
ICAM-1 Binding Rhinoviruses Enter HeLa Cells via Multiple Pathways and Travel to Distinct Intracellular Compartments for Uncoating


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Virus Propagation, and Reagents
2.2. Measuring the Effect of Inhibitors on RV Productive Uncoating
2.3. Internalization of RVs in the Presence of Filipin
2.4. Measuring Uncoating at 20 °C
2.5. Determination of the Generation of Modified Viral Particles upon Internalization at 20 °C
2.6. Uncoating from the Plasma Membrane
2.7. Effect of RV Entry on the Actin Cytoskeleton
2.8. Effect of Overexpression of Dominant-Negative Mutants of the C-Terminal Domain of AP-180 (AP180-C) and the SH3 Domain of Amphiphysin (amp-SH3) on Viral Uptake and Infectivity
3. Results
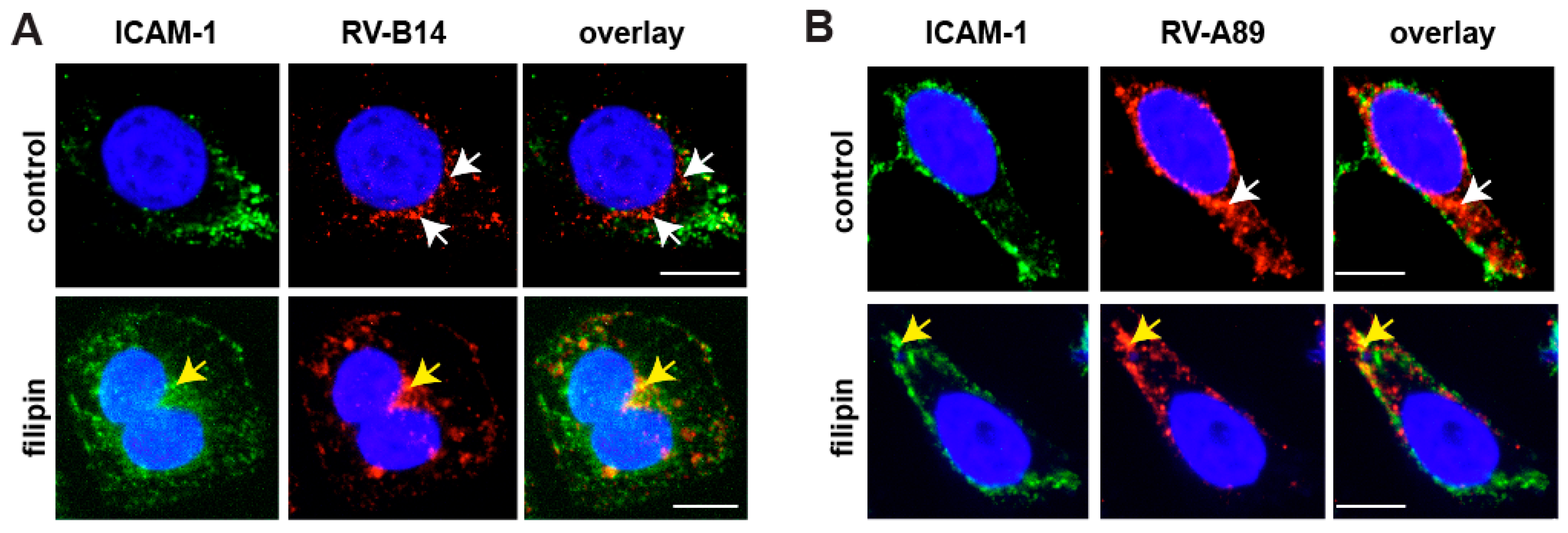
3.1. Internalization of RV-B14 and RV-A89 Occurs via Multiple Pathways
3.2. Internalization of the Major Receptor Group RV-B3, RV-B14, RV-A16, and RV-A89 Is Dependent on Actin
3.3. RV-A89, but None of the Three Other Major Group Viruses Studied Uncoats at 20 °C
3.4. RV-B14 and RV-A89 Cannot Uncoat at the Plasma Membrane
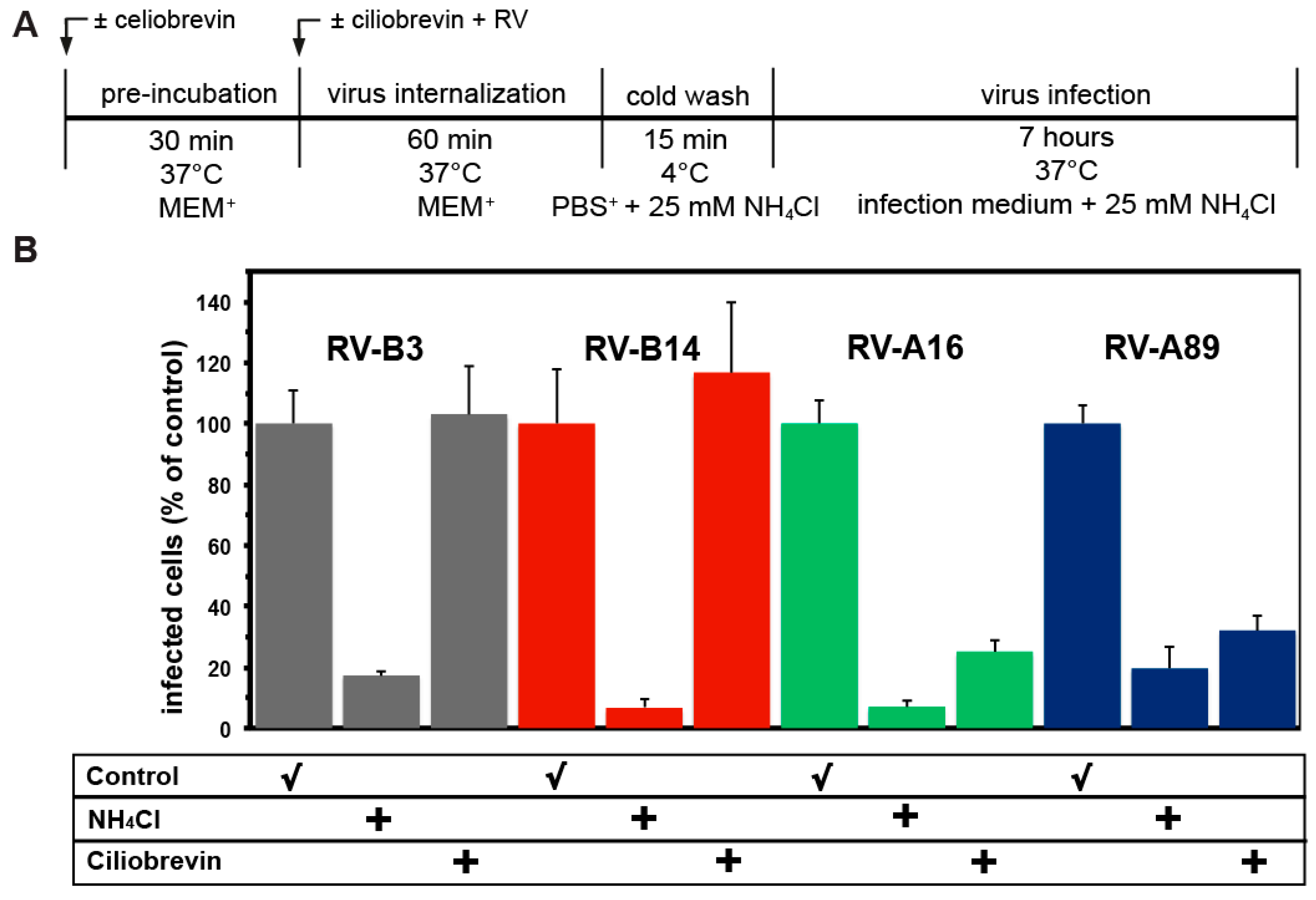
3.5. Uncoating of RV-B3 and RV-B14 Depends on Functional Dynein
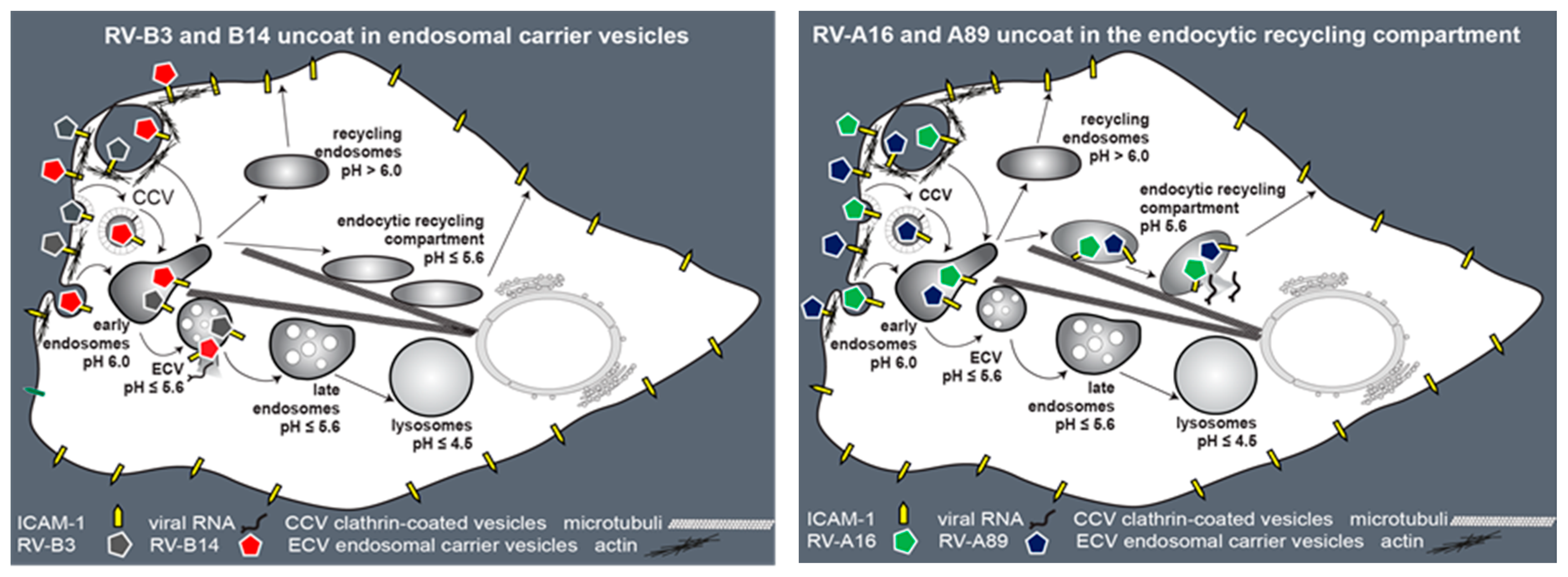
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Uncapher, C.R.; Dewitt, C.M.; Colonno, R.J. The major and minor group receptor families contain all but one human rhinovirus serotype. Virology 1991, 180, 814–817. [Google Scholar] [CrossRef]
- Vlasak, M.; Roivainen, M.; Reithmayer, M.; Goesler, I.; Laine, P.; Snyers, L.; Hovi, T.; Blaas, D. The minor receptor group of human rhinovirus (HRV) includes HRV23 and HRV25, but the presence of a lysine in the VP1 HI loop is not sufficient for receptor binding. J. Virol. 2005, 79, 7389–7395. [Google Scholar] [CrossRef] [PubMed]
- Palmenberg, A.C.; Spiro, D.; Kuzmickas, R.; Wang, S.; Djikeng, A.; Rathe, J.A.; Fraser-Liggett, C.M.; Liggett, S.B. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution. Science 2009, 324, 55–59. [Google Scholar] [CrossRef] [PubMed]
