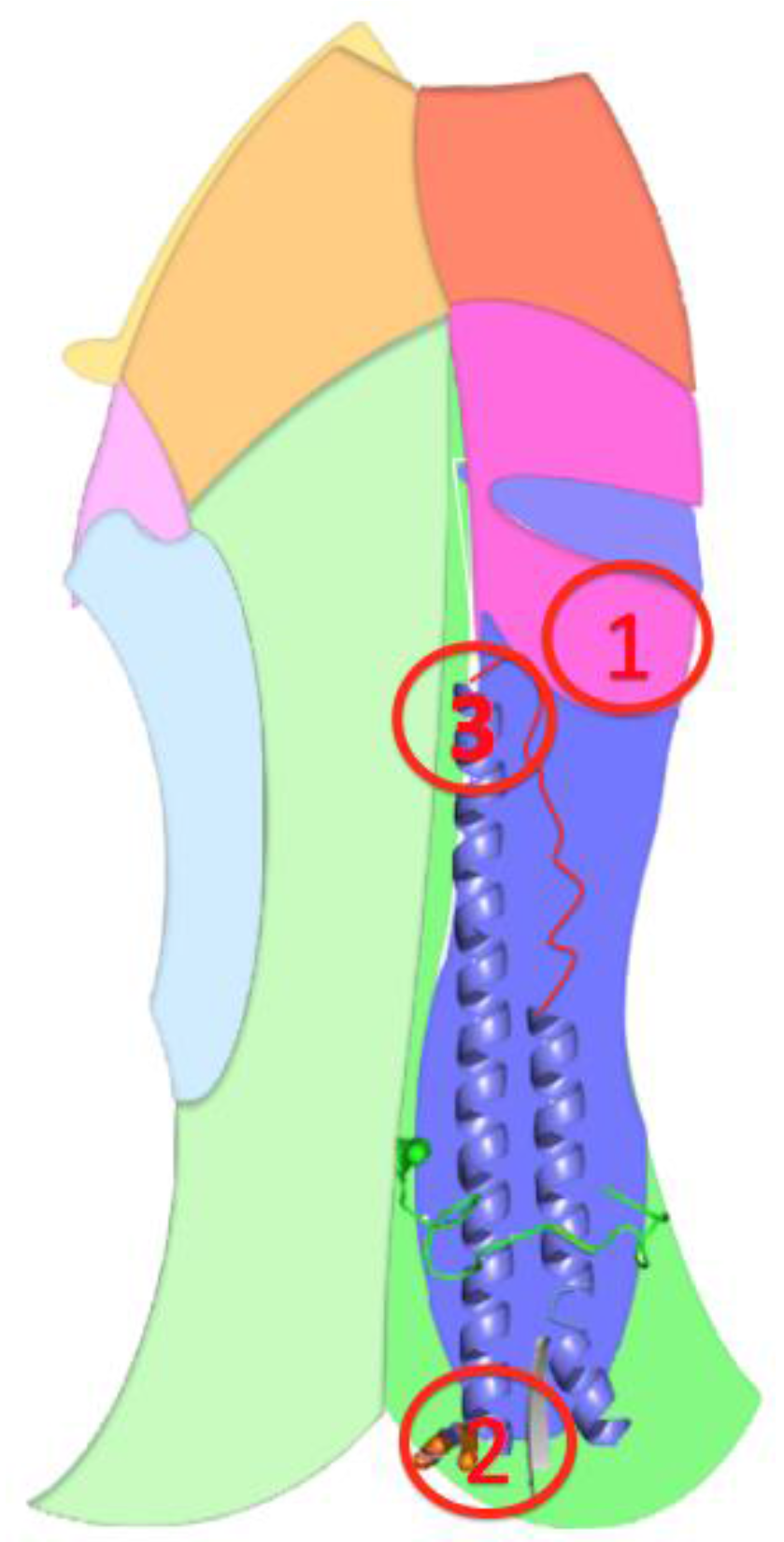
3.2.2. The Role of Hemagglutinin Domains
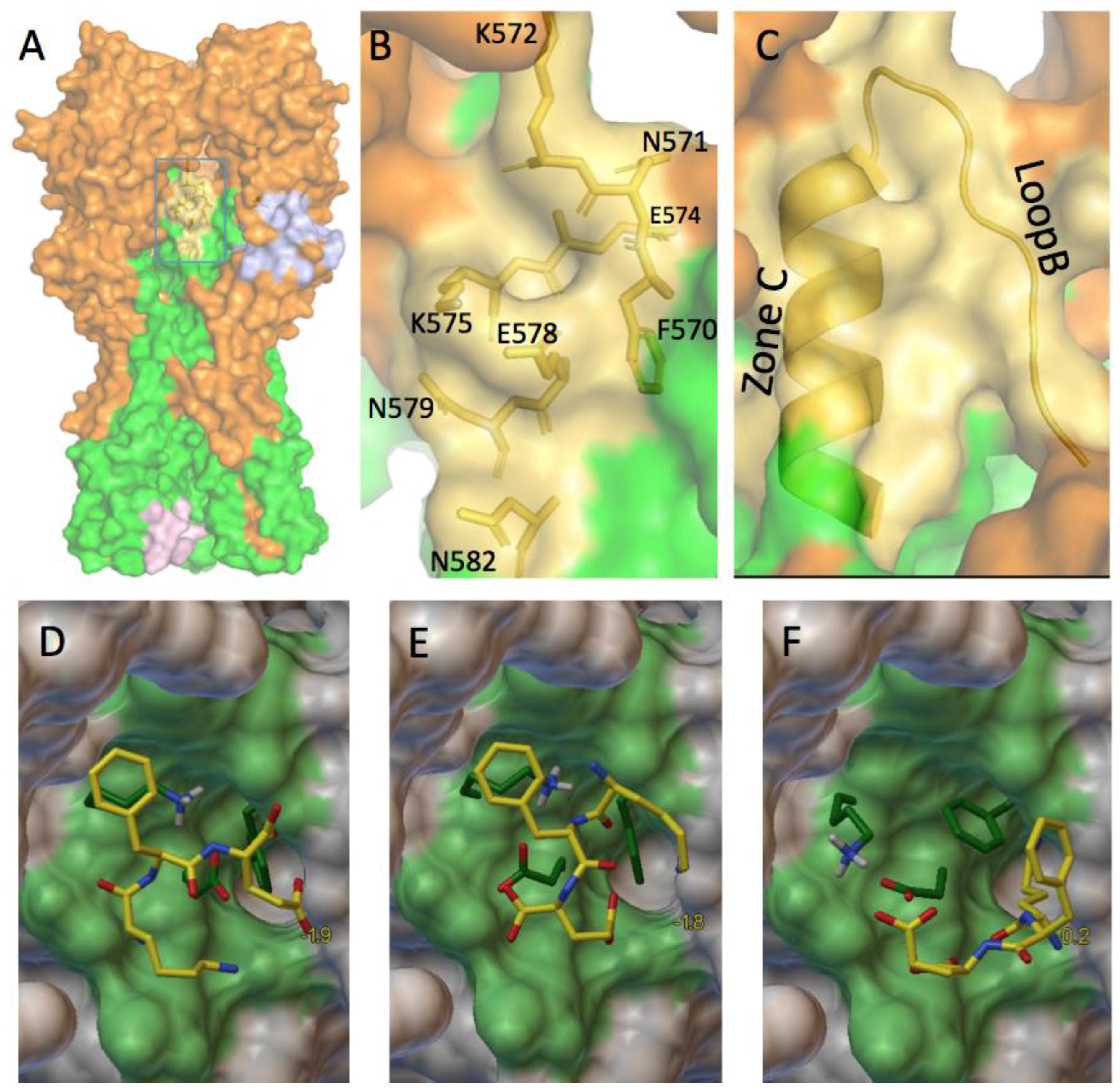
3.2.3. A Function for the Described Pockets?
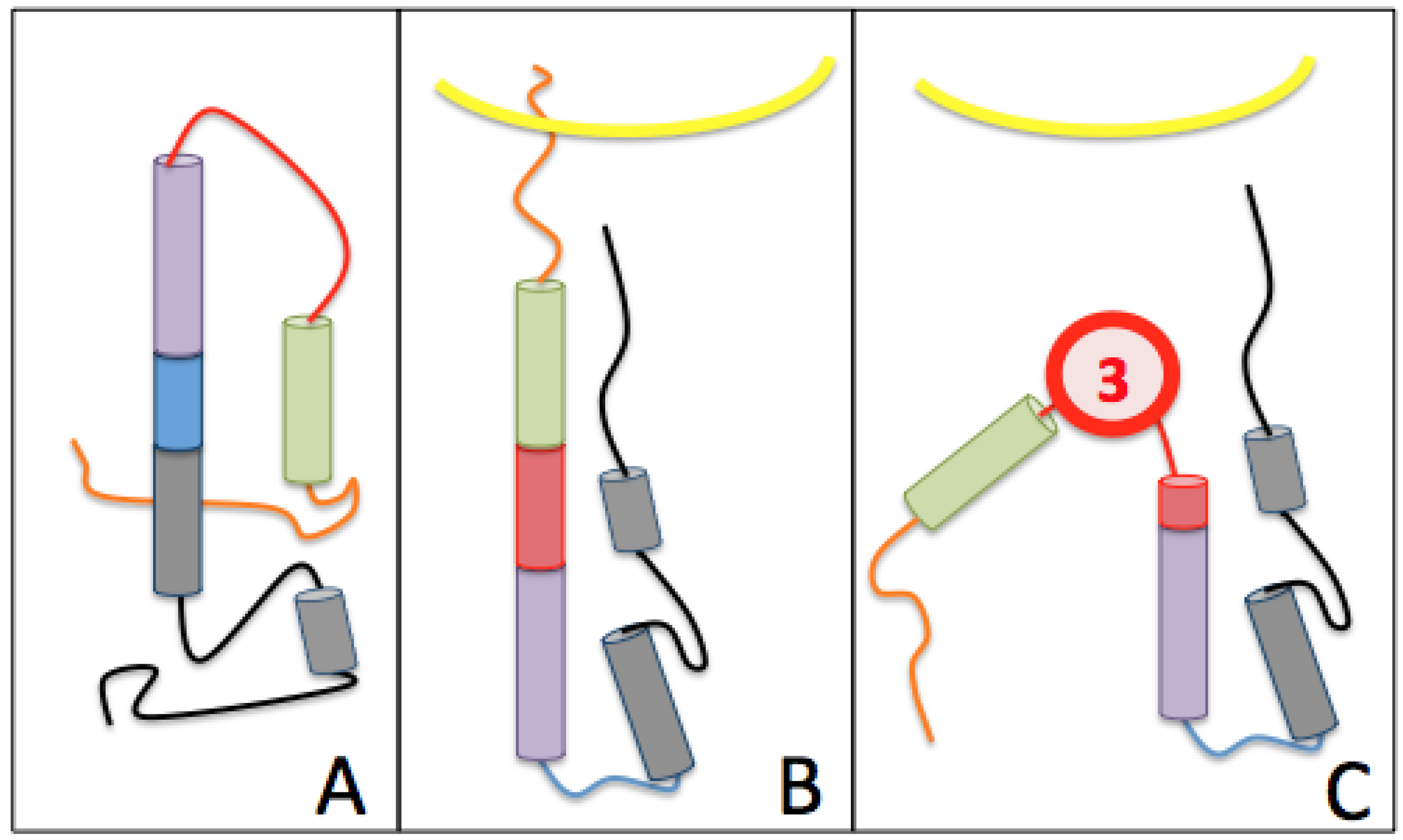
The Third Pocket Could Block the Spring-Loaded Transition
The First Pocket Could Block Structural Changes
The Second Pocket Could Block the Fusion Mechanism
3.3. Comparison of H1N1 and H3N2 Strains
3.4. Which Ligand Could Bind This Third Target?
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Steinhauer, D.A.; Wharton, S.A.; Skehel, J.J.; Wiley, D.C.; Hay, A.J. Amantadine selection of a mutant influenza virus containing an acid-stable hemagglutinin glycoprotein: Evidence for virus-specific regulation of the ph of glycoprotein transport vesicles. Proc. Natl. Acad. Sci. USA 1991, 88, 11525–11529. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Antiviral Drug Resistance among Influenza Viruses. Available online: http://www.cdc.gov/flu/professionals/antivirals/antiviral-drug-resistance.htm (accessed on 9 March 2016).
- Hayden, F.G.; de Jong, M.D. Emerging influenza antiviral resistance threats. J. Infect. Dis. 2011, 203, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.I.; Simonsen, L.; Viboud, C.; Miller, M.A.; Holmes, E.C. The origin and global emergence of adamantane resistant a/h3n2 influenza viruses. Virology 2009, 388, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Sheu, T.G.; Fry, A.M.; Garten, R.J.; Deyde, V.M.; Shwe, T.; Bullion, L.; Peebles, P.J.; Li, Y.; Klimov, A.I.; Gubareva, L.V. Dual resistance to adamantanes and oseltamivir among seasonal influenza a(H1N1) viruses: 2008–2010. J. Infect. Dis. 2011, 203, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Enserink, M. Drug resistance. A ‘wimpy’ flu strain mysteriously turns scary. Science 2009, 323, 1162–1163. [Google Scholar] [CrossRef] [PubMed]
