
Viral Protein Kinetics of Piscine Orthoreovirus Infection in Atlantic Salmon Blood Cells


 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction and Expression of Recombinant Piscine orthoreovirus (PRV) μNS
2.2. Construction and Expression of Recombinant PRV λ1
2.3. Protein Purification
2.4. Immunization of Rabbits
2.5. Specificity of Antisera
2.6. Experimental Challenge of Salmon
2.7. RNA Isolation and Reverse Transcription Quantiative Polymerase Chain Reaction (RT-qPCR)
2.8. Flow Cytometry
2.9. Immunofluorescence Microscopy
2.10. Transmission Electron Microscopy (TEM)
2.11. Western Blotting (WB)
2.12. Immunoprecipitation (IP)
2.13. Liquid Chromatography–Mass Spectrometry (LC–MS)
2.14. Computational Analysis
2.15. Statistical Analysis
3. Results
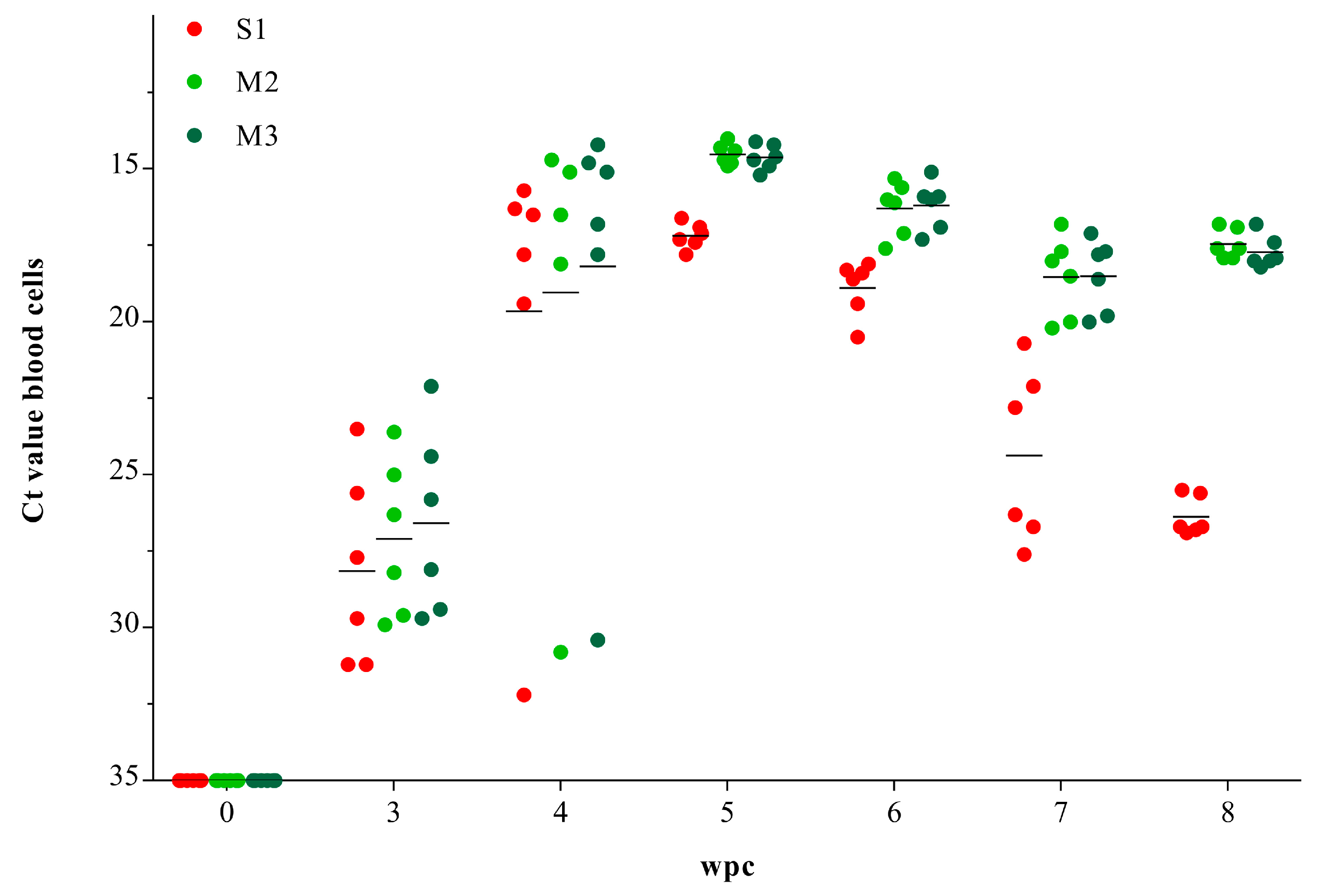
3.1. Viral RNA Load in Blood Cells
3.2. Expression of Innate Antiviral Genes in PRV Infected Blood Cells
3.3. Flow Cytometry Indicates a Transient Peak in Blood Cells
3.4. Viral Factories Observed in Blood Cells
3.5. TEM of PRV Infected Blood Cells
3.6. µNS Protein Expression in Individual Fish Correlate with viral RNA only during the Acute Phase of Infection
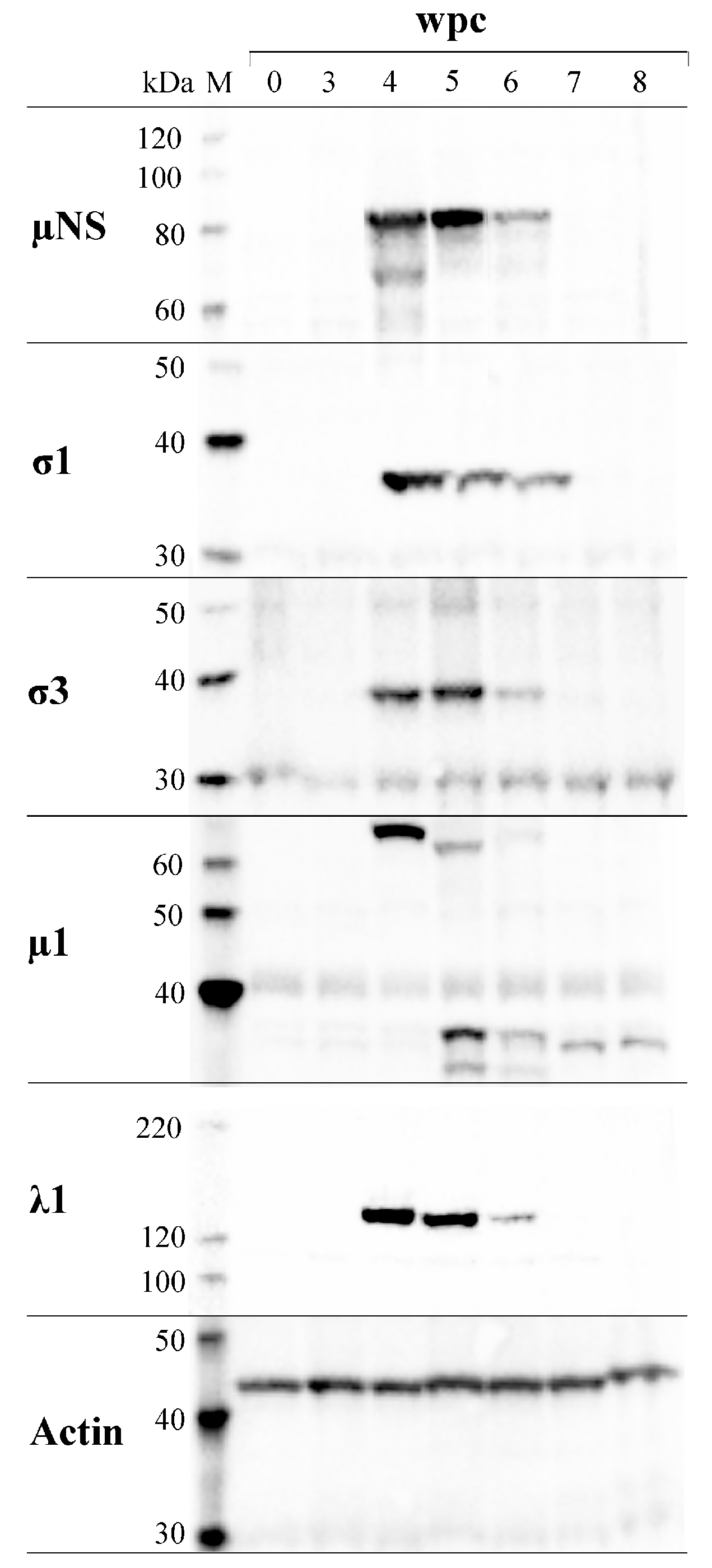
3.7. PRV Protein Levels Display a Transient Peak in Blood Cells
3.8. PRV Proteins Interact with µNS
3.9. µNS Exists in Two Forms
3.10. µ1 Has Two Putative Proteolytic Cleavage Sites
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Palacios, G.; Lovoll, M.; Tengs, T.; Hornig, M.; Hutchison, S.; Hui, J.; Kongtorp, R.T.; Savji, N.; Bussetti, A.V.; Solovyov, A.; et al. Heart and skeletal muscle inflammation of farmed salmon is associated with infection with a novel reovirus. PLoS ONE 2010, 5, e11487. [Google Scholar] [CrossRef] [PubMed]
- Markussen, T.; Dahle, M.K.; Tengs, T.; Lovoll, M.; Finstad, O.W.; Wiik-Nielsen, C.R.; Grove, S.; Lauksund, S.; Robertsen, B.; Rimstad, E. Sequence analysis of the genome of piscine orthoreovirus (PRV) associated with heart and skeletal muscle inflammation (HSMI) in Atlantic salmon (Salmo salar). PLoS ONE 2013, 8, e70075. [Google Scholar] [CrossRef]