- McErlean, P.; Shackelton, L.A.; Andrews, E.; Webster, D.R.; Lambert, S.B.; Nissen, M.D.; Sloots, T.P.; Mackay, I.M. Distinguishing molecular features and clinical characteristics of a putative new rhinovirus species, human rhinovirus C (HRV C). PLoS ONE 2008, 3, e1847. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, Y.A.; Watters, K.; Ashraf, S.; Griggs, T.F.; Devries, M.K.; Jackson, D.J.; Palmenberg, A.C.; Gern, J.E. Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus C binding and replication. Proc. Natl. Acad. Sci. USA 2015, 112, 5485–5490. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, R.; Blaas, D. Productive entry pathways of human rhinoviruses. Adv. Virol. 2012, 2012, 826301. [Google Scholar] [CrossRef] [PubMed]
- Snyers, L.; Zwickl, H.; Blaas, D. Human rhinovirus type 2 is internalized by clathrin-mediated endocytosis. J. Virol. 2003, 77, 5360–5369. [Google Scholar] [CrossRef] [PubMed]
- Prchla, E.; Kuechler, E.; Blaas, D.; Fuchs, R. Uncoating of human rhinovirus serotype 2 from late endosomes. J. Virol. 1994, 68, 3713–3723. [Google Scholar] [PubMed]
- Bayer, N.; Schober, D.; Prchla, E.; Murphy, R.F.; Blaas, D.; Fuchs, R. Effect of bafilomycin A1 and nocodazole on endocytic transport in HeLa cells: Implications for viral uncoating and infection. J. Virol. 1998, 72, 9645–9655. [Google Scholar] [PubMed]
- Hoover-Litty, H.; Greve, J.M. Formation of rhinovirus-soluble ICAM-1 complexes and conformational changes in the virion. J. Virol. 1993, 67, 390–397. [Google Scholar] [PubMed]
- Nurani, G.; Lindqvist, B.; Casasnovas, J.M. Receptor priming of major group human rhinoviruses for uncoating and entry at mild low-pH environments. J. Virol. 2003, 77, 11985–11991. [Google Scholar] [CrossRef] [PubMed]
- Bayer, N.; Prchla, E.; Schwab, M.; Blaas, D.; Fuchs, R. Human rhinovirus HRV14 uncoats from early endosomes in the presence of bafilomycin. FEBS Lett. 1999, 463, 175–178. [Google Scholar] [CrossRef]
- Schober, D.; Kronenberger, P.; Prchla, E.; Blaas, D.; Fuchs, R. Major and minor receptor group human rhinoviruses penetrate from endosomes by different mechanisms. J. Virol. 1998, 72, 1354–1364. [Google Scholar] [PubMed]
- Conzemius, R.; Ganjian, H.; Blaas, D.; Fuchs, R. ICAM-1 binding rhinoviruses A89 and B14 uncoat in different endosomal compartments. J. Virol. 2016, 90, 7934–7942. [Google Scholar] [CrossRef] [PubMed]
- Blake, K.; O’Connell, S. Virus culture. In Virology Labfax; Harper, D.R., Ed.; Blackwell Scientific Publications: London, UK, 1993; pp. 81–122. [Google Scholar]
