
Non-Canonical Roles of Dengue Virus Non-Structural Proteins


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
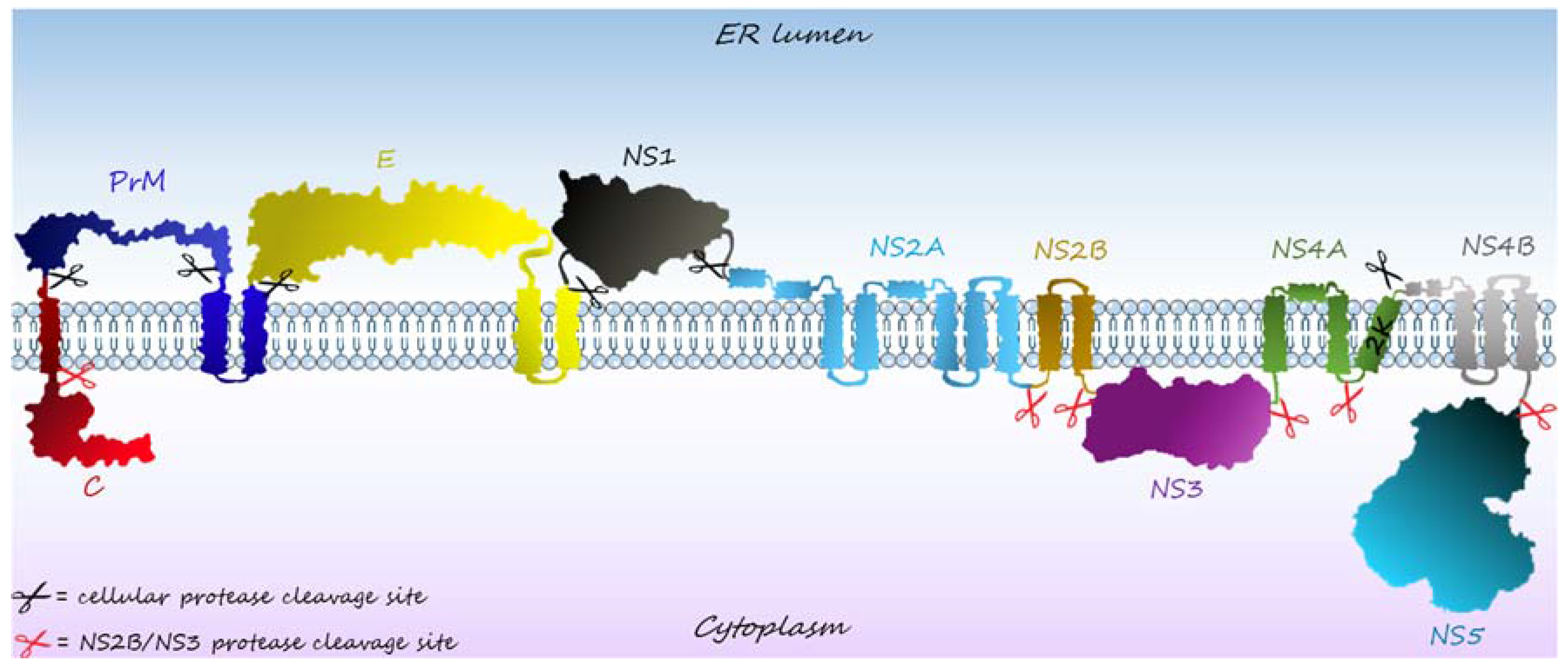
2. The Canonical Roles of Flaviviruses’ NSPs
3. Participation of DENV NSPs in Dengue Physiopathology
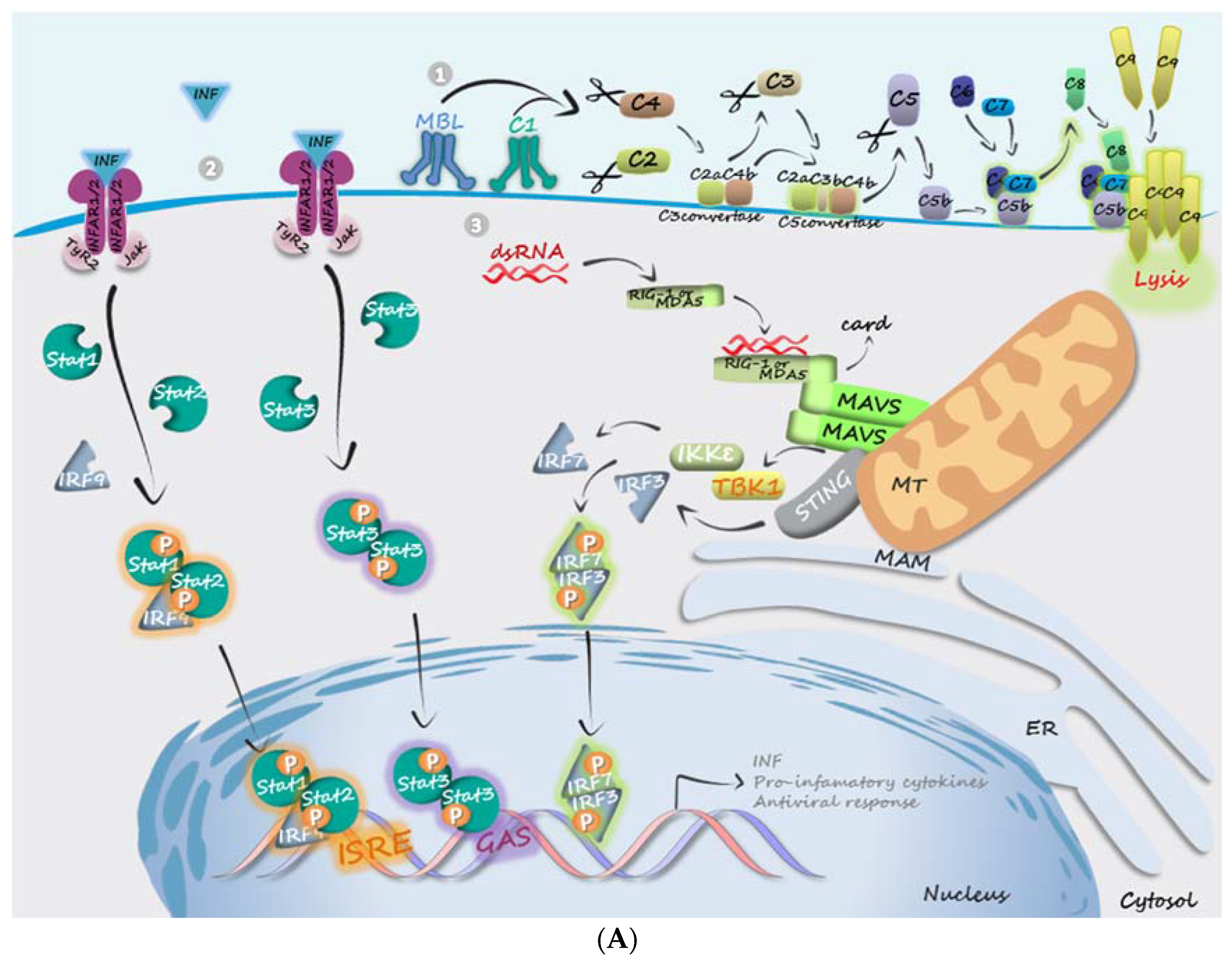
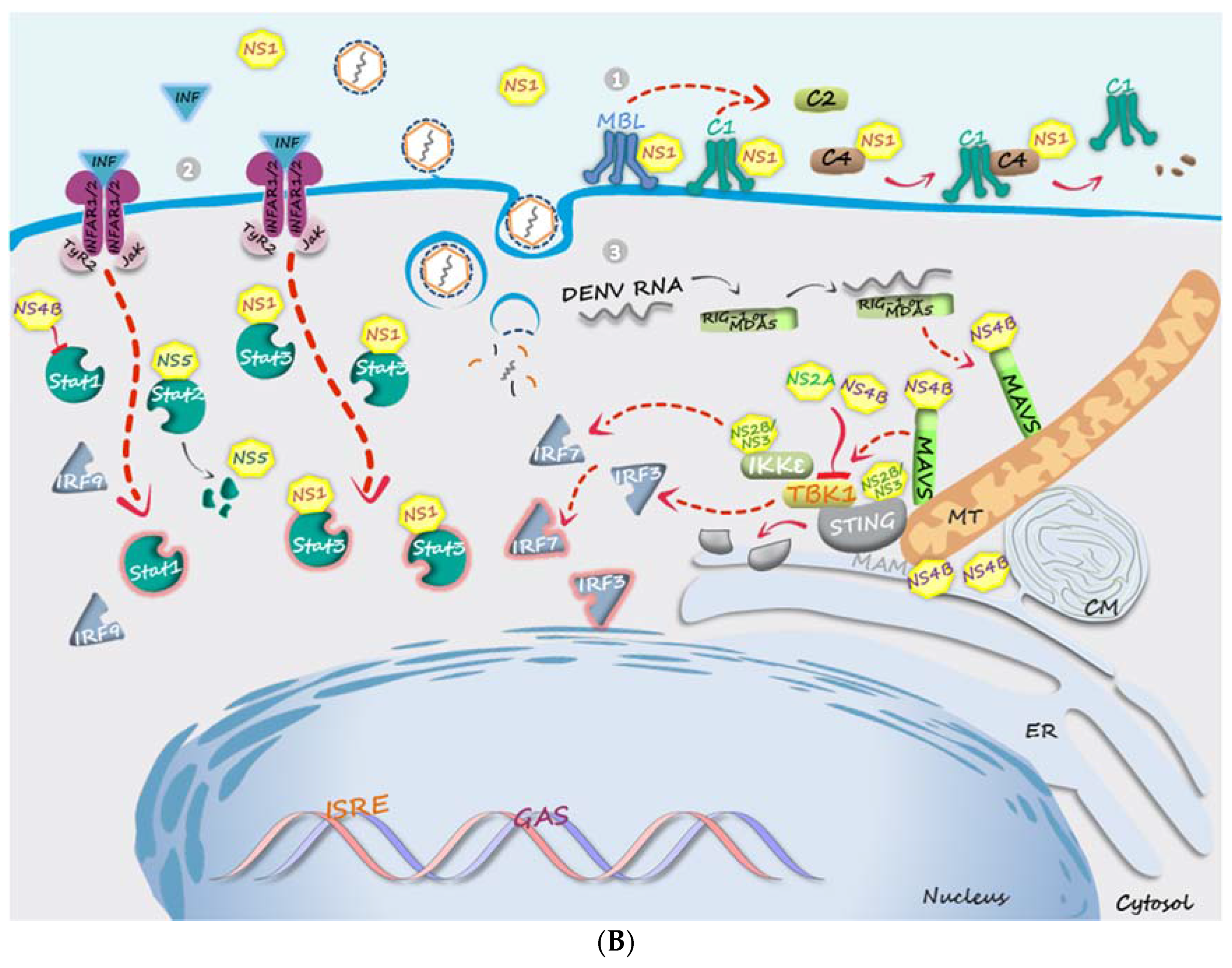
4. Evasion from Host Innate Immune Response
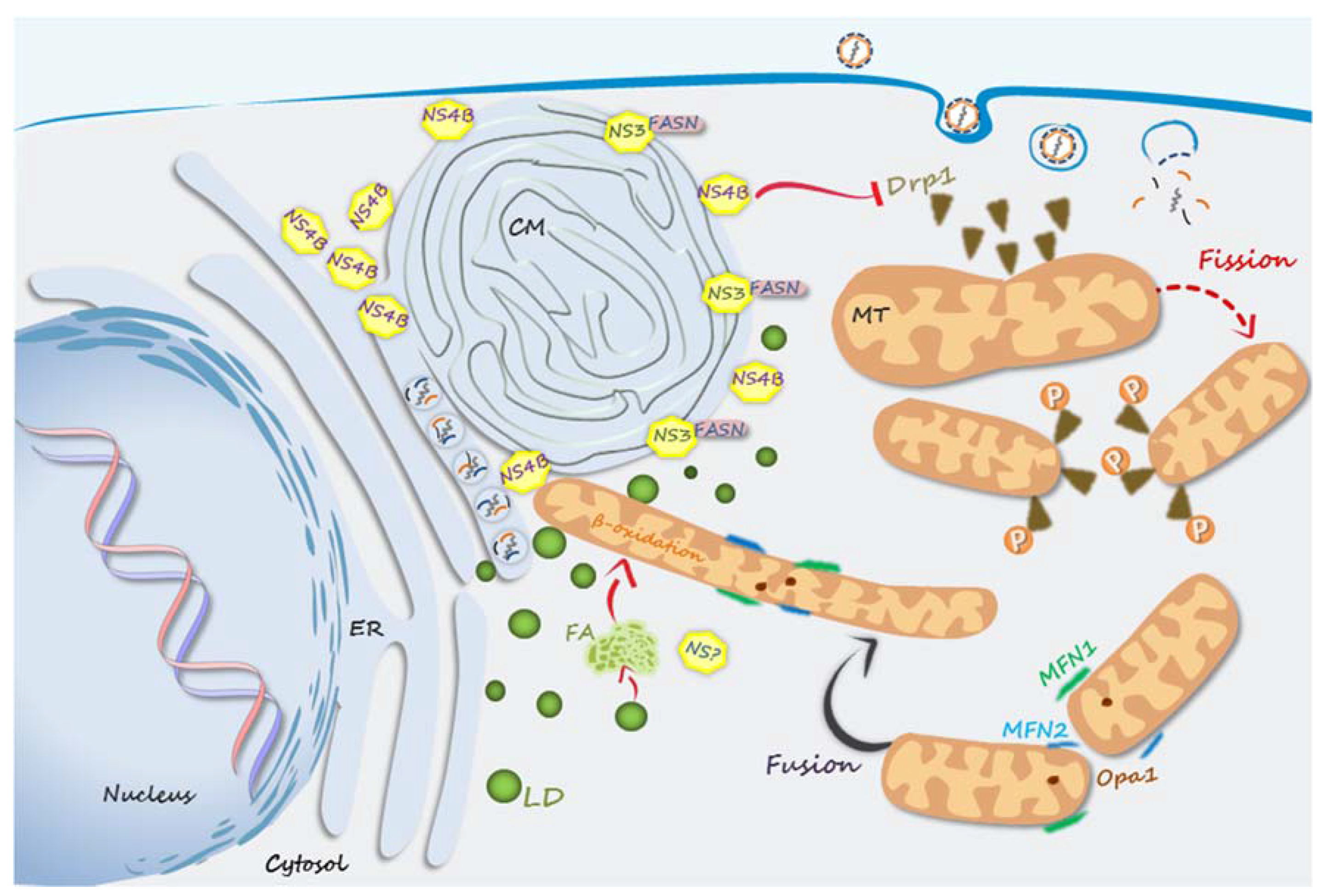
5. Metabolic Alterations
6. NSP-Induced Re-Localization of Host Proteins
Acknowledgments
Conflicts of Interest
References
- Apte-Sengupta, S.; Sirohi, D.; Kuhn, R.J. Coupling of replication and assembly in flaviviruses. Curr. Opin. Virol. 2014, 9, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Avirutnan, P.; Fuchs, A.; Hauhart, R.E.; Somnuke, P.; Youn, S.; Diamond, M.S.; Atkinson, J.P. Antagonism