- Day, J.M. The diversity of the orthoreoviruses: Molecular taxonomy and phylogentic divides. Infect. Genet. Evol. 2009, 9, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Finstad, O.W.; Falk, K.; Lovoll, M.; Evensen, O.; Rimstad, E. Immunohistochemical detection of piscine reovirus (PRV) in hearts of Atlantic salmon coincide with the course of heart and skeletal muscle inflammation (HSMI). Vet. Res 2012, 43, 27. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.B.; Hjortaas, M.; Tengs, T.; Hellberg, H.; Johansen, R. First description of a new disease in Rainbow Trout (Oncorhynchus mykiss (Walbaum)) similar to Heart and skeletal muscle inflammation (HSMI) and detection of a gene sequence related to Piscine Orthoreovirus (PRV). PLoS ONE 2015, 10, e0131638. [Google Scholar] [CrossRef] [PubMed]
- Sibley, S.D.; Finley, M.A.; Baker, B.B.; Puzach, C.; Armien, A.G.; Giehtbrock, D.; Goldberg, T.L. Novel reovirus associated with epidemic mortality in wild Largemouth Bass (Micropterus salmoides). J. Gen. Virol. 2016, 97, 2482–2487. [Google Scholar] [PubMed]
- Takano, T.; Nawata, A.; Sakai, T.; Matsuyama, T.; Ito, T.; Kurita, J.; Terashima, S.; Yasuike, M.; Nakamura, Y.; Fujiwara, A.; et al. Full-Genome sequencing and confirmation of the causative agent of Erythrocytic inclusion body syndrome in Coho Salmon identifies a new type of Piscine Orthoreovirus. PLoS ONE 2016, 11, e0165424. [Google Scholar] [CrossRef] [PubMed]
- Kibenge, M.J.; Iwamoto, T.; Wang, Y.; Morton, A.; Godoy, M.G.; Kibenge, F.S. Whole-genome analysis of piscine reovirus (PRV) shows PRV represents a new genus in family Reoviridae and its genome segment S1 sequences group it into two separate sub-genotypes. Virol. J. 2013, 10, 230. [Google Scholar] [CrossRef] [PubMed]
- Garver, K.A.; Johnson, S.C.; Polinski, M.P.; Bradshaw, J.C.; Marty, G.D.; Snyman, H.N.; Morrison, D.B.; Richard, J. Piscine Orthoreovirus from Western North America is transmissible to Atlantic Salmon and Sockeye Salmon but fails to cause Heart and skeletal muscle inflammation. PLoS ONE 2016, 11, e0146229. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, H.W.; Kongtorp, R.T.; Taksdal, T.; Graham, D.; Falk, K. An outbreak of disease resembling heart and skeletal muscle inflammation in Scottish farmed salmon, Salmo salar L., with observations on myocardial regeneration. J. Fish Dis. 2005, 28, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Lovoll, M.; Alarcon, M.; Bang Jensen, B.; Taksdal, T.; Kristoffersen, A.B.; Tengs, T. Quantification of Piscine reovirus (PRV) at different stages of Atlantic salmon Salmo salar production. Dis. Aquat. Organ. 