- Jurgeit, A.; Moese, S.; Roulin, P.; Dorsch, A.; Lotzerich, M.; Lee, W.M.; Greber, U.F. An RNA replication-center assay for high content image-based quantifications of human rhinovirus and coxsackievirus infections. Virol. J. 2010, 7, 264. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.G.; Pickl-Herk, A.; Gajdzik, L.; Marlovits, T.C.; Fuchs, R.; Blaas, D. Entry of a heparan sulphate-binding HRV8 variant strictly depends on dynamin but not on clathrin, caveolin, and flotillin. Virology 2011, 412, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Skern, T.; Neubauer, C.; Frasel, L.; Grundler, P.; Sommergruber, W.; Zorn, M.; Kuechler, E.; Blaas, D. A neutralizing epitope on human rhinovirus type 2 includes amino acid residues between 153 and 164 of virus capsid protein VP2. J. Gen. Virol. 1987, 68, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.J.; Wigge, P.; Vallis, Y.; Moore, J.D.; Evans, P.R.; McMahon, H.T. Crystal structure of the amphiphysin-2 SH3 domain and its role in the prevention of dynamin ring formation. EMBO J. 1998, 17, 5273–5285. [Google Scholar] [CrossRef] [PubMed]
- Nichols, B.J. A distinct class of endosome mediates clathrin-independent endocytosis to the Golgi complex. Nat. Cell Biol. 2002, 4, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Macia, E.; Ehrlich, M.; Massol, R.; Boucrot, E.; Brunner, C.; Kirchhausen, T. Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 2006, 10, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Rothberg, K.; Anderson, R. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J. Cell Biol. 1993, 123, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- McGookey, D.J.; Fagerberg, K.; Anderson, R.G. Filipin-cholesterol complexes form in uncoated vesicle membrane derived from coated vesicles during receptor-mediated endocytosis of low density lipoprotein. J. Cell Biol. 1983, 96, 1273–1278. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Helenius, A. Gulping rather than sipping: Macropinocytosis as a way of virus entry. Curr. Opin. Microbiol. 2012, 15, 490–499. [Google Scholar] [CrossRef] [PubMed]
- DeTulleo, L.; Kirchhausen, T. The clathrin endocytic pathway in viral infection. EMBO J. 1998, 17, 4585–4593. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.G.; Pickl-Herk, A.; Gajdzik, L.; Marlovits, T.C.; Fuchs, R.; Blaas, D. Human rhinovirus 14 enters rhabdomyosarcoma cells expressing icam-1 by a clathrin-, caveolin-, and flotillin-independent pathway. J. Virol. 2010, 84, 3984–3992. [Google Scholar] [CrossRef] [PubMed]
