
Virological Surveillance of Influenza A Subtypes Isolated in 2014 from Clinical Outbreaks in Canadian Swine

Abstract
:1. Introduction
2. Material and Methods
2.1. Viruses
2.2. Genomic Sequencing and Sequence Assembly
2.3. Sequence Analysis
3. Results
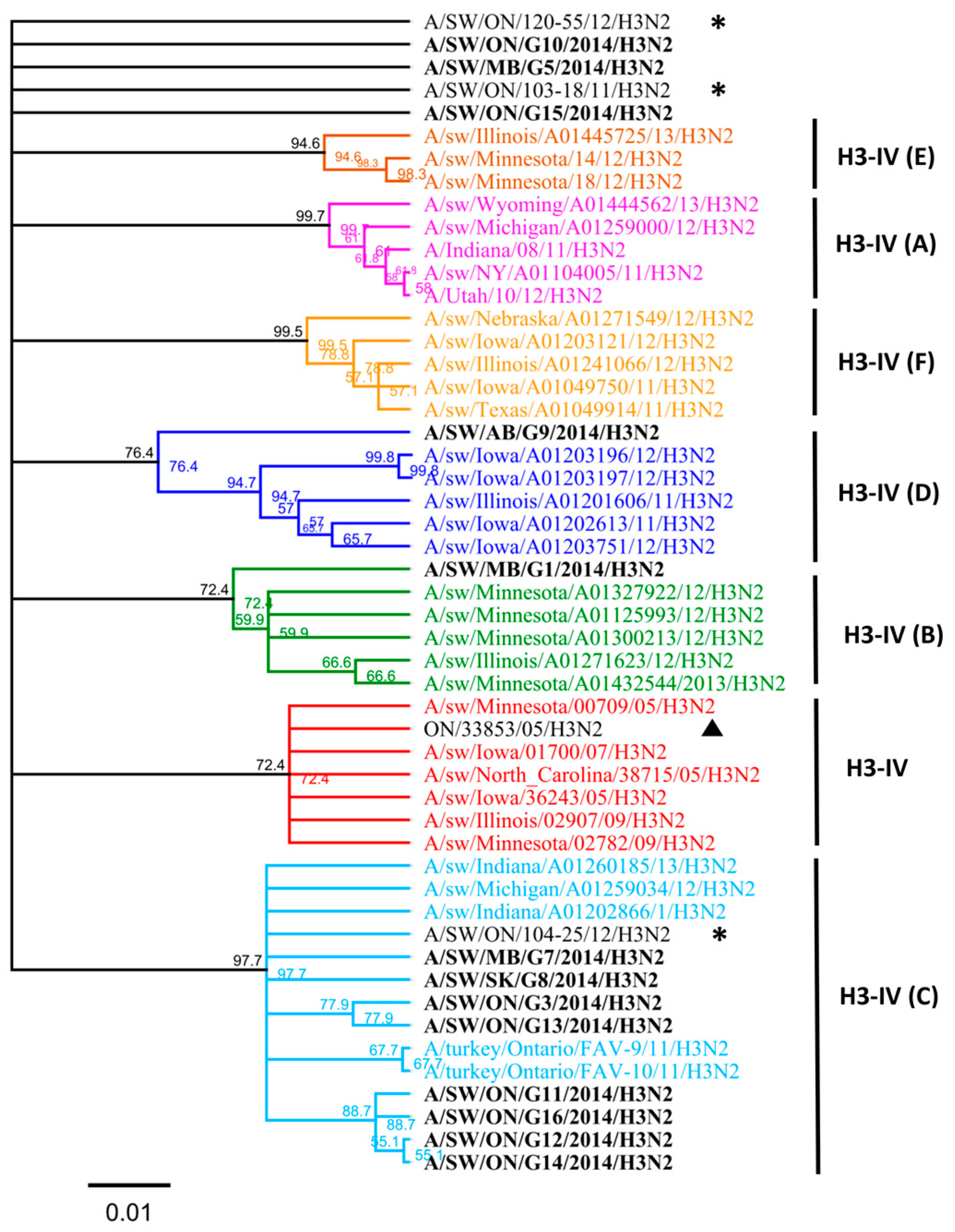
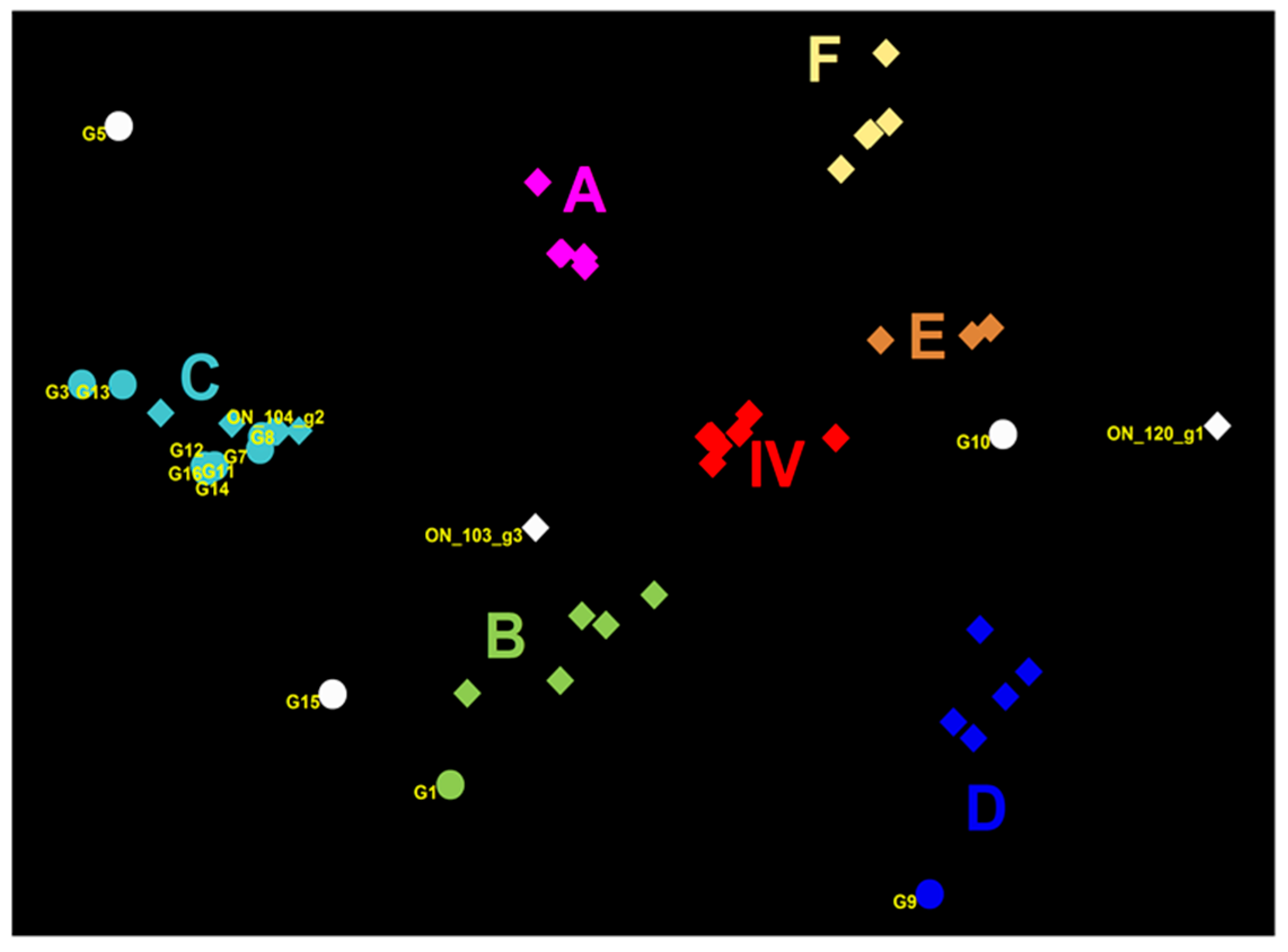
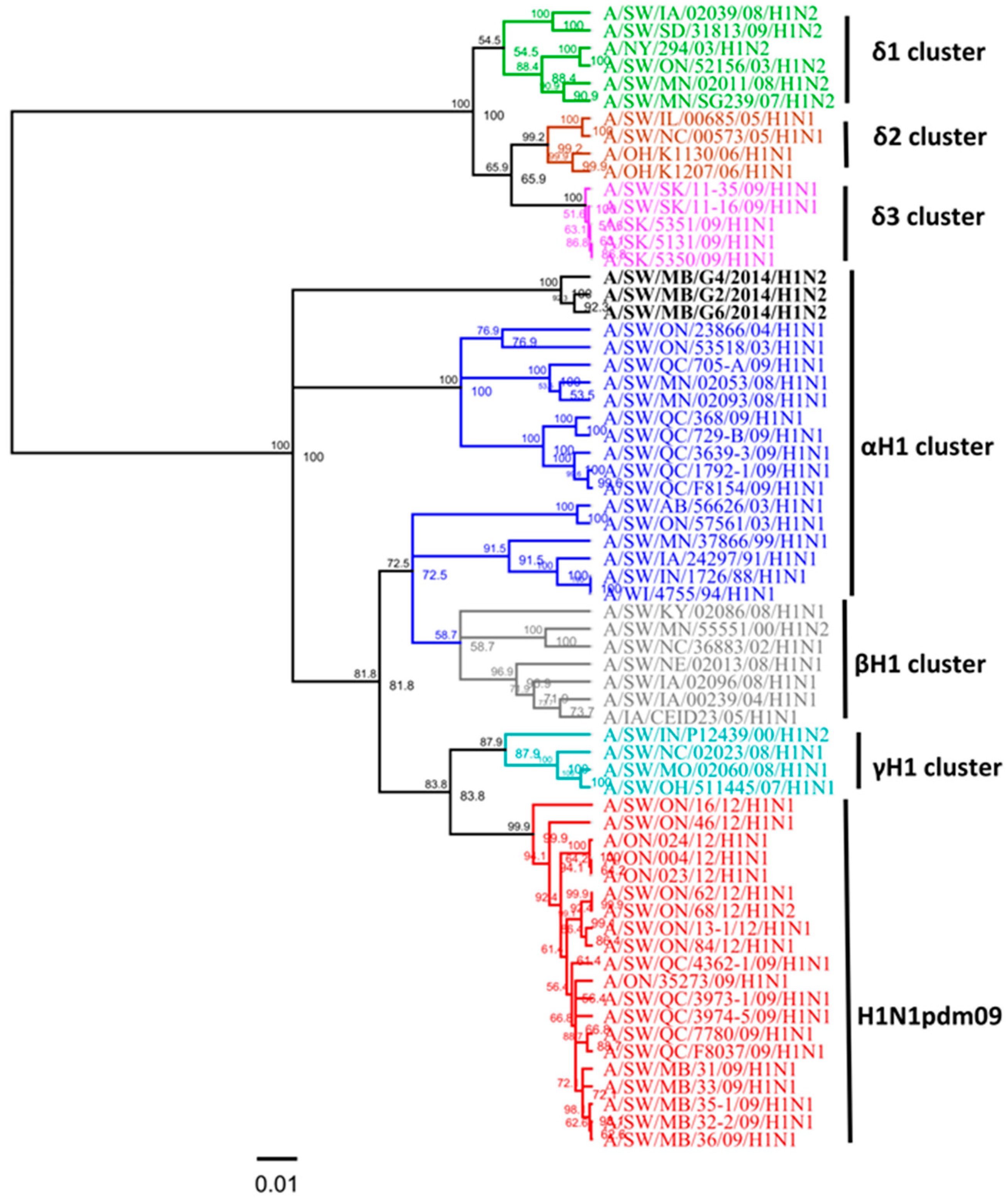
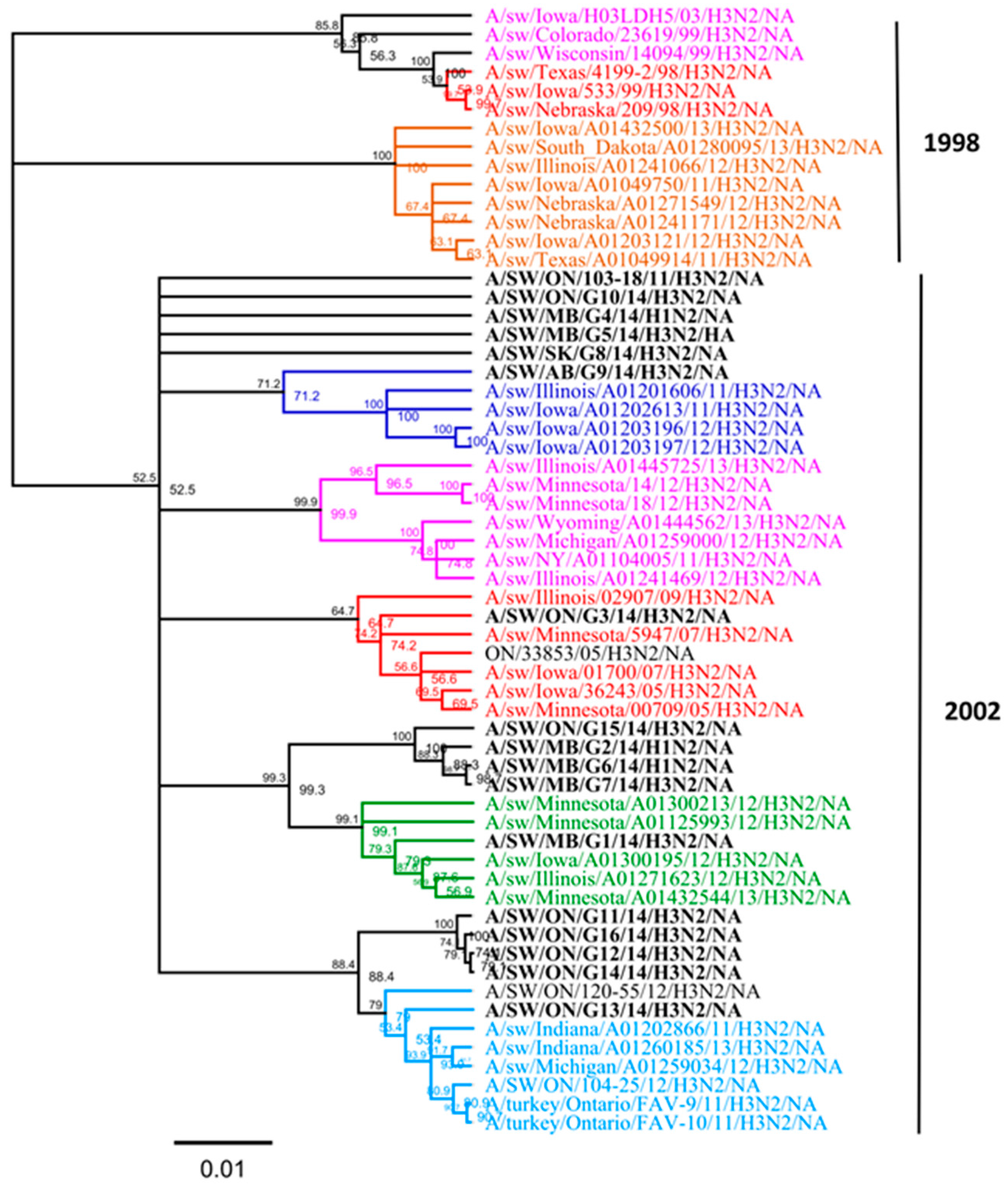
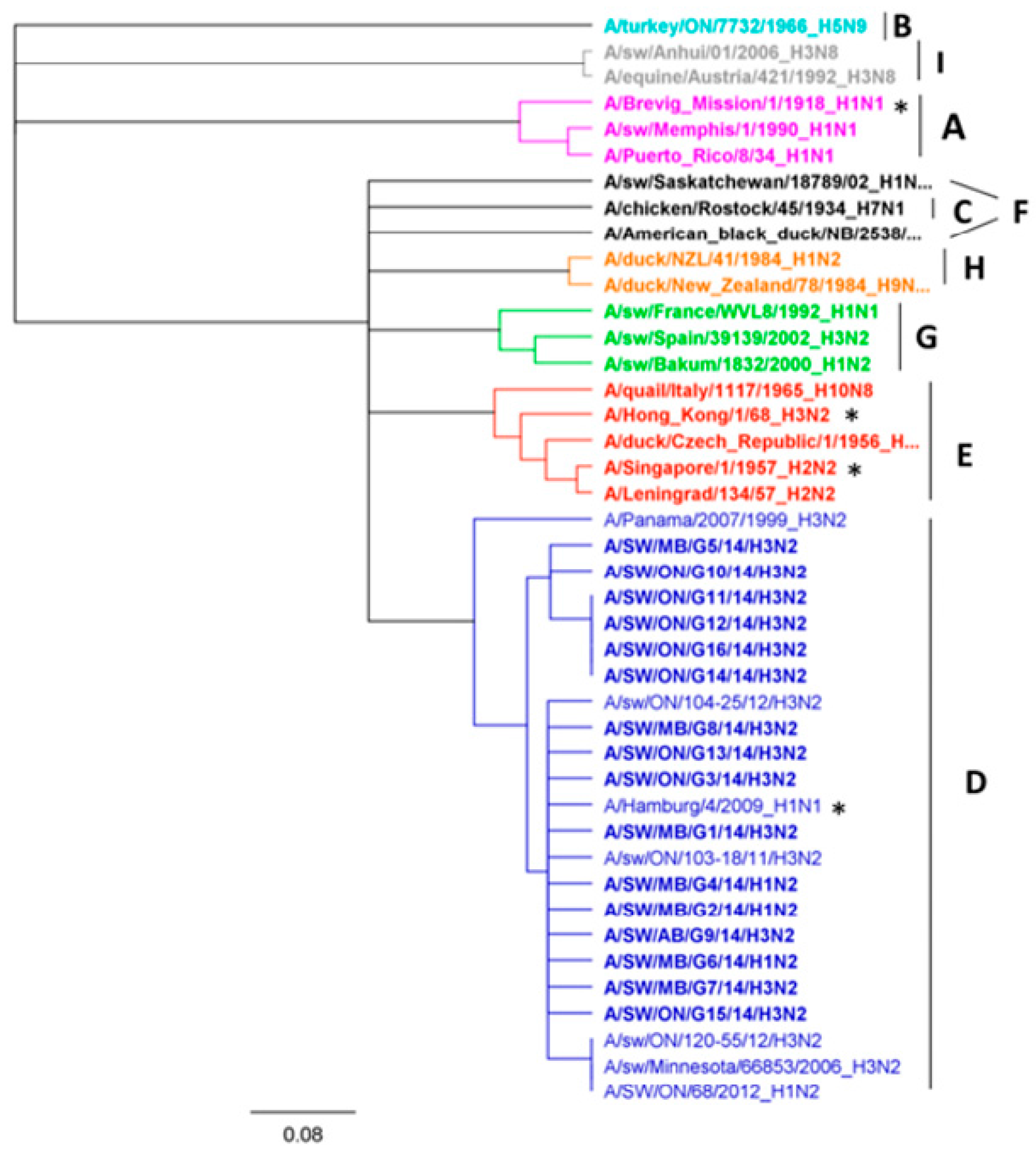
3.1. Genetic Characterization of IAV Isolated from Clinical Outbreaks in Canadian Swine
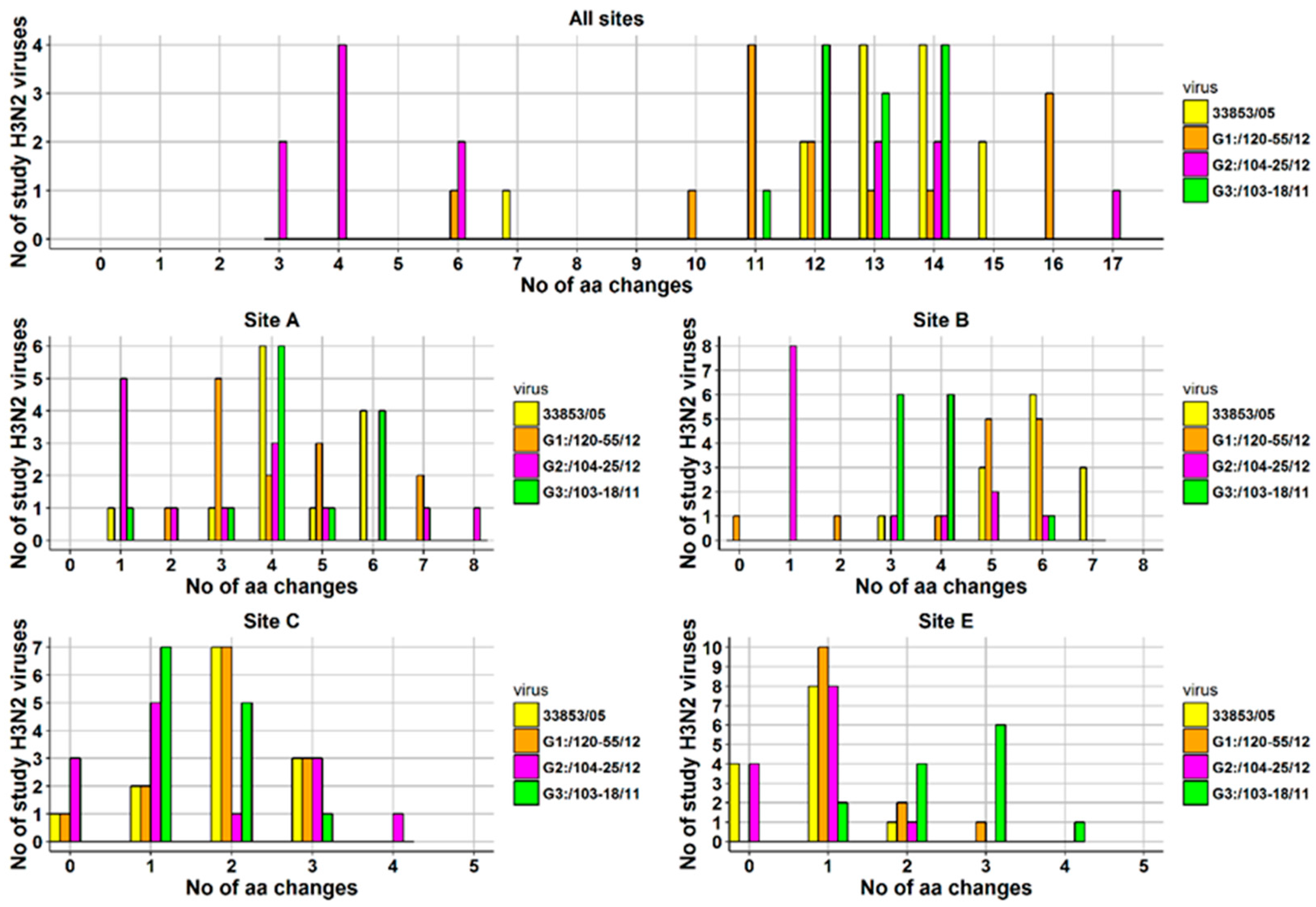
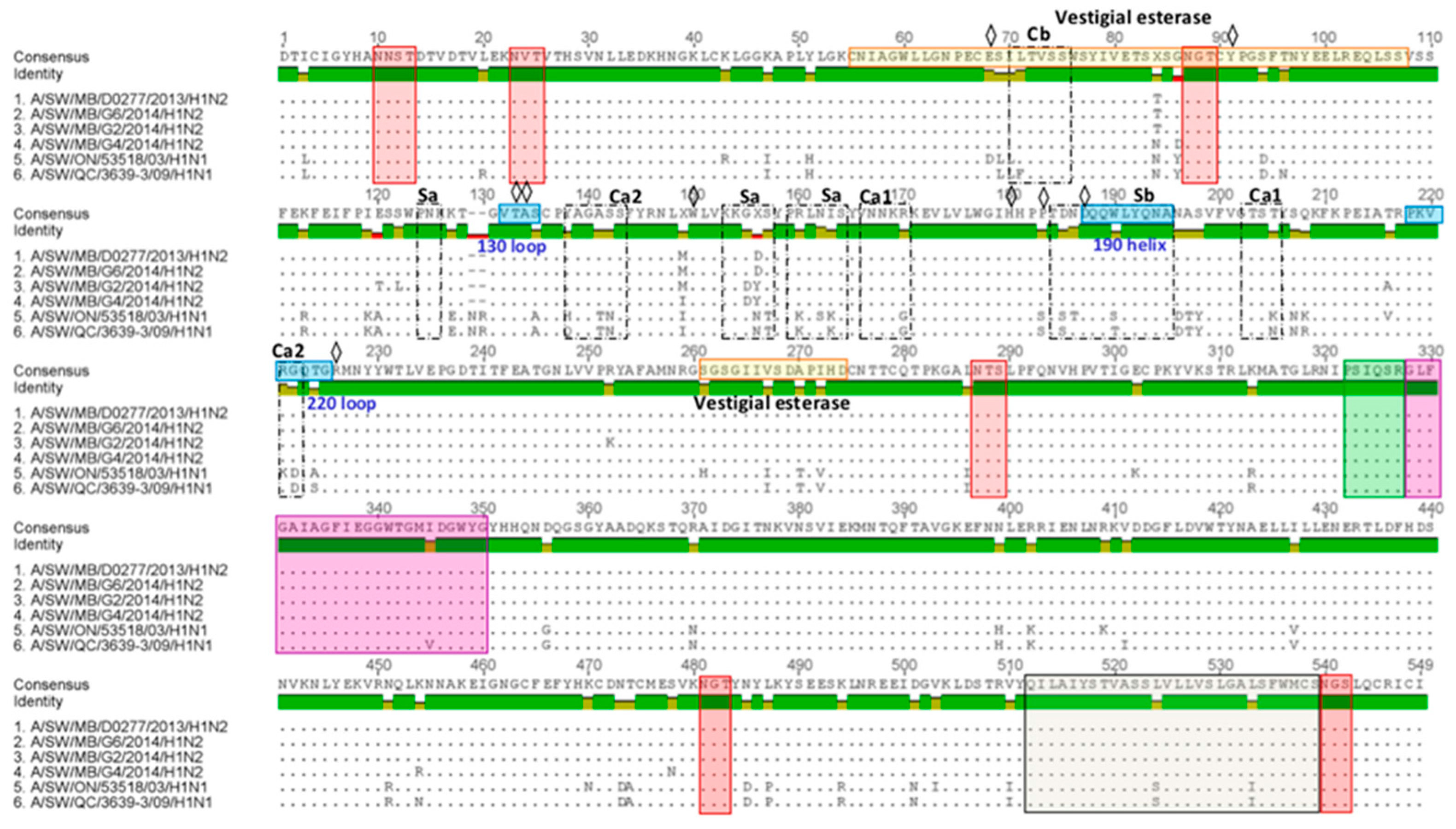
3.2. Amino Acid Variations in Antigenic Sites of the HA1 Region of 13 H3N2 Viruses
3.3. Amino Acid Variations in Antigenic Sites of the HA1 Region of Three H1N2 Viruses
3.4. Amino Acids and Phylogenetic Analysis of the PB1-F2 Protein
3.5. Resistance-Associated Mutations