- Baz, M.; Abed, Y.; Simon, P.; Hamelin, M.E.; Boivin, G. Effect of the neuraminidase mutation h274y conferring resistance to oseltamivir on the replicative capacity and virulence of old and recent human influenza a(H1N1) viruses. J. Infect. Dis. 2010, 201, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, J.E.; Prabu-Jeyabalan, M.; Cooper, D.; Henderson, G.J.; Harris, J.; Swanstrom, R.; Schiffer, C.A. Role of invariant thr80 in human immunodeficiency virus type 1 protease structure, function, and viral infectivity. J. Virol. 2006, 80, 6906–6916. [Google Scholar] [CrossRef] [PubMed]
- Cotter, C.R.; Jin, H.; Chen, Z. A single amino acid in the stalk region of the H1N1pdm influenza virus ha protein affects viral fusion, stability and infectivity. PLoS Pathog. 2014, 10, e1003831. [Google Scholar] [CrossRef] [PubMed]
- Brouillet, S.; Valere, T.; Ollivier, E.; Marsan, L.; Vanet, A. Co-lethality studied as an asset against viral drug escape: The hiv protease case. Biol. Direct. 2010, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, M.; Badel, A.; Veitia, R.A.; Vanet, A. Synthetic lethals in hiv: Ways to avoid drug resistance: Running title: Preventing hiv resistance. Biol. Direct. 2015, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Kuiken, H.J.; Beijersbergen, R.L. Exploration of synthetic lethal interactions as cancer drug targets. Future Oncol. 2010, 6, 1789–1802. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Paik, S. Genomics of adjuvant therapy for breast cancer. Cancer J. 2011, 17, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Bazin, C.; Coupaye, R.; Middendorp, S.; Vanet, A. Between compensatory mutations ans synthetic lethals: Genetic mutations, a new challenge for tomorrow’s medicine. Sci. Postprint 2014, 11, e00035. [Google Scholar]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef] [PubMed]
- Sriwilaijaroen, N.; Suzuki, Y. Molecular basis of the structure and function of h1 hemagglutinin of influenza virus. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 226–249. [Google Scholar] [CrossRef] [PubMed]
- Wiley, D.C.; Skehel, J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 1987, 56, 365–394. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.I.; Viboud, C.; Simonsen, L.; Bennett, R.T.; Griesemer, S.B.; St George, K.; Taylor, J.; Spiro, D.J.; Sengamalay, N.A.; Ghedin, E.; et al. Multiple reassortment events in the evolutionary history of H1N1 influenza a virus since 1918. PLoS Pathog. 2008, 4, e1000012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, A.H.; Fanning, T.G.; Janczewski, T.A.; Taubenberger, J.K. Characterization of the 1918 “spanish” influenza virus neuraminidase gene. Proc. Natl. Acad. Sci. USA 2000, 97, 6785–6790. [Google Scholar] [CrossRef] [PubMed]
- Verhoeyen, M.; Fang, R.; Jou, W.M.; Devos, R.; Huylebroeck, D.; Saman, E.; Fiers, W. Antigenic drift between the haemagglutinin of the hong kong influenza strains a/aichi/2/68 and a/victoria/3/75. Nature 1980, 286, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Bean, W.J.; Schell, M.; Katz, J.; Kawaoka, Y.; Naeve, C.; Gorman, O.; Webster, R.G. Evolution of the h3 influenza virus hemagglutinin from human and nonhuman hosts. J. Virol. 1992, 66, 1129–1138. [Google Scholar] [PubMed]