of the complement component C4 by flavivirus nonstructural protein NS1. J. Exp. Med. 2010, 207, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Somnuke, P.; Hauhart, R.E.; Atkinson, J.P.; Diamond, M.S.; Avirutnan, P. N-linked glycosylation of dengue virus NS1 protein modulates secretion, cell-surface expression, hexamer stability, and interactions with human complement. Virology 2011, 413, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Avirutnan, P.; Hauhart, R.E.; Somnuke, P.; Blom, A.M.; Diamond, M.S.; Atkinson, J.P. Binding of flavivirus nonstructural protein NS1 to C4b binding protein modulates complement activation. J. Immunol. 2011, 187, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.M.; Conde, J.N.; Allonso, D.; Nogueira, M.L.; Mohana-Borges, R. Mapping the interactions of dengue virus NS1 protein with human liver proteins using a yeast two-hybrid system: Identification of C1q as an interacting partner. PLoS ONE 2013, 8, e57514. [Google Scholar]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Liu, J.; Cheng, G. Vaccines and immunization strategies for dengue prevention. Emerg. Microbes Infect. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.B. Critique of World Health Organization Recommendation of a Dengue Vaccine. J. Infect. Dis. 2016, 214, 1793–1795. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.G.; Harris, E. Dengue. Lancet 2015, 385, 453–465. [Google Scholar] [CrossRef]
- Dengue: Guidelines for Diagnosis, Treatment, Prevention and Control, new ed.; World Health Organization: Geneva, Switzerland, 2009.
