
Mechanisms of LTR‐Retroelement Transposition: Lessons from Drosophila melanogaster

Abstract
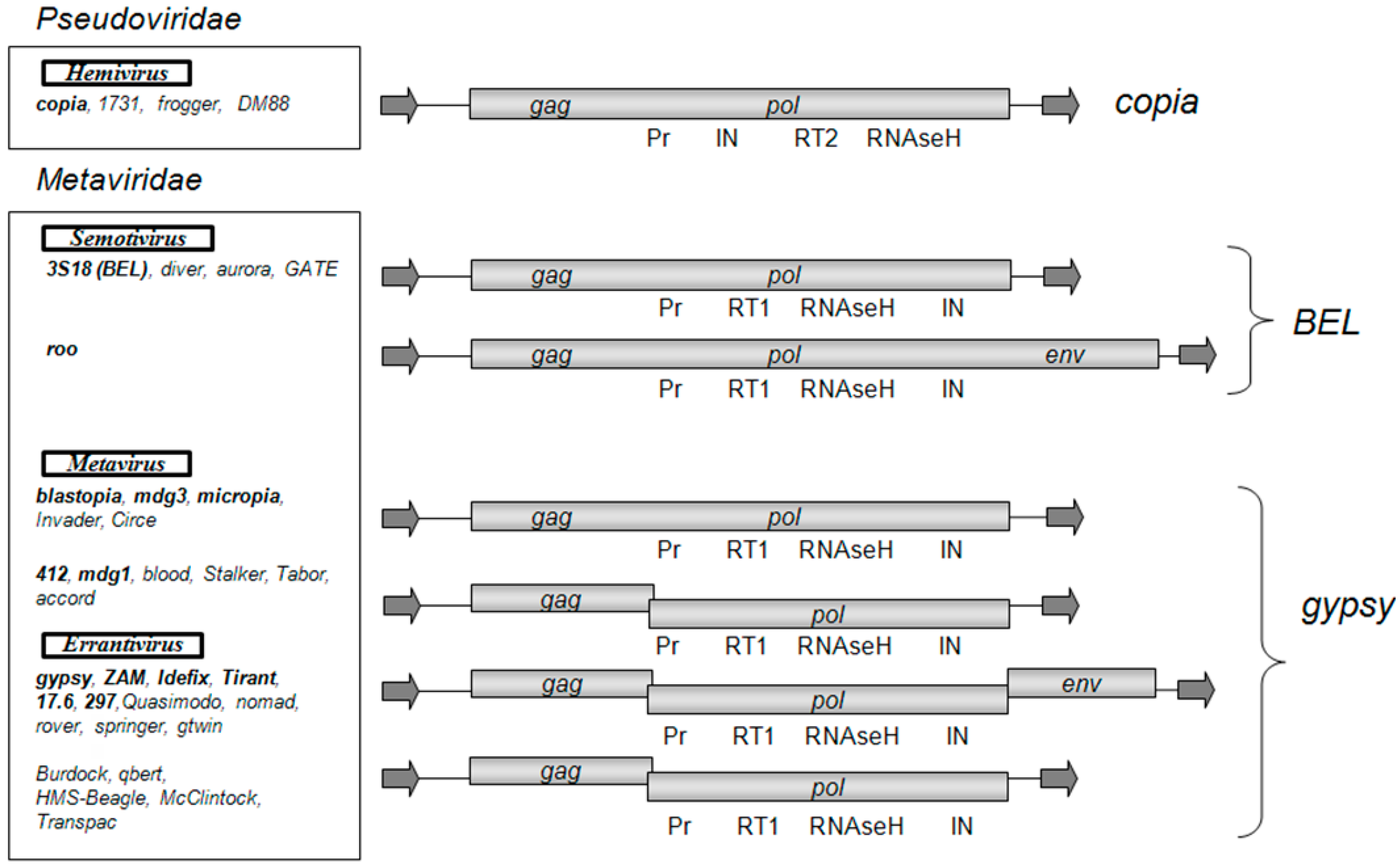
:1. Introduction
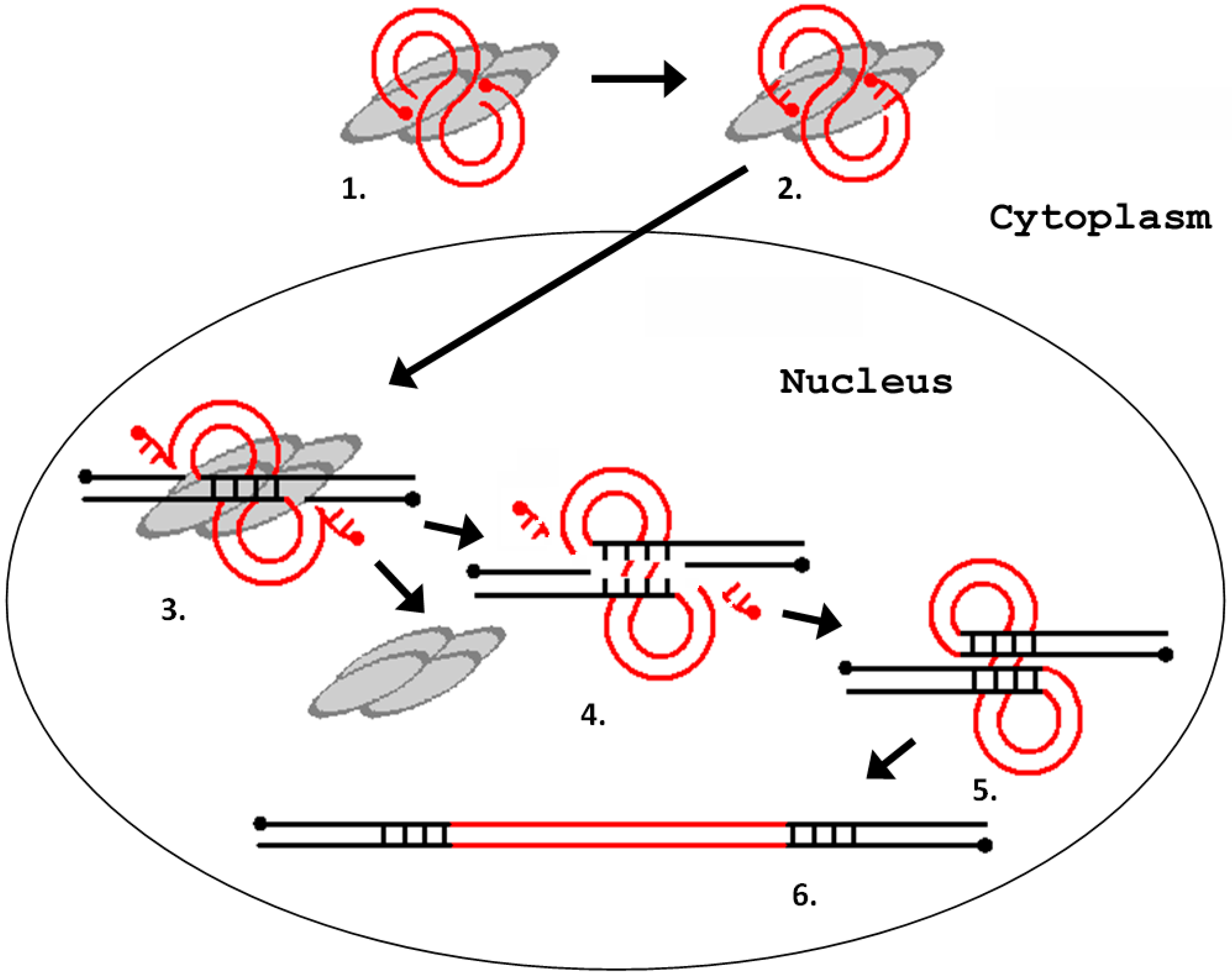
2. Errantiviruses Specifically Integrate into the Target DNA
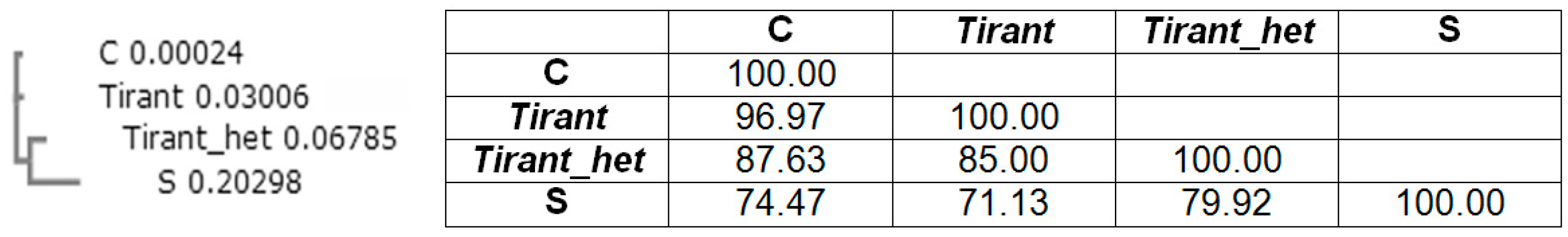
3. Repeats in the 5′-UTR Can Direct Heterochromatic Localization of Errantiviruses
4. Specific Terminal Nucleotides of Errantivirus Long Terminal Repeats Are Involved in the Interaction with Integrase
5. LTR Retrotransposons of the Metavirus Genus Have a Chromodomain in the Integrase Structure
6. LTR Retrotransposons of the Metavirus Genus Are Able to Transfer Horizontally
7. Consequences of the Retroelement Transposition
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Koning, A.P.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef] [PubMed]
- Bergman, C.M.; Quesneville, H. Discovering and detecting transposable elements in genome sequences. Brief. Bioinform. 2007, 8, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.A.; Fiston-Lavier, A.S.; Lipatov, M.; Lenkov, K.; González, J. Population genomics of transposable elements in Drosophila melanogaster. Mol. Biol. Evol. 2011, 28, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Terzian, С.; Santamaria, P.; Pélisson, A.; Prud’homme, N.; Bucheton, A. Retroviruses in vertebrates: The gypsy retrotransposon is apparently an infectious retrovirus of Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 1994, 91, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, P.; Desset, S.; Giorgi, F.; Taddei, A.R.; Fausto, A.M.; Mazzini, M.; Dastugue, B.; Vaury, C. Life cycle of an endogenous retrovirus, ZAM, in Drosophila melanogaster. J. Virol. 2000, 74, 10658–10669. [Google Scholar] [CrossRef] [PubMed]
- Bowen, N.J.; McDonald, J.F. Drosophila euchromatic LTR retrotransposons are much younger than the host species in which they reside. Genome Res. 2001, 11, 1527–1540. [Google Scholar] [CrossRef] [PubMed]
- Kaminker, J.S.; Bergman, C.M.; Kronmiller, B.; Carlson, J.; Svirskas, R.; Patel, S.; Frise, E.; Wheeler, D.A.; Lewis, S.E.; Rubin, G.M.; et al. The transposable elements of the Drosophila melanogaster euchromatin: A genomics perspective. Genome Biol. 2002, 3. [Google Scholar] [CrossRef]