- Weis, W.I.; Brunger, A.T.; Skehel, J.J.; Wiley, D.C. Refinement of the influenza virus hemagglutinin by simulated annealing. J. Mol. Biol. 1990, 212, 737–761. [Google Scholar] [CrossRef]
- Gamblin, S.J.; Haire, L.F.; Russell, R.J.; Stevens, D.J.; Xiao, B.; Ha, Y.; Vasisht, N.; Steinhauer, D.A.; Daniels, R.S.; Elliot, A.; et al. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science 2004, 303, 1838–1842. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.J.; Kerry, P.S.; Stevens, D.J.; Steinhauer, D.A.; Martin, S.R.; Gamblin, S.J.; Skehel, J.J. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. USA 2008, 105, 17736–17741. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Antanasijevic, A.; Wang, M.; Li, B.; Mills, D.M.; Ames, J.A.; Nash, P.J.; Williams, J.D.; Peet, N.P.; Moir, D.T.; et al. New small molecule entry inhibitors targeting hemagglutinin-mediated influenza a virus fusion. J. Virol. 2014, 88, 1447–1460. [Google Scholar] [CrossRef] [PubMed]
- Richmond, T.J. Solvent accessible surface area and excluded volume in proteins. Analytical equations for overlapping spheres and implications for the hydrophobic effect. J. Mol. Biol. 1984, 178, 63–89. [Google Scholar] [CrossRef]
- Alland, C.; Moreews, F.; Boens, D.; Carpentier, M.; Chiusa, S.; Lonquety, M.; Renault, N.; Wong, Y.; Cantalloube, H.; Chomilier, J.; et al. Rpbs: A web resource for structural bioinformatics. Nucleic Acids Res. 2005, 33, W44–W49. [Google Scholar] [CrossRef] [PubMed]