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Koen, J.S. A practical method for field diagnosis of swine diseases. Am. J. Vet. Med. 1919, 14, 468–470. [Google Scholar]
- Shope, R.E. Swine Influenza: III. Filtration Experiments and Etiology. J. Exp. Med. 1931, 54, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.N.; Senne, D.A.; Landgraf, J.S.; Swenson, S.L.; Erickson, G.; Rossow, K.; Liu, L.; Yoon, K.; Krauss, S.; Webster, R.G. Genetic reassortment of avian, swine, and human influenza A viruses in American pigs. J. Virol. 1999, 73, 8851–8856. [Google Scholar] [PubMed]
- Lorusso, A.; Vincent, A.L.; Harland, M.L.; Alt, D.; Bayles, D.O.; Swenson, S.L.; Gramer, M.R.; Russell, C.A.; Smith, D.J.; Lager, K.M.; et al. Genetic and antigenic characterization of H1 influenza viruses from United States swine from 2008. J. Gen. Virol. 2011, 92, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.L.; Ma, W.; Lager, K.M.; Gramer, M.R.; Richt, J.A.; Janke, B.H. Characterization of a newly emerged genetic cluster of H1N1 and H1N2 swine influenza virus in the United States. Virus Genes 2009, 39, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.K.; Campbell, B.A.; Nelson, M.I.; Lewis, N.S.; Janas-Martindale, A.; Killian, M.L.; Vincent, A.L. Characterization of co-circulating swine influenza A viruses in North America and the identification of a novel H1 genetic clade with antigenic significance. Virus Res. 2015, 201, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.S.; Anderson, T.K.; Kitikoon, P.; Skepner, E.; Burke, D.F.; Vincent, A.L. Substitutions near the hemagglutinin receptor-binding site determine the antigenic evolution of influenza A H3N2 viruses in U.S. swine. J. Virol. 2014, 88, 4752–4763. [Google Scholar] [CrossRef] [PubMed]