- King, A.M.Q.; Adams, M.J.; Carsten, E.B.; Lefkowitz, E. Virus Taxonomy, Ninth Report of the International Committee for the Taxonomy of Viruses, 1st ed.; Elsevier: San Diego, CA, USA, 2012; 1338p. [Google Scholar]
- Nefedova, L.N.; Kim, A.I. Molecular phylogeny and systematics of Drosophila retrotransposons and retroviruses. Mol. Biol. 2009, 43, 747–756. [Google Scholar] [CrossRef]
- Llorens, C.; Muñoz-Pomer, A.; Bernad, L.; Botella, H.; Moya, A. Network dynamics of eukaryotic LTR retroelements beyond phylogenetic trees. Biol. Direct. 2009, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Llorens, C.; Fares, M.A.; Moya, A. Relationships of gag-pol diversity between Ty3/Gypsy and Retroviridae LTR retroelements and the three kings hypothesis. BMC Evol. Biol. 2008, 8, 276. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Gai, X.; Zhu, Y.; Zappulla, D.C.; Sternglanz, R.; Voytas, D.F. Targeting of the yeast Ty5 retrotransposon to silent chromatin is mediated by interactions between integrase and Sir4p. Mol. Cell. Biol. 2001, 21, 6606–6614. [Google Scholar] [CrossRef] [PubMed]
- Aye, M.; Dildine, S.L.; Claypool, J.A.; Jourdain, S.; Sandmeyer, S.B. A truncation mutant of the 95-kilodalton subunit of transcription factor IIIC reveals asymmetry in Ty3 integration. Mol. Cell. Biol. 2001, 21, 7839–7851. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.; Ma, L.; Chan, P.H.; Hu, H.L.; Mayor, T.; Chen, H.T.; Measday, V. Ty1 integrase interacts with RNA polymerase III-specific subcomplexes to promote insertion of Ty1 elements upstream of polymerase (Pol)III-transcribed genes. J. Biol. Chem. 2016, 291, 6396–6411. [Google Scholar] [CrossRef] [PubMed]
- Shun, M.-C.; Raghavendra, N.K.; Vandegraaff, N.; Daigle, J.E.; Hughes, S.; Kellam, P.; Cherepanov, P.; Engelman, A. LEDGF/p75 functions downstream from preintegration complex formation to effect genespecific HIV-1 integration. Genes Dev. 2007, 21, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.S.; Maetzig, T.; Maertens, G.N.; Sharif, A.; Rothe, M.; Weidner-Glunde, M.; Galla, M.; Schambach, A.; Cherepanov, P.; Schulz, T.F. Bromo-and extraterminal domain chromatin regulators serve as cofactors for murine leukemia virus integration. J. Virol. 