2012, 99, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Bjorgen, H.; Wessel, O.; Fjelldal, P.G.; Hansen, T.; Sveier, H.; Saebo, H.R.; Enger, K.B.; Monsen, E.; Kvellestad, A.; Rimstad, E.; et al. Piscine orthoreovirus (PRV) in red and melanised foci in white muscle of Atlantic salmon (Salmo salar). Vet. Res. 2015, 46, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kongtorp, R.T.; Halse, M.; Taksdal, T.; Falk, K. Longitudinal study of a natural outbreak of heart and skeletal muscle inflammation in Atlantic salmon, Salmo salar L. J. Fish Dis. 2006, 29, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Wiik-Nielsen, C.R.; Ski, P.M.; Aunsmo, A.; Lovoll, M. Prevalence of viral RNA from piscine reovirus and piscine myocarditis virus in Atlantic salmon, Salmo salar L., broodfish and progeny. J. Fish Dis. 2012, 35, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Johansen, L.H.; Dahle, M.K.; Wessel, O.; Timmerhaus, G.; Lovoll, M.; Rosaeg, M.; Jorgensen, S.M.; Rimstad, E.; Krasnov, A. Differences in gene expression in Atlantic salmon parr and smolt after challenge with Piscine orthoreovirus (PRV). Mol. Immunol. 2016, 73, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Wessel, O.; Olsen, C.M.; Rimstad, E.; Dahle, M.K. Piscine orthoreovirus (PRV) replicates in Atlantic salmon (Salmo salar L.) erythrocytes ex vivo. Vet. Res. 2015, 46, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morera, D.; Roher, N.; Ribas, L.; Balasch, J.C.; Donate, C.; Callol, A.; Boltana, S.; Roberts, S.; Goetz, G.; Goetz, F.W.; et al. RNA-Seq reveals an integrated immune response in nucleated erythrocytes. PLoS ONE 2011, 6, e26998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahle, M.K.; Wessel, O.; Timmerhaus, G.; Nyman, I.B.; Jorgensen, S.M.; Rimstad, E.; Krasnov, A. Transcriptome analyses of Atlantic salmon (Salmo salar L.) erythrocytes infected with piscine orthoreovirus (PRV). Fish Shellfish Immunol. 