- Zou, J.; Xie, X.; Wang, Q.Y.; Dong, H.; Lee, M.Y.; Kang, C.; Yuan, Z.; Shi, P.Y. Characterization of dengue virus NS4A and NS4B protein interaction. J. Virol. 2015, 89, 3455–3470. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Lee le, T.; Wang, Q.Y.; Xie, X.; Lu, S.; Yau, Y.H.; Yuan, Z.; Geifman Shochat, S.; Kang, C.; Lescar, J.; et al. Mapping the Interactions between the NS4B and NS3 proteins of dengue virus. J. Virol. 2015, 89, 3471–3483. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J. Wrapping things up about virus RNA replication. Traffic 2005, 6, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Salonen, A.; Ahola, T.; Kaariainen, L. Viral RNA replication in association with cellular membranes. Curr. Top. Microbiol. Immunol. 2005, 285, 139–173. [Google Scholar] [PubMed]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell. Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Winkler, G.; Maxwell, S.E.; Ruemmler, C.; Stollar, V. Newly synthesized dengue-2 virus nonstructural protein NS1 is a soluble protein but becomes partially hydrophobic and membrane-associated after dimerization. Virology 1989, 171, 302–305. [Google Scholar] [CrossRef]
- Winkler, G.; Randolph, V.B.; Cleaves, G.R.; Ryan, T.E.; Stollar, V. Evidence that the mature form of the flavivirus nonstructural protein NS1 is a dimer. Virology 1988, 162, 187–196. [Google Scholar] [CrossRef]
- Parrish, C.R.; Woo, W.S.; Wright, P.J. Expression of the NS1 gene of dengue virus type 2 using vaccinia virus. Dimerisation of the NS1 glycoprotein. Arch. Virol. 1991, 117, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Flamand, M.; Megret, F.; Mathieu, M.; Lepault, J.; Rey, F.A.; Deubel, V. Dengue virus type 1 nonstructural glycoprotein NS1 is secreted from mammalian cells as a soluble hexamer in a glycosylation-dependent fashion. J. Virol. 1999, 73, 6104–6110. [Google Scholar] [PubMed]
- Mackenzie, J.M.; Jones, M.K.; Young, P.R. Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology 1996, 220, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Suthar, M.S.; Diamond, M.S.; Gale, M., Jr. West Nile virus infection and immunity. Nat. Rev. Microbiol. 2013, 11, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.A.; Young, P.R. The flavivirus NS1 protein: Molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker. Antivir. Res. 2013, 98, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Clum, S.; You, S.; Ebner, K.E.; Padmanabhan, R. The serine protease and RNA-stimulated nucleoside triphosphatase and RNA helicase functional domains of dengue virus type 2 NS3 converge within a region of 20 amino acids. J. Virol. 1999, 73, 3108–3116. [Google Scholar] [PubMed]
- Akey, D.L.; Brown, W.C.; Dutta, S.; Konwerski, J.; Jose, J.; Jurkiw, T.J.; DelProposto, J.; Ogata, C.M.; Skiniotis, G.; Kuhn, R.J.; et al. Flavivirus NS1 structures reveal surfaces for associations with membranes and the immune system. Science 2014, 343, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Krishna, V.D.; Rangappa, M.; Satchidanandam, V. Virus-specific cytolytic antibodies to nonstructural protein 1 of Japanese encephalitis virus effect reduction of virus output from infected cells. J. Virol. 2009, 83, 4766–4777. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, M.P.; Chambers, J.A.; Pankhong, P.; Chattergoon, M.; Attatippaholkun, W.; Dang, K.; Shah, N.; Weiner, D.B. Host cell killing by the West Nile Virus NS2B-NS3 proteolytic complex: NS3 alone is sufficient to recruit caspase-8-based apoptotic pathway. Virology 2006, 345, 56–72. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.Y.; Chang, T.H.; Liang, J.J.; Chiang, R.L.; Lee, Y.L.; Liao, C.L.; Lin, Y.L. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog. 2012, 8, e1002780. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.Y.; Li, Y.; Diao, F.; Lei, C.; He, X.; Zhang, L.; Tien, P.; et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 2008, 29, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Egloff, M.P.; Decroly, E.; Malet, H.; Selisko, B.; Benarroch, D.; Ferron, F.; Canard, B. Structural and functional analysis of methylation and 5′-RNA sequence requirements of short capped RNAs by the methyltransferase domain of dengue virus NS5. J. Mol. Biol. 2007, 372, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Chang, D.C.; Hua, M.H.; Lim, S.P.; Chionh, Y.H.; Hia, F.; Lee, Y.H.; Kukkaro, P.; Lok, S.M.; Dedon, P.C.; et al. 2′-O methylation of internal adenosine by flavivirus NS5 methyltransferase. PLoS Pathog. 2012, 8, e1002642. [Google Scholar] [CrossRef] [PubMed]