- Grunert, H.P.; Wolf, K.U.; Langner, K.D.; Sawitzky, D.; Habermehl, K.O.; Zeichhardt, H. Internalization of human rhinovirus 14 into HeLa and ICAM-1-transfected BHK cells. Med. Microbiol. Immunol. 1997, 186, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pfanzagl, B.; Andergassen, D.; Edlmayr, J.; Niespodziana, K.; Valenta, R.; Blaas, D. Entry of human rhinovirus 89 via ICAM-1 into HeLa epithelial cells is inhibited by actin skeleton disruption and by bafilomycin. Arch. Virol. 2013, 159, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, L.M.; Roth, R.; Heuser, J.E.; Schmid, S.L. Actin assembly plays a variable, but not obligatory role in receptor-mediated endocytosis. Traffic 2000, 1, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Heiska, L.; Alfthan, K.; Gronholm, M.; Vilja, P.; Vaheri, A.; Carpen, O. Association of ezrin with intercellular adhesion molecule-1 and -2 (ICAM-1 and ICAM-2). Regulation by phosphatidylinositol 4, 5-bisphosphate. J. Biol. Chem. 1998, 273, 21893–21900. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lau, C.; Wiehler, S.; Pow, A.; Mazzulli, T.; Gutierrez, C.; Proud, D.; Chow, C.W. Syk is downstream of intercellular adhesion molecule-1 and mediates human rhinovirus activation of p38 MAPK in airway epithelial cells. J. Immunol. 2006, 177, 6859–6870. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.; Wang, X.; Song, L.; North, M.; Wiehler, S.; Proud, D.; Chow, C.W. Syk associates with clathrin and mediates phosphatidylinositol 3-kinase activation during human rhinovirus internalization. J. Immunol. 2008, 180, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.I.; Bleck, C.K.; Helenius, A.; Mercer, J. Vaccinia extracellular virions enter cells by macropinocytosis and acid-activated membrane rupture. EMBO J. 2011, 30, 3647–3661. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, S.; Kowalski, H.; Blaas, D. The rhinovirus subviral a-particle exposes 3′-terminal sequences of its genomic RNA. J. Virol. 2014, 88, 6307–6317. [Google Scholar] [CrossRef] [PubMed]