2015, 45, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Finstad, O.W.; Dahle, M.K.; Lindholm, T.H.; Nyman, I.B.; Lovoll, M.; Wallace, C.; Olsen, C.M.; Storset, A.K.; Rimstad, E. Piscine orthoreovirus (PRV) infects Atlantic salmon erythrocytes. Vet. Res. 2014, 45, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kongtorp, R.T.; Taksdal, T.; Lyngoy, A. Pathology of heart and skeletal muscle inflammation (HSMI) in farmed Atlantic salmon Salmo salar. Dis. Aquat. Organ. 2004, 59, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Netherton, C.; Moffat, K.; Brooks, E.; Wileman, T. A guide to viral inclusions, membrane rearrangements, factories, and viroplasm produced during virus replication. Adv. Virus Res. 2007, 70, 101–182. [Google Scholar] [PubMed]
- Knipe, D.M.; Howley, P.M. Fields Virology, 5th ed.; Wolters Kluwer/Lippincott Williams & Wilkins Health: Philadelphia, PA, USA, 2007; pp. 1854–1858. [Google Scholar]
- Lee, P.W.; Hayes, E.C.; Joklik, W.K. Protein sigma 1 is the reovirus cell attachment protein. Virology 1981, 108, 156–163. [Google Scholar] [CrossRef]
- Nibert, M.L.; Fields, B.N. A carboxy-terminal fragment of protein mu 1/mu 1C is present in infectious subvirion particles of mammalian reoviruses and is proposed to have a role in penetration. J. Virol. 1992, 66, 6408–6418. [Google Scholar] [PubMed]
- Thete, D.; Snyder, A.J.; Mainou, B.A.; Danthi, P. Reovirus mu1 protein affects infectivity by altering virus-receptor interactions. J. Virol. 2016, 90, 10951–10962. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.M.; Peters, T.R.; Dermody, T.S. Reovirus ơNS and µNS proteins form cytoplasmic inclusion structures in the absence of viral infection. J. Virol. 2003, 77, 5948–5963. [Google Scholar] [CrossRef] [PubMed]
- Schiff, L.A.; Nibert, M.L.; Tyler, K.L. Orthoreoviruses and their replication. In Fields virology, 5th ed.; Knipe, D.M., Howley, P.M., Fields, B.N., Eds.; Wolters Kluwer/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 2, pp. 1853–1915. [Google Scholar]
- Jayasuriya, A.K.; Nibert, M.L.; Fields, B.N. Complete nucleotide sequence of the M2 gene segment of reovirus type 3 dearing and analysis of its protein product mu 1. Virology 1988, 163, 591–602. [Google Scholar] [CrossRef]
- Duncan, R. The low pH-dependent entry of avian reovirus is accompanied by two specific cleavages of the major outer capsid protein mu 2C. Virology 1996, 219, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Wiener, J.R.; Joklik, W.K. Evolution of reovirus genes: A comparison of serotype 1, 2, and 3 M2 genome segments, which encode the major structural capsid protein mu 1C. Virology 1988, 163, 603–613. [Google Scholar] [CrossRef]
- Haatveit, H.M.; Nyman, I.B.; Markussen, T.; Wessel, O.; Dahle, M.K.; Rimstad, E. The non-structural protein µNS of piscine orthoreovirus (PRV) forms viral factory-like structures. Vet. Res. 2016, 47, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.S.; Broering, T.J.; Kim, J.; Higgins, D.E.; Nibert, M.L. Reovirus core protein mu2 determines the filamentous morphology of viral inclusion bodies by interacting with and stabilizing microtubules. J. Virol. 2002, 76, 4483–4496. [Google Scholar] [CrossRef] [PubMed]
- Busch, L.K.; Rodriguez-Grille, J.; Casal, J.I.; Martinez-Costas, J.; Benavente, J. Avian and mammalian reoviruses use different molecular mechanisms to synthesize their microNS isoforms. J. Gen. Virol. 2011, 92, 2566–2574. [Google Scholar] [CrossRef] [PubMed]
- Touris-Otero, F.; Martinez-Costas, J.; Vakharia, V.N.; Benavente, J. Avian reovirus nonstructural protein microNS forms viroplasm-like inclusions and recruits protein sigmaNS to these structures. Virology 2004, 319, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Wiener, J.R.; Bartlett, J.A.; Joklik, W.K. The sequences of reovirus serotype 3 genome segments M1 and M3 encoding the minor protein mu 2 and the major nonstructural protein mu NS, respectively. Virology 1989, 169, 293–304. [Google Scholar] [CrossRef]
- McCutcheon, A.M.; Broering, T.J.; Nibert, M.L. Mammalian reovirus M3 gene sequences and conservation of coiled-coil motifs near the carboxyl terminus of the microNS protein. Virology 1999, 264, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Koehler, C.J.; Bollineni, R.C.; Thiede, B. Application of the half decimal place rule to increase the peptide identification rate. Rapid Commun. Mass Spectrom. 2016, 31, 227–233. [Google Scholar] [CrossRef] [PubMed]
- ExPASy Bioinformatics Resource Portal. Available online: http://web.expasy.org/compute_pi/ (accessed on 1 April 2016).
- UCL Department Of Computer Science. Available online: http://bioinf.cs.ucl.ac.uk/psipred/ (accessed on 1 April 2016).
- Su, J.; Zhang, R.; Dong, J.; Yang, C. Evaluation of internal control genes for qRT-PCR normalization in tissues and cell culture for antiviral studies of grass carp (Ctenopharyngodon idella). Fish Shellfish Immunol. 2011, 30, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Lovoll, M.; Wiik-Nielsen, J.; Grove, S.; Wiik-Nielsen, C.R.; Kristoffersen, A.B.; Faller, R.; Poppe, T.; Jung, J.; Pedamallu, C.S.; Nederbragt, A.J.; et al. A novel totivirus and piscine reovirus (PRV) in Atlantic salmon (Salmo salar) with cardiomyopathy syndrome (CMS). Virol. J. 2010, 7, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marty, G.D.; Morrison, D.B.; Bidulka, J.; Joseph, T.; Siah, A. Piscine reovirus in wild and farmed salmonids in British Columbia, Canada: 1974–2013. J. Fish Dis. 2015, 38, 713–728. [Google Scholar] [CrossRef] [PubMed]
- Julin, K.; Johansen, L.H.; Sommer, A.I.; Jorgensen, J.B. Persistent infections with infectious pancreatic necrosis virus (IPNV) of different virulence in Atlantic salmon, Salmo salar L. J. Fish Dis. 2015, 38, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Gjessing, M.C.; Kvellestad, A.; Ottesen, K.; Falk, K. Nodavirus provokes subclinical encephalitis and retinochoroiditis in adult farmed Atlantic cod, Gadus morhua L. J. Fish Dis. 2009, 32, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Amend, D.F. Detection and transmission of infectious hematopoietic necrosis virus in rainbow trout. J. Wildl. Dis. 1975, 11, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Neukirch, M. Demonstration of persistent viral haemorrhagic septicaemia (VHS) virus in rainbow trout after experimental waterborne infection. Zentralbl Veterinarmed B 1986, 33, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, P.K.; Gregg, J.L.; Grady, C.A.; Taylor, L.; Winton, J.R. Chronic and persistent viral hemorrhagic septicemia virus infections in Pacific herring. Dis. Aquat. Organ. 2010, 93, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Dalet, A.; Gatti, E.; Pierre, P. Integration of PKR-dependent translation inhibition with innate immunity is required for a coordinated anti-viral response. FEBS Lett. 2015, 589, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Durfee, L.A.; Lyon, N.; Seo, K.; Huibregtse, J.M. The ISG15 conjugation system broadly targets newly synthesized proteins: Implications for the antiviral function of ISG15. Mol. Cell 2010, 38, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Touris-Otero, F.; Cortez-San Martin, M.; Martinez-Costas, J.; Benavente, J. Avian reovirus morphogenesis occurs within viral factories and begins with the selective recruitment of sigmaNS and lambdaA to microNS inclusions. J. Mol. Biol. 2004, 341, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Carroll, K.; Hastings, C.; Miller, C.L. Amino acids 78 and 79 of Mammalian Orthoreovirus protein microNS are necessary for stress granule localization, core protein lambda 2 interaction, and de novo virus replication. Virology 2014, 448, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Burlingame, A.L.; Bradshaw, R.A. Western blots versus selected reaction monitoring assays: time to turn the tables? Mol. Cell. Proteomics 2013, 12, 2381–2382. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Grille, J.; Busch, L.K.; Martinez-Costas, J.; Benavente, J. Avian reovirus-triggered apoptosis enhances both virus spread and the processing of the viral nonstructural muNS protein. Virology 2014, 462–463, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Zhang, J.; Guo, H.; Yan, S.; Chen, Q.; Zhang, F.; Fang, Q. Aquareovirus NS80 initiates efficient viral replication by retaining core proteins within replication-associated viral inclusion bodies. PLoS ONE 2015, 10, e0126127. [Google Scholar] [CrossRef] [PubMed]
- Nibert, M.L.; Odegard, A.L.; Agosto, M.A.; Chandran, K.; Schiff, L.A. Putative autocleavage of reovirus mu1 protein in concert with outer-capsid disassembly and activation for membrane permeabilization. J. Mol. Biol. 2005, 345, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Liemann, S.; Chandran, K.; Baker, T.S.; Nibert, M.L.; Harrison, S.C. Structure of the reovirus membrane-penetration protein, Mu1, in a complex with is protector protein, Sigma3. Cell 2002, 108, 283–295. [Google Scholar] [CrossRef]
- Coffey, C.M.; Sheh, A.; Kim, I.S.; Chandran, K.; Nibert, M.L.; Parker, J.S. Reovirus outer capsid protein micro1 induces apoptosis and associates with lipid droplets, endoplasmic reticulum, and mitochondria. J. Virol. 2006, 80, 8422–8438. [Google Scholar] [CrossRef] [PubMed]
- Danthi, P.; Coffey, C.M.; Parker, J.S.; Abel, T.W.; Dermody, T.S. Independent regulation of reovirus membrane penetration and apoptosis by the mu1 phi domain. PLoS Pathog. 2008, 4, e1000248. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| * Band Excised from SDS-PAGE (kDa) | Identified PRV Proteins | Unique Peptides | Theoretical PRV Protein Size (kDa) |
|---|---|---|---|
| 140 (5 wpc) | µNS | 1 | 83.5 |
| 130 (5 wpc) | λ3 | 2 | 144.5 |
| λ2 | 7 | 143.7 | |
| λ1 | 14 | 141.5 | |
| µNS | 9 | 83.5 | |
| σ1 | 1 | 34.6 | |
| 80 (5 wpc) | λ1 | 11 | 141.5 |
| µNS | 24 | 83.5 | |
| 70 (4 wpc) | µNS | 16 | 83.5 |
| 35 (5 wpc) | µNS | 4 | 83.5 |
| δ † | 3 | 37.7 | |
| σNS | 1 | 39.1 | |
| σ1 | 2 | 34.6 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haatveit, H.M.; Wessel, Ø.; Markussen, T.; Lund, M.; Thiede, B.; Nyman, I.B.; Braaen, S.; Dahle, M.K.; Rimstad, E. Viral Protein Kinetics of Piscine Orthoreovirus Infection in Atlantic Salmon Blood Cells. Viruses 2017, 9, 49. https://doi.org/10.3390/v9030049
Haatveit HM, Wessel Ø, Markussen T, Lund M, Thiede B, Nyman IB, Braaen S, Dahle MK, Rimstad E. Viral Protein Kinetics of Piscine Orthoreovirus Infection in Atlantic Salmon Blood Cells. Viruses. 2017; 9(3):49. https://doi.org/10.3390/v9030049
Chicago/Turabian StyleHaatveit, Hanne Merethe, Øystein Wessel, Turhan Markussen, Morten Lund, Bernd Thiede, Ingvild Berg Nyman, Stine Braaen, Maria Krudtaa Dahle, and Espen Rimstad. 2017. "Viral Protein Kinetics of Piscine Orthoreovirus Infection in Atlantic Salmon Blood Cells" Viruses 9, no. 3: 49. https://doi.org/10.3390/v9030049
APA StyleHaatveit, H. M., Wessel, Ø., Markussen, T., Lund, M., Thiede, B., Nyman, I. B., Braaen, S., Dahle, M. K., & Rimstad, E. (2017). Viral Protein Kinetics of Piscine Orthoreovirus Infection in Atlantic Salmon Blood Cells. Viruses, 9(3), 49. https://doi.org/10.3390/v9030049