- Egloff, M.P.; Benarroch, D.; Selisko, B.; Romette, J.L.; Canard, B. An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: Crystal structure and functional characterization. EMBO J. 2002, 21, 2757–2768. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Padmanabhan, R. De novo synthesis of RNA by the dengue virus RNA-dependent RNA polymerase exhibits temperature dependence at the initiation but not elongation phase. J. Biol. Chem. 2001, 276, 39926–39937. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, J.F.; Rossignol, E.; Bullitt, E.; Kirkegaard, K. Enzymatic and nonenzymatic functions of viral RNA-dependent RNA polymerases within oligomeric arrays. RNA 2010, 16, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Buhler, S.; Bartenschlager, R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.E.; Wudzinska, A.; Datan, E.; Quaglino, D.; Zakeri, Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J. Biol. Chem. 2011, 286, 22147–22159. [Google Scholar] [CrossRef] [PubMed]
- Umareddy, I.; Chao, A.; Sampath, A.; Gu, F.; Vasudevan, S.G. Dengue virus NS4B interacts with NS3 and dissociates it from single-stranded RNA. J. Gen. Virol. 2006, 87, 2605–2614. [Google Scholar] [CrossRef] [PubMed]
- Young, P.R.; Hilditch, P.A.; Bletchly, C.; Halloran, W. An antigen capture enzyme-linked immunosorbent assay reveals high levels of the dengue virus protein NS1 in the sera of infected patients. J. Clin. Microbiol. 2000, 38, 1053–1057. [Google Scholar] [PubMed]
- Libraty, D.H.; Young, P.R.; Pickering, D.; Endy, T.P.; Kalayanarooj, S.; Green, S.; Vaughn, D.W.; Nisalak, A.; Ennis, F.A.; Rothman, A.L. High circulating levels of the dengue virus nonstructural protein NS1 early in dengue illness correlate with the development of dengue hemorrhagic fever. J. Infect. Dis. 2002, 186, 1165–1168. [Google Scholar] [CrossRef] [PubMed]
- Alcon, S.; Talarmin, A.; Debruyne, M.; Falconar, A.; Deubel, V.; Flamand, M. Enzyme-linked immunosorbent assay specific to Dengue virus type 1 nonstructural protein NS1 reveals circulation of the antigen in the blood during the acute phase of disease in patients experiencing primary or secondary infections. J. Clin. Microbiol. 2002, 40, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Gutsche, I.; Coulibaly, F.; Voss, J.E.; Salmon, J.; d’Alayer, J.; Ermonval, M.; Larquet, E.; Charneau, P.; Krey, T.; Megret, F.; et al. Secreted dengue virus nonstructural protein NS1 is an atypical barrel-shaped high-density lipoprotein. Proc. Natl. Acad. Sci. USA 2011, 108, 8003–8008. [Google Scholar] [CrossRef] [PubMed]
- Alcon-LePoder, S.; Drouet, M.T.; Roux, P.; Frenkiel, M.P.; Arborio, M.; Durand-Schneider, A.M.; Maurice, M.; Le Blanc, I.; Gruenberg, J.; Flamand, M. The secreted form of dengue virus nonstructural protein NS1 is endocytosed by hepatocytes and accumulates in late endosomes: Implications for viral infectivity. J. Virol. 2005, 79, 11403–11411. [Google Scholar] [CrossRef] [PubMed]
- Avirutnan, P.; Punyadee, N.; Noisakran, S.; Komoltri, C.; Thiemmeca, S.; Auethavornanan, K.; Jairungsri, A.; Kanlaya, R.; Tangthawornchaikul, N.; Puttikhunt, C.; et al. Vascular leakage in severe dengue virus infections: A potential role for the nonstructural viral protein NS1 and complement. J. Infect. Dis. 2006, 193, 1078–1088. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.F.; Chiu, S.C.; Hsiao, Y.L.; Wan, S.W.; Lei, H.Y.; Shiau, A.L.; Liu, H.S.; Yeh, T.M.; Chen, S.H.; Liu, C.C.; et al. Expression of cytokine, chemokine, and adhesion molecules during endothelial cell activation induced by antibodies against dengue virus nonstructural protein 1. J. Immunol. 2005, 174, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.F.; Lei, H.Y.; Shiau, A.L.; Liu, C.C.; Liu, H.S.; Yeh, T.M.; Chen, S.H.; Lin, Y.S. Antibodies from dengue patient sera cross-react with endothelial cells and induce damage. J. Med. Virol. 2003, 69, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Indian J. Anaesthesia 2014, 58, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.W.; Chuang, Y.C.; Lin, Y.S.; Lei, H.Y.; Liu, H.S.; Yeh, T.M. Dengue virus nonstructural protein NS1 binds to prothrombin/thrombin and inhibits prothrombin activation. J. Infect. 2012, 64, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Modhiran, N.; Watterson, D.; Muller, D.A.; Panetta, A.K.; Sester, D.P.; Liu, L.; Hume, D.A.; Stacey, K.J.; Young, P.R. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Beatty, P.R.; Puerta-Guardo, H.; Killingbeck, S.S.; Glasner, D.R.; Hopkins, K.; Harris, E. Dengue virus NS1 triggers endothelial permeability and vascular leak that is prevented by NS1 vaccination. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Puerta-Guardo, H.; Glasner, D.R.; Harris, E. Dengue virus NS1 disrupts the endothelial glycocalyx, leading to hyperpermeability. PLoS Pathog. 2016, 12, e1005738. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.G.; Robinson, P.J.; Bletchly, C.; Mackenzie, J.M.; Young, P.R. Dengue virus nonstructural protein 1 is expressed in a glycosyl-phosphatidylinositol-linked form that is capable of signal transduction. FASEB J. 2000, 14, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Mayor, S.; Riezman, H. Sorting GPI-anchored proteins. Nat. Rev. Mol. Cell Biol. 2004, 5, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Shenoy-Scaria, A.M.; Kwong, J.; Fujita, T.; Olszowy, M.W.; Shaw, A.S.; Lublin, D.M. Signal transduction through decay-accelerating factor. Interaction of glycosyl-phosphatidylinositol anchor and protein tyrosine kinases p56lck and p59fyn 1. J. Immunol. 1992, 149, 3535–3541. [Google Scholar] [PubMed]
- Lund-Johansen, F.; Olweus, J.; Symington, F.W.; Arli, A.; Thompson, J.S.; Vilella, R.; Skubitz, K.; Horejsi, V. Activation of human monocytes and granulocytes by monoclonal antibodies to glycosylphosphatidylinositol-anchored antigens. Eur. J. Immunol. 1993, 23, 2782–2791. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.C.; Yarwood, H.; Tarnok, A.; Sugars, K.; Harrison, A.A.; Robinson, P.J.; Haskard, D.O. Human Thy-1 is cytokine-inducible on vascular endothelial cells and is a signaling molecule regulated by protein kinase C. J. Immunol. 1996, 157, 874–883. [Google Scholar] [PubMed]
- Medin, C.L.; Fitzgerald, K.A.; Rothman, A.L. Dengue virus nonstructural protein NS5 induces interleukin-8 transcription and secretion. J. Virol. 2005, 79, 11053–11061. [Google Scholar] [CrossRef] [PubMed]
- Kelley, J.F.; Kaufusi, P.H.; Volper, E.M.; Nerurkar, V.R. Maturation of dengue virus nonstructural protein 4B in monocytes enhances production of dengue hemorrhagic fever-associated chemokines and cytokines. Virology 2011, 418, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Kelley, J.F.; Kaufusi, P.H.; Nerurkar, V.R. Dengue hemorrhagic fever-associated immunomediators induced via maturation of dengue virus nonstructural 4B protein in monocytes modulate endothelial cell adhesion molecules and human microvascular endothelial cells permeability. Virology 2012, 422, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Khunchai, S.; Junking, M.; Suttitheptumrong, A.; Kooptiwut, S.; Haegeman, G.; Limjindaporn, T.; Yenchitsomanus, P.T. NF-κB is required for dengue virus NS5-induced RANTES expression. Virus Res. 2015, 197, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Khunchai, S.; Junking, M.; Suttitheptumrong, A.; Yasamut, U.; Sawasdee, N.; Netsawang, J.; Morchang, A.; Chaowalit, P.; Noisakran, S.; Yenchitsomanus, P.T.; et al. Interaction of dengue virus nonstructural protein 5 with Daxx modulates RANTES production. Biochem. Biophys. Res. Commun. 2012, 423, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Makhluf, H.; Shresta, S. Innate antiviral immunity against dengue virus. Crit. Rev. Immunol. 2015, 35, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.; Aguirre, S.; Fernandez-Sesma, A. Innate immunity evasion by Dengue virus. Viruses 2012, 4, 397–413. [Google Scholar] [CrossRef] [PubMed]
- Stoermer, K.A.; Morrison, T.E. Complement and viral pathogenesis. Virology 2011, 411, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Thiemmeca, S.; Tamdet, C.; Punyadee, N.; Prommool, T.; Songjaeng, A.; Noisakran, S.; Puttikhunt, C.; Atkinson, J.P.; Diamond, M.S.; Ponlawat, A.; et al. Secreted NS1 protects dengue virus from mannose-binding lectin-mediated neutralization. J. Immunol. 2016, 197, 4053–4065. [Google Scholar] [CrossRef] [PubMed]