- Poljak, Z.; Dewey, C.E.; Martin, S.W.; Christensen, J.; Carman, S.; Friendship, R.M. Prevalence of and risk factors for influenza in southern Ontario swine herds in 2001 and 2003. Can. J. Vet. Res. 2008, 72, 7–17. [Google Scholar] [PubMed]
- Olsen, C.W.; Karasin, A.I.; Carman, S.; Li, Y.; Bastien, N.; Ojkic, D.; Alves, D.; Charbonneau, G.; Henning, B.M.; Low, D.E.; et al. Triple reassortant H3N2 influenza A viruses, Canada, 2005. Emerg. Infect. Dis. 2006, 12, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Howden, K.J.; Brockhoff, E.J.; Caya, F.D.; McLeod, L.J.; Lavoie, M.; Ing, J.D.; Bystrom, J.M.; Alexandersen, S.; Pasick, J.M.; Berhane, Y.; et al. An investigation into human pandemic influenza virus (H1N1) 2009 on an Alberta swine farm. Can. Vet. J. 2009, 50, 1153–1161. [Google Scholar] [PubMed]
- Nfon, C.K.; Berhane, Y.; Hisanaga, T.; Zhang, S.; Handel, K.; Kehler, H.; Labrecque, O.; Lewis, N.S.; Vincent, A.L.; Copps, J.; et al. Characterization of H1N1 swine influenza viruses circulating in Canadian pigs in 2009. J. Virol. 2011, 85, 8667–8679. [Google Scholar] [CrossRef] [PubMed]