- Noivirt, O.; Eisenstein, M.; Horovitz, A. Detection and reduction of evolutionary noise in correlated mutation analysis. Protein Eng. Des. Sel. 2005, 18, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Lewontin, R.C. The interaction of selection and linkage. I. General considerations; heterotic models. Genetics 1964, 49, 49–67. [Google Scholar] [PubMed]
- Lewontin, R.C. On measures of gametic disequilibrium. Genetics 1988, 120, 849–852. [Google Scholar] [PubMed]
- Wang, Q.; Lee, C. Distinguishing functional amino acid covariation from background linkage disequilibrium in hiv protease and reverse transcriptase. PLoS ONE 2007, 2, e814. [Google Scholar] [CrossRef] [PubMed]
- King, D.; Cherry, R.; Hu, W. Covariation of mutation pairs expressed in hiv-1 protease and reverse transcriptase genes subjected to varying treatments. J. Biomed. Sci. Eng. 2010, 3, 291–299. [Google Scholar] [CrossRef]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Carr, C.M.; Kim, P.S. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 1993, 73, 823–832. [Google Scholar] [CrossRef]
- Ginting, T.E.; Shinya, K.; Kyan, Y.; Makino, A.; Matsumoto, N.; Kaneda, S.; Kawaoka, Y. Amino acid changes in hemagglutinin contribute to the replication of oseltamivir-resistant H1N1 influenza viruses. J. Virol. 2012, 86, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wilson, I.A. Structural characterization of an early fusion intermediate of influenza virus hemagglutinin. J. Virol. 2011, 85, 5172–5182. [Google Scholar] [CrossRef] [PubMed]