- Fullam, A.; Schroder, M. DExD/H-box RNA helicases as mediators of anti-viral innate immunity and essential host factors for viral replication. Biochim. Biophys. Acta 2013, 1829, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, Z.J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 2014, 32, 461–488. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Da Conceicao, T.M.; Rust, N.M.; Berbel, A.C.; Martins, N.B.; do Nascimento Santos, C.A.; Da Poian, A.T.; de Arruda, L.B. Essential role of RIG-I in the activation of endothelial cells by dengue virus. Virology 2013, 435, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Anglero-Rodriguez, Y.I.; Pantoja, P.; Sariol, C.A. Dengue virus subverts the interferon induction pathway via NS2B/3 protease-IkappaB kinase epsilon interaction. Clin. Vaccine Immunol. 2014, 21, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Dalrymple, N.A.; Cimica, V.; Mackow, E.R. Dengue virus NS proteins inhibit RIG-I/MAVS signaling by blocking TBK1/IRF3 phosphorylation: Dengue virus serotype 1 NS4A is a unique interferon-regulating virulence determinant. mBio 2015, 6, e00553-15. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Ye, W.; Yang, J.; Han, P.; Wang, Y.; Ye, C.; Weng, D.; Zhang, F.; Xu, Z.; Lei, Y. DDX21 translocates from nucleus to cytoplasm and stimulates the innate immune response due to dengue virus infection. Biochem. Biophys. Res. Commun. 2016, 473, 648–653. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhu, X.; Wen, W.; Yuan, J.; Hu, Y.; Chen, J.; An, S.; Dong, X.; Lin, C.; Yu, J.; et al. Dengue virus subverts host innate immunity by targeting adaptor protein MAVS. J. Virol. 2016, 90, 7219–7230. [Google Scholar] [CrossRef] [PubMed]
- Chatel-Chaix, L.; Cortese, M.; Romero-Brey, I.; Bender, S.; Neufeldt, C.J.; Fischl, W.; Scaturro, P.; Schieber, N.; Schwab, Y.; Fischer, B.; et al. Dengue virus perturbs mitochondrial morphodynamics to dampen innate immune responses. Cell. Host Microbe 2016, 20, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Chua, J.J.; Bhuvanakantham, R.; Chow, V.T.; Ng, M.L. Recombinant non-structural 1 (NS1) protein of dengue-2 virus interacts with human STAT3beta protein. Virus Res. 2005, 112, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Jordan, J.L.; Laurent-Rolle, M.; Ashour, J.; Martinez-Sobrido, L.; Ashok, M.; Lipkin, W.I.; Garcia-Sastre, A. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J. Virol. 2005, 79, 8004–8013. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Jordan, J.L.; Sanchez-Burgos, G.G.; Laurent-Rolle, M.; Garcia-Sastre, A. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. USA 2003, 100, 14333–14338. [Google Scholar] [CrossRef] [PubMed]
- Le Breton, M.; Meyniel-Schicklin, L.; Deloire, A.; Coutard, B.; Canard, B.; de Lamballerie, X.; Andre, P.; Rabourdin-Combe, C.; Lotteau, V.; Davoust, N. Flavivirus NS3 and NS5 proteins interaction network: A high-throughput yeast two-hybrid screen. BMC Microbiol. 2011, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, J.; Laurent-Rolle, M.; Maestre, A.M.; Rajsbaum, R.; Pisanelli, G.; Simon, V.; Mulder, L.C.; Fernandez-Sesma, A.; Garcia-Sastre, A. Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog. 2013, 9, e1003265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crabtree, H.G. The carbohydrate metabolism of certain pathological overgrowths. Biochem. J. 1928, 22, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- El-Bacha, T.; Da Poian, A.T. Virus-induced changes in mitochondrial bioenergetics as potential targets for therapy. Int. J. Biochem. Cell Biol. 2013, 45, 41–46. [Google Scholar] [CrossRef] [PubMed]
- El-Bacha, T.; Midlej, V.; Pereira da Silva, A.P.; Silva da Costa, L.; Benchimol, M.; Galina, A.; Da Poian, A.T. Mitochondrial and bioenergetic dysfunction in human hepatic cells infected with dengue 2 virus. Biochim. Biophys. Acta 2007, 1772, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, K.A.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Dengue virus induces and requires glycolysis for optimal replication. J. Virol. 2015, 89, 2358–2366. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Randall, G. Dengue virus-induced autophagy regulates lipid metabolism. Cell. Host Microbe 2010, 8, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Bartenschlager, R. Flaviviridae replication organelles: Oh, what a tangled web we weave. Annu. Rev. Virol. 2015, 2, 289–310. [Google Scholar] [CrossRef] [PubMed]
- Den Boon, J.A.; Diaz, A.; Ahlquist, P. Cytoplasmic viral replication complexes. Cell. Host Microbe 2010, 8, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Perera, R.; Berger, K.L.; Khadka, S.; Lacount, D.J.; Kuhn, R.J.; Randall, G. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 17345–17350. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.C.; Lin, R.J.; Liao, C.L.; Lin, Y.L. Rab18 facilitates dengue virus infection by targeting fatty acid synthase to sites of viral replication. J. Virol. 2014, 88, 6793–6804. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, S.; Cheng, J.; Tauchi-Sato, K.; Hatano, N.; Taniguchi, H.; Fujimoto, T. Rab18 localizes to lipid droplets and induces their close apposition to the endoplasmic reticulum-derived membrane. J. Cell Sci. 2005, 118, 2601–2611. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- Barbier, V.; Lang, D.; Valois, S.; Rothman, A.L.; Medin, C.L. Dengue virus induces mitochondrial elongation through impairment of Drp1-triggered mitochondrial fission. Virology 2017, 500, 149–160. [Google Scholar] [CrossRef]