- Baravalle, G.; Schober, D.; Huber, M.; Bayer, N.; Murphy, R.F.; Fuchs, R. Transferrin recycling and dextran transport to lysosomes is differentially affected by bafilomycin, nocodazole, and low temperature. Cell Tissue Res. 2005, 320, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Brabec, M.; Baravalle, G.; Blaas, D.; Fuchs, R. Conformational changes, plasma membrane penetration, and infection by human rhinovirus type 2: Role of receptors and low pH. J. Virol. 2003, 77, 5370–5377. [Google Scholar] [CrossRef] [PubMed]
- Ohlin, A.; Hoover-Litty, H.; Sanderson, G.; Paessens, A.; Johnston, S.L.; Holgate, S.T.; Huguenel, E.; Greve, J.M. Spectrum of activity of soluble intercellular adhesion molecule-1 against rhinovirus reference strains and field isolates. Antimicrob. Agents Chemother. 1994, 38, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Reischl, A.; Reithmayer, M.; Winsauer, G.; Moser, R.; Gosler, I.; Blaas, D. Viral evolution toward change in receptor usage: Adaptation of a major group human rhinovirus to grow in ICAM-1-negative cells. J. Virol. 2001, 75, 9312–9319. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, S.; Feng, Y.; Nebioglu, F.; Heilig, R.; Picotti, P.; Helenius, A. Stepwise priming by acidic pH and a high K+ concentration is required for efficient uncoating of influenza A virus cores after penetration. J. Virol. 2014, 88, 13029–13046. [Google Scholar] [CrossRef] [PubMed]







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganjian, H.; Zietz, C.; Mechtcheriakova, D.; Blaas, D.; Fuchs, R. ICAM-1 Binding Rhinoviruses Enter HeLa Cells via Multiple Pathways and Travel to Distinct Intracellular Compartments for Uncoating. Viruses 2017, 9, 68. https://doi.org/10.3390/v9040068
Ganjian H, Zietz C, Mechtcheriakova D, Blaas D, Fuchs R. ICAM-1 Binding Rhinoviruses Enter HeLa Cells via Multiple Pathways and Travel to Distinct Intracellular Compartments for Uncoating. Viruses. 2017; 9(4):68. https://doi.org/10.3390/v9040068
Chicago/Turabian StyleGanjian, Haleh, Christin Zietz, Diana Mechtcheriakova, Dieter Blaas, and Renate Fuchs. 2017. "ICAM-1 Binding Rhinoviruses Enter HeLa Cells via Multiple Pathways and Travel to Distinct Intracellular Compartments for Uncoating" Viruses 9, no. 4: 68. https://doi.org/10.3390/v9040068
APA StyleGanjian, H., Zietz, C., Mechtcheriakova, D., Blaas, D., & Fuchs, R. (2017). ICAM-1 Binding Rhinoviruses Enter HeLa Cells via Multiple Pathways and Travel to Distinct Intracellular Compartments for Uncoating. Viruses, 9(4), 68. https://doi.org/10.3390/v9040068