- Grgić, H.; Costa, M.; Friendship, R.M.; Carman, S.; Nagy, E.; Poljak, Z. Genetic Characterization of H1N1 and H1N2 Influenza A Viruses Circulating in Ontario Pigs in 2012. PLoS ONE 2015, 10, e0127840. [Google Scholar] [CrossRef] [PubMed]
- Redies, L.L.; Culhane, M.R.; Detmer, S.S. An emerging H1N2 sub-cluster within the alpha H1 cluster of influenza A viruses of swine. In Proceeding of the 47th Annual Meeting of the AASV, New Orleans, LS, USA, 27 February–1 March 2016.
- Tremblay, D.; Allard, V.; Doyon, J.F.; Bellehumeur, C.; Spearman, J.G.; Harel, J.; Gagnon, C.A. Emergence of a new swine H3N2 and pandemic (H1N1) 2009 influenza A virus reassortant in two Canadian animal populations, mink and swine. J. Clin. Microbiol. 2011, 49, 4386–4390. [Google Scholar] [CrossRef] [PubMed]
- Grgić, H.; Costa, M.; Friendship, R.M.; Carman, S.; Nagy, E.; Wideman, G.; Weese, S.; Poljak, Z. Molecular characterization of H3N2 influenza A viruses isolated from Ontario swine in 2011 and 2012. Virol. J. 2014, 11, 194. [Google Scholar] [CrossRef] [PubMed]
- Nfon, C.; Berhane, Y.; Zhang, S.; Handel, K.; Labrecque, O.; Pasick, J. Molecular and antigenic characterization of triple-reassortant H3N2 swine influenza viruses isolated from pigs, turkey and quail in Canada. Transbound. Emerg. Dis. 2011, 58, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Holtkamp, D.; Rotto, H.; Garcia, R. Economic cost of major health challenges in large US swine production systems. In Proceeding of the 38th Annual Meeting of the AASV, Orlando, FL, USA, 3–6 March 2007; pp. 85–89.
- World Health Organization. WHO manual on animal influenza diagnosis and surveillance. 2002.5 Rev.1. Available online: http://www.who.int/csr/resources/publications/influenza/en/whocdscsrncs20025rev.pdf (accessed on 15 July 2015).
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony (and Other Methods); Sinauer Associaates: Sunderland, MA, USA, 2002. [Google Scholar]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, J.; Mair, P. Multidimensional Scaling Using Majorization: SMACOF in R. J. Stat. Softw. 2009, 31, 1–30. [Google Scholar] [CrossRef]
- Koel, B.F.; Burke, D.F.; Bestebroer, T.M.; van der Vliet, S.; Zondag, G.C.M.; Vervaet, G.; Skepner, E.; Lewis, N.S.; Spronken, M.I.J.; Russell, C.A.; et al. Substitutions Near the Receptor Binding Site Determine Major Antigenic Change During Influenza Virus Evolution. Science 2013, 342, 976–979. [Google Scholar] [CrossRef] [PubMed]
- Matrosovich, M.; Tuzikov, A.; Bovin, N.; Gambaryan, A.; Klimov, A.; Castrucci, M.R.; Donatelli, I.; Kawaoka, Y. Early alterations of the receptor-binding properties of H1, H2, and H3 avian influenza virus hemagglutinins after their introduction into mammals. J. Virol. 2000, 74, 8502–8512. [Google Scholar] [CrossRef] [PubMed]
- Neumann, G.; Kawaoka, Y. Host range restriction and pathogenicity in the context of influenza pandemic. Emerg. Infect. Dis. 2006, 12, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S.; Mihara, K.; Koshiba, T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014, 5, 4713. [Google Scholar] [CrossRef] [PubMed]
- Krumbholz, A.; Philipps, A.; Oehring, H.; Schwarzer, K.; Eitner, A.; Wutzler, P.; Zell, R. Current knowledge on PB1-F2 of influenza A viruses. Med. Microbiol. Immunol. 2011, 200, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26 (Suppl. 4), D49–D53. [Google Scholar] [CrossRef] [PubMed]
- Wise, H.M.; Foeglein, A.; Sun, J.; Dalton, R.M.; Patel, S.; Howard, W.; Anderson, E.C.; Barclay, W.S.; Digard, P. A complicated message: Identification of a novel PB1-related protein translated from influenza A virus segment 2 mRNA. J. Virol. 2009, 83, 8021–8031. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.F.; Neumann, G.; Kawaoka, Y. Orthomyxoviruses. In Fields Virology; Knipe, D.M.H., Peter, M., Cohen, J.I., Griffin, D.E., Lamb, R.A., Martin, M.A., Racaniello, V.R., Roizman, B., Eds.; Wolters Kluwer, Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume I, pp. 1186–1243. [Google Scholar]
- Michaelis, M.; Doerr, H.W.; Cinatl, J. Novel swine-origin influenza A virus in humans: Another pandemic knocking at the door. Med. Microbiol. Immunol. 2009, 198, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Belshe, R.B.; Smith, M.H.; Hall, C.B.; Betts, R.; Hay, A.J. Genetic basis of resistance to rimantadine emerging during treatment of influenza virus infection. J. Virol. 1988, 62, 1508–1512. [Google Scholar] [PubMed]
- Hay, A.J.; Zambon, M.C.; Wolstenholme, A.J.; Skehel, J.J.; Smith, M.H. Molecular basis of resistance of influenza A viruses to amantadine. J. Antimicrob. Chemother. 1986, 18 (Suppl. B), 19–29. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, C.A.; Spearman, G.; Hamel, A.; Godson, D.L.; Fortin, A.; Fontaine, G.; Tremblay, D. Characterization of a Canadian mink H3N2 influenza A virus isolate genetically related to triple reassortant swine influenza virus. J. Clin. Microbiol. 2009, 47, 796–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducatez, M.F.; Hause, B.; Stigger-Rosser, E.; Darnell, D.; Corzo, C.; Juleen, K.; Simonson, R.; Brockwell-Staats, C.; Rubrum, A.; Wang, D.; et al. Multiple reassortment between pandemic (H1N1) 2009 and endemic influenza viruses in pigs, United States. Emerg. Infect. Dis. 2011, 17, 1624–1629. [Google Scholar] [CrossRef] [PubMed]
- Kitikoon, P.; Nelson, M.I.; Killian, M.L.; Anderson, T.K.; Koster, L.; Culhane, M.R.; Vincent, A.L. Genotype patterns of contemporary reassorted H3N2 virus in US swine. J. Gen. Virol. 2013, 94, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.M.; Oxford, J.S.; Dunleavy, U.; Newman, R.W.; Major, D.; Robertson, J.S. Influenza A (H1N1) vaccine efficacy in animal models is influenced by two amino acid substitutions in the hemagglutinin molecule. Virology 1989, 171, 214–221. [Google Scholar] [CrossRef]
- Kodihalli, S.; Justewicz, D.M.; Gubareva, L.V.; Webster, R.G. Selection of a single amino acid substitution in the hemagglutinin molecule by chicken eggs can render influenza A virus (H3) candidate vaccine ineffective. J. Virol. 1995, 69, 4888–4897. [Google Scholar] [PubMed]
- Choi, Y.K.; Goyal, S.M.; Joo, H.S. Prevalence of swine influenza virus subtypes on swine farms in the United States. Arch. Virol. 2002, 147, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.Q.; Xu, Y.F.; Harris, J.; Sun, H.L.; Bowman, A.S.; Cunningham, F.; Cardona, C.; Yoon, K.J.; Slemons, R.D.; Wan, X.F. Mutation from arginine to lysine at the position 189 of hemagglutinin contributes to the antigenic drift in H3N2 swine influenza viruses. Virology 2013, 446, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Suzuki, Y. Evidence for N-glycan shielding of antigenic sites during evolution of human influenza A virus hemagglutinin. J. Virol. 2012, 86, 3446–3451. [Google Scholar] [CrossRef] [PubMed]