- Hiscox, J.A. The interaction of animal cytoplasmic RNA viruses with the nucleus to facilitate replication. Virus Res. 2003, 95, 13–22. [Google Scholar] [CrossRef]
- Teo, C.S.; Chu, J.J. Cellular vimentin regulates construction of dengue virus replication complexes through interaction with NS4A protein. J. Virol. 2014, 88, 1897–1913. [Google Scholar] [CrossRef] [PubMed]
- Styers, M.L.; Salazar, G.; Love, R.; Peden, A.A.; Kowalczyk, A.P.; Faundez, V. The endo-lysosomal sorting machinery interacts with the intermediate filament cytoskeleton. Mol. Biol. Cell 2004, 15, 5369–5382. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Salazar, M.; Angel-Ambrocio, A.H.; Soto-Acosta, R.; Bautista-Carbajal, P.; Hurtado-Monzon, A.M.; Alcaraz-Estrada, S.L.; Ludert, J.E.; Del Angel, R.M. Dengue virus NS1 protein interacts with the ribosomal protein RPL18: This interaction is required for viral translation and replication in Huh-7 cells. Virology 2015, 484, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Allonso, D.; Andrade, I.S.; Conde, J.N.; Coelho, D.R.; Rocha, D.C.; da Silva, M.L.; Ventura, G.T.; Silva, E.M.; Mohana-Borges, R. Dengue virus NS1 protein modulates cellular energy metabolism by increasing glyceraldehyde-3-phosphate dehydrogenase activity. J. Virol. 2015, 89, 11871–11883. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Liu, M.L.; Tien, C.F.; Chou, S.J.; Chang, R.Y. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) interaction with 3′ ends of Japanese encephalitis virus RNA and colocalization with the viral NS5 protein. J. Biomed. Sci. 2009, 16. [Google Scholar] [CrossRef] [PubMed]
- De, B.P.; Gupta, S.; Zhao, H.; Drazba, J.A.; Banerjee, A.K. Specific interaction in vitro and in vivo of glyceraldehyde-3-phosphate dehydrogenase and LA protein with cis-acting RNAs of human parainfluenza virus type 3. J. Biol. Chem. 1996, 271, 24728–24735. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.E.; Hardin, C.C.; Lemon, S.M. Specific interaction of glyceraldehyde 3-phosphate dehydrogenase with the 5′-nontranslated RNA of hepatitis A virus. J. Biol. Chem. 1996, 271, 14134–14142. [Google Scholar] [PubMed]
- Petrik, J.; Parker, H.; Alexander, G.J. Human hepatic glyceraldehyde-3-phosphate dehydrogenase binds to the poly(U) tract of the 3′ non-coding region of hepatitis C virus genomic RNA. J. Gen. Virol. 1999, 80, 3109–3113. [Google Scholar] [CrossRef] [PubMed]
- Sirover, M.A. New insights into an old protein: The functional diversity of mammalian glyceraldehyde-3-phosphate dehydrogenase. Biochim. Biophys. Acta 1999, 1432, 159–184. [Google Scholar] [CrossRef]
- Prasanth, K.R.; Huang, Y.W.; Liou, M.R.; Wang, R.Y.; Hu, C.C.; Tsai, C.H.; Meng, M.; Lin, N.S.; Hsu, Y.H. Glyceraldehyde 3-phosphate dehydrogenase negatively regulates the replication of Bamboo mosaic virus and its associated satellite RNA. J. Virol. 2011, 85, 8829–8840. [Google Scholar] [CrossRef] [PubMed]
- Chua, J.J.; Ng, M.M.; Chow, V.T. The non-structural 3 (NS3) protein of dengue virus type 2 interacts with human nuclear receptor binding protein and is associated with alterations in membrane structure. Virus Res. 2004, 102, 151–163. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeidler, J.D.; Fernandes-Siqueira, L.O.; Barbosa, G.M.; Da Poian, A.T. Non-Canonical Roles of Dengue Virus Non-Structural Proteins. Viruses 2017, 9, 42. https://doi.org/10.3390/v9030042
Zeidler JD, Fernandes-Siqueira LO, Barbosa GM, Da Poian AT. Non-Canonical Roles of Dengue Virus Non-Structural Proteins. Viruses. 2017; 9(3):42. https://doi.org/10.3390/v9030042
Chicago/Turabian StyleZeidler, Julianna D., Lorena O. Fernandes-Siqueira, Glauce M. Barbosa, and Andrea T. Da Poian. 2017. "Non-Canonical Roles of Dengue Virus Non-Structural Proteins" Viruses 9, no. 3: 42. https://doi.org/10.3390/v9030042
APA StyleZeidler, J. D., Fernandes-Siqueira, L. O., Barbosa, G. M., & Da Poian, A. T. (2017). Non-Canonical Roles of Dengue Virus Non-Structural Proteins. Viruses, 9(3), 42. https://doi.org/10.3390/v9030042