- Mair, C.M.; Ludwig, K.; Herrmann, A.; Sieben, C. Receptor binding and ph stability—How influenza a virus hemagglutinin affects host-specific virus infection. Biochim. Biophys. Acta 2014, 1838, 1153–1168. [Google Scholar] [CrossRef] [PubMed]
- Kemble, G.W.; Bodian, D.L.; Rose, J.; Wilson, I.A.; White, J.M. Intermonomer disulfide bonds impair the fusion activity of influenza virus hemagglutinin. J. Virol. 1992, 66, 4940–4950. [Google Scholar] [PubMed]
- Masurel, N.; Marine, W.M. Recycling of asian and hong kong influenza a virus hemagglutinins in man. Am J. Epidemiol. 1973, 97, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Laver, W.G.; Webster, R.G. Studies on the origin of pandemic influenza. 3. Evidence implicating duck and equine influenza viruses as possible progenitors of the hong kong strain of human influenza. Virology 1973, 51, 383–391. [Google Scholar] [CrossRef]
- Kida, H.; Kawaoka, Y.; Naeve, C.W.; Webster, R.G. Antigenic and genetic conservation of h3 influenza virus in wild ducks. Virology 1987, 159, 109–119. [Google Scholar] [CrossRef]
- Kida, H.; Shortridge, K.F.; Webster, R.G. Origin of the hemagglutinin gene of h3n2 influenza viruses from pigs in china. Virology 1988, 162, 160–166. [Google Scholar] [CrossRef]
- Pielak, R.M.; Chou, J.J. Flu channel drug resistance: A tale of two sites. Protein Cell 2010, 1, 246–258. [Google Scholar] [CrossRef] [PubMed]
- World-Health-Organization. Influenza a (H1N1) Virus Resistance to Oseltamivir—Last Quarter 2007 to 4 April 2008. Available online: http://www.who.int/influenza/resources/documents/H1N1webupdate20090318_ed_ns.pdf (accessed on 18 March 2009).