- Pentiah, K.; Lees, W.D.; Moss, D.S.; Shepherd, A.J. N-linked glycans on influenza A H3N2 hemagglutinin constrain binding of host antibodies, but shielding is limited. Glycobiology 2015, 25, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Berhane, Y.; Kehler, H.; Handel, K.; Hisanaga, T.; Xu, W.; Ojkic, D.; Pasick, J. Molecular and antigenic characterization of reassortant H3N2 viruses from turkeys with a unique constellation of pandemic H1N1 internal genes. PLoS ONE 2012, 7, e32858. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Calvo, P.A.; Malide, D.; Gibbs, J.; Schubert, U.; Bacik, I.; Basta, S.; O’Neill, R.; Schickli, J.; Palese, P.; et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 2001, 7, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Krumbholz, A.; Eitner, A.; Krieg, R.; Halbhuber, K.J.; Wutzler, P. Prevalence of PB1-F2 of influenza A viruses. J. Gen. Virol. 2007, 88, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Pasricha, G.; Mishra, A.C.; Chakrabarti, A.K. Comprehensive global amino acid sequence analysis of PB1F2 protein of influenza A H5N1 viruses and the influenza A virus subtypes responsible for the 20th-century pandemics. Influ. Other Respir Viruses 2013, 7, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.K.; Pasricha, G. An insight into the PB1F2 protein and its multifunctional role in enhancing the pathogenicity of the influenza A viruses. Virology 2013, 440, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Hurt, A.C.; Ho, H.T.; Barr, I. Resistance to anti-influenza drugs: Adamantanes and neuraminidase inhibitors. Expert Rev. Anti Infect. Ther. 2006, 4, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.H.; Lamb, R.A. The M2 proton channels of influenza A and B viruses. J. Biol. Chem. 2006, 281, 8997–9000. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Peng, C.; Luo, J.; Wang, C.; Han, L.; Wu, B.; Ji, G.; He, H. Adamantane-resistant influenza A viruses in the world (1902–2013): Frequency and distribution of M2 gene mutations. PLoS ONE 2015, 10, e0119115. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Tang, J.W.T.; Loh, T.P.; Hurt, A.C.; Oon, L.L.E.; Koay, E.S.C. Molecular Surveillance of Antiviral Drug Resistance of Influenza A/H3N2 Virus in Singapore, 2009–2013. PLoS ONE 2015, 10, e0117822. [Google Scholar] [CrossRef] [PubMed]
- Bright, R.A.; Shay, D.K.; Shu, B.; Cox, N.J.; Klimov, A.I. Adamantane resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. JAMA 2006, 295, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Krumbholz, A.; Schmidtke, M.; Bergmann, S.; Motzke, S.; Bauer, K.; Stech, J.; Durrwald, R.; Wutzler, P.; Zell, R. High prevalence of amantadine resistance among circulating European porcine influenza A viruses. J. Gen. Virol. 2009, 90, 900–908. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | Subtype |
|---|---|
| A/SW/MB/G1/2014 | H3N2 |
| A/SW/MB/G2/2014 | H1N2 |
| A/SW/ON/G3/2014 | H3N2 |
| A/SW/MB/G4/2014 | H1N2 |
| A/SW/MB/G5/2014 | H3N2 |
| A/SW/MB/G6/2014 | H1N2 |
| A/SW/MB/G7/2014 | H3N2 |
| A/SW/SK/G8/2014 | H3N2 |
| A/SW/AB/G9/2014 | H3N2 |
| A/SW/ON/G10/2014 | H3N2 |
| A/SW/ON/G11/2014 | H3N2 |
| A/SW/ON/G12/2014 | H3N2 |
| A/SW/ON/G13/2014 | H3N2 |
| A/SW/ON/G14/2014 | H3N2 |
| A/SW/ON/G15/2014 | H3N2 |
| A/SW/ON/G16/2014 | H3N2 |
| Virus * | HA | NA | PA | PB1 | PB2 | NP | M | NS |
|---|---|---|---|---|---|---|---|---|
| /SW/MB/G1; /SW/MB/G5, /SW/SK/G8, /SW/AB/G9 | ||||||||
| /SW/MB/G2, /SW/MB/G4, /SW/MB/G6 | αH1 | |||||||
| /SW/ON/G3 | ||||||||
| /SW/MB/G7, /SW/ON/G15 | ||||||||
| /SW/ON/G10 | ||||||||
| /SW/ON/G11; /SW/ON/G12; /SW/ON/G13; /SW/ON/G14; /SW/ON/G16 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grgić, H.; Gallant, J.; Poljak, Z. Virological Surveillance of Influenza A Subtypes Isolated in 2014 from Clinical Outbreaks in Canadian Swine. Viruses 2017, 9, 55. https://doi.org/10.3390/v9030055
Grgić H, Gallant J, Poljak Z. Virological Surveillance of Influenza A Subtypes Isolated in 2014 from Clinical Outbreaks in Canadian Swine. Viruses. 2017; 9(3):55. https://doi.org/10.3390/v9030055
Chicago/Turabian StyleGrgić, Helena, Jackie Gallant, and Zvonimir Poljak. 2017. "Virological Surveillance of Influenza A Subtypes Isolated in 2014 from Clinical Outbreaks in Canadian Swine" Viruses 9, no. 3: 55. https://doi.org/10.3390/v9030055
APA StyleGrgić, H., Gallant, J., & Poljak, Z. (2017). Virological Surveillance of Influenza A Subtypes Isolated in 2014 from Clinical Outbreaks in Canadian Swine. Viruses, 9(3), 55. https://doi.org/10.3390/v9030055