2013, 87, 12721–12736. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.P.; Varmus, H.E. DNA bending creates favored sites for retroviral integration, an explanation for preferred insertion sites in nucleosomes. EMBO J. 1994, 13, 4704–4714. [Google Scholar] [PubMed]
- Kitamura, Y.; Lee, Y.M.H.; Coffin, J.M. Nonrandom integration of retroviral DNA in vitro, Effect of CpG methylation. Proc. Natl. Acad. Sci. USA 1992, 89, 5532–5536. [Google Scholar] [CrossRef] [PubMed]
- Pryciak, P.M.; Varmus, H.E. Nucleosomes, DNA-binding proteins, and DNA sequence modulate retroviral integration target site selection. Cell 1992, 69, 769–780. [Google Scholar] [CrossRef]
- Pruss, D.; Reeves, R.; Bushman, F.D.; Wolffe, A.P. The influence of DNA and nucleosome structure on integration events directed by HIV integrase. J. Biol. Chem. 1994, 269, 25031–25041. [Google Scholar] [PubMed]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA integration, ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004, 2, e234. [Google Scholar] [CrossRef] [PubMed]
- Derse, D.; Crise, C.; Li, Y.; Princler, G.; Lum, N.; Stewart, C.; McGrath, C.F.; Hughes, S.H.; Munroe, D.J.; Wu, X. Human T-Cell Leukemia Virus Type 1 Integration Target Sites in the Human Genome: Comparison with Those of Other Retroviruses. J. Virol. 2007, 81, 6731–6741. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M.; Munroe, D.J. Weak palindromic consensus sequences are a common feature found at the integration target sites of many retroviruses. J. Virol. 2005, 79, 5211–5214. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.F.; Ishibashi, T.; Nomoto, A.; Masuda, M. Isolation and analysis of retroviral integration targets by solo long terminal repeat inverse PCR. J. Virol. 2002, 76, 5540–5547. [Google Scholar] [CrossRef] [PubMed]