{kind=link}
{kind=link}
{kind=link}
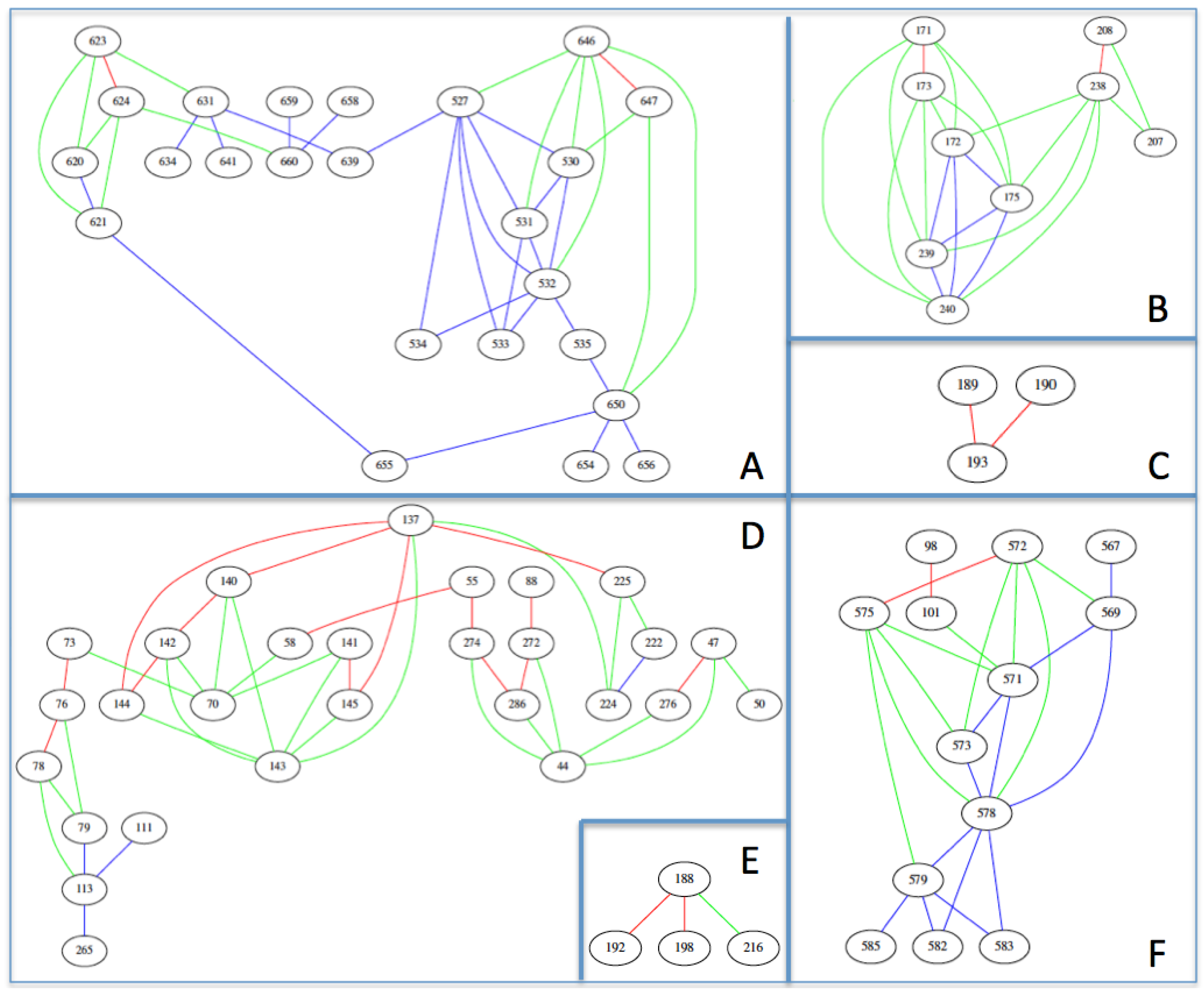{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AAi; AAj | Nobs | Nex | ξ | AAi; AAj | Nobs | Nex | ξ | |||
|---|---|---|---|---|---|---|---|---|---|---|
| i = 47; j = 276 | KD | 49 | 133.66 | −53.63 | i = 189; j = 193 | ES | 0 | 106.39 | −106.39 | |
| i = 55; j = 58 | HN | 9 | 99.86 | −82.67 | AA | 0 | 83.22 | −83.22 | ||
| i = 55; j = 274 | HQ | 4 | 5.87 | −0.60 | GS | 2 | 82.96 | −79.01 | ||
| HP | 10,722 | 10,507.82 | 4.37 | RS | 0 | 5.86 | −5.86 | |||
| i = 73; j = 76 | PT | 20 | 26.41 | −1.56 | SS | 3 | 5.86 | −1.39 | ||
| i = 76; j = 78 | TK | 0 | 100.76 | −100.76 | AN | 17 | 22.00 | −1.14 | ||
| TE | 2 | 74.34 | −70.40 | AS | 10,535 | 10,323.53 | 4.33 | |||
| PS | 3 | 15.60 | −10.17 | i = 190; j = 193 | NS | 3 | 7.81 | −2.96 | ||
| TS | 10,757 | 10,545.06 | 4.26 | i = 208; j = 238 | RK | 32 | 40.81 | −1.90 | ||
| i = 88; j = 272 | DD | 3 | 5.87 | −1.40 | i = 272; j = 286 | DL | 0 | 6.84 | −6.84 | |
| SD | 10,505 | 10,290.91 | 4.45 | EK | 3 | 5.66 | −1.25 | |||
| i = 98; j = 101 | YN | 0 | 42.02 | −42.02 | DE | 1810 | 1771.36 | 0.84 | ||
| NN | 5 | 13.93 | −5.72 | DK | 8927 | 8744.39 | 3.81 | |||
| i = 137; j = 140 | VP | 3 | 11.67 | −6.44 | i = 274; j = 286 | PL | 0 | 6.79 | −6.79 | |
| i = 137; j = 144 | VA | 6 | 11.67 | −2.75 | TK | 20 | 16.18 | 0.90 | ||
| i = 137; j = 145 | VK | 3 | 11.70 | −6.47 | PE | 1805 | 1758.74 | 1.22 | ||
| i = 137; j = 225 | TE | 0 | 5.61 | −5.61 | PK | 8852 | 8682.10 | 3.32 | ||
| i = 140; j = 142 | PK | 0 | 95.33 | −95.33 | i = 572; j = 574 | HR | 9 | 94.94 | −77.79 | |
| PN | 0 | 77.82 | −77.82 | i = 623; j = 624 | RT | 5 | 22.44 | −13.56 | ||
| PS | 3 | 5.84 | −1.38 | i = 646; j = 647 | DT | 32 | 36.77 | −0.62 | ||
| i = 141; j = 145 | YK | 11 | 29.25 | −11.38 | ||||||
| i = 142; j = 144 | KA | 0 | 95.28 | −95.28 | ||||||
| NA | 0 | 77.78 | −77.78 | |||||||
| i = 171; j = 173 | DE | 17 | 198.23 | −165.68 | ||||||
| i = 188; j = 192 | SR | 0 | 71.14 | −71.14 | ||||||
| SK | 0 | 68.38 | −68.38 | |||||||
| TR | 0 | 29.32 | −29.32 | |||||||
| TK | 0 | 28.18 | −28.18 | |||||||
| Pockets | SL + Inv | SL | Inv | Targets | Volume (Å3) |
|---|---|---|---|---|---|
| #9 | 47, 274, 286, 44, 55 | 4 | 1 | D | 488 |
| #32 | 621, 656, 655, 658, 624 | 1 | 4 | A | 175 |
| #45 | 575, 571, 578, 572, 579 | 2 | 3 | F | 98 |
| #67 | 659, 658, 660, 624, 656 | 1 | 4 | A | 166 |
| #83 | 572, 571, 578, 575, 582, 579 | 2 | 4 | F | 221 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lao, J.; Vanet, A. A New Strategy to Reduce Influenza Escape: Detecting Therapeutic Targets Constituted of Invariance Groups. Viruses 2017, 9, 38. https://doi.org/10.3390/v9030038
Lao J, Vanet A. A New Strategy to Reduce Influenza Escape: Detecting Therapeutic Targets Constituted of Invariance Groups. Viruses. 2017; 9(3):38. https://doi.org/10.3390/v9030038
Chicago/Turabian StyleLao, Julie, and Anne Vanet. 2017. "A New Strategy to Reduce Influenza Escape: Detecting Therapeutic Targets Constituted of Invariance Groups" Viruses 9, no. 3: 38. https://doi.org/10.3390/v9030038
APA StyleLao, J., & Vanet, A. (2017). A New Strategy to Reduce Influenza Escape: Detecting Therapeutic Targets Constituted of Invariance Groups. Viruses, 9(3), 38. https://doi.org/10.3390/v9030038