- Nefedova, L.N.; Mannanova, M.M.; Kim, A.I. Integration specificity of LTR-retrotransposons and retroviruses in the Drosophila melanogaster genome. Virus Genes 2011, 42, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Linheiro, R.S.; Bergman, C.M. Whole genome resequencing reveals natural target site preferences of transposable elements in Drosophila melanogaster. PLoS ONE 2012, 7, e30008. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Wang, J.; Theurkauf, W.; Weng, Z. TEMP: A computational method for analyzing transposable element polymorphism in populations. Nucleic Acids Res. 2014, 42, 6826–6838. [Google Scholar] [CrossRef] [PubMed]
- Fiston-Lavier, A.S.; Barrón, M.G.; Petrov, D.A.; González, J. T-lex2: Genotyping, frequency estimation and re-annotation of transposable elements using single or pooled next-generation sequencing data. Nucleic Acids Res. 2015, 43, e22. [Google Scholar] [CrossRef] [PubMed]
- Crooks, C.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Kirk, P.D.; Huvet, M.; Melamed, A.; Maertens, G.N.; Bangham, C.R. Retroviruses integrate into a shared, non-palindromic DNA motif. Nat. Microbiol. 2016, 2, 16212. [Google Scholar] [CrossRef] [PubMed]
- Gramates, L.S.; Marygold, S.J.; dos Santos, G.; Urbano, J.-M.; Antonazzo, G.; Matthews, B.B.; Rey, A.J.; Tabone, C.J.; Crosby, M.A.; Emmert, D.B.; et al. FlyBase at 25: Looking to the future. Nucleic Acids Res. 2017, 5(D1), D663–D671. [Google Scholar] [CrossRef] [PubMed]
- Minervini, C.; Marsano, R.; Casieri, P.; Fanti, L.; Caizzi, R.; Pimpinelli, S.; Mariano, R.; Luigi, V. Heterochromatin protein 1 interacts with 5′UTR of transposable element ZAM in a sequence-specific fashion. Gene 2007, 393, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fablet, M.; McDonald, J.F.; Biemont, C.; Vieira, C. Ongoing loss of the tirant transposable element in natural populations of Drosophila simulans. Gene 2006, 375, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Fablet, M.; Lerat, E.; Rebollo, R.; Horard, B.; Burlet, N.; Martinez, S.; Brasset, E.; Gilson, E.; Vaury, C.; Vieira, C. Genomic environment influences the dynamics of the tirant LTR retrotransposon in Drosophila. FASEBJ 2009, 23, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.R.; Johnson, W.E. Plastic proteins and monkey blocks, how lentiviruses evolved to replicate in the presence of primate restriction factors. PLoS Pathog. 2014, 10, e1004017. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Butler, S.L.; Bushman, F. Human immunodeficiency virus type 1 integrase, arrangement of protein domains in active cDNA complexes. EMBO J. 2001, 20, 3565–3576. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.E.; Chen, H.; Engelman, A. Structure-based mutagenesis of the human immunodeficiency virus type 1 DNA attachment site, effects on integration and cDNA synthesis. J. Virol. 1999, 73, 9011–9020. [Google Scholar] [PubMed]
- LaFemina, R.L.; Callahan, P.L.; Cordingley, M.G. Substrate specificity of recombinant human immunodeficiency virus integrase protein. J. Virol. 1991, 65, 5624–5630. [Google Scholar] [PubMed]
- Mashkova, T.D.; Oparina, N.Y.; Lacroix, M.H.; Fedorova, L.I.; Tumeneva, G.; Zinovieva, O.L.; Kisselev, L.L. Structural rearrangements and insertions of dispersed elements in pericentromeric alpha satellites occur preferably at kinkable DNA sites. J. Mol. Biol. 2001, 305, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Delelis, O.; Carayon, K.; Saib, A.; Deprez, E.; Mouscadet, J.F. Integrase and integration, biochemical activities of HIV-1 integrase. Retrovirology 2008, 5, 114. [Google Scholar] [CrossRef] [PubMed]
- Dyda, F.; Hickman, A.B.; Jenkins, T.M.; Engelman, A.; Craigie, R.; Davies, D.R. Crystal structure of the catalytic domain of HIV-1 integrase, similarity to other polynucleotidyl transferases. Science 1994, 266, 1981–1986. [Google Scholar] [CrossRef] [PubMed]
- Katzman, M.; Sudol, M. Mapping viral DNA specificity to the central region of integrase by using functional human immunodeficiency virus type 1/Visna virus chimeric proteins. J. Virol. 1998, 72, 1744–1753. [Google Scholar]
- Appa, R.S.; Shin, C.G.; Lee, P.; Chow, S.A. Role of the nonspecific DNA-binding region and alpha helices within the core domain of retroviral integrase in selecting target DNA sites for integration. J. Biol. Chem. 2001, 276, 45848–45855. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Jenkins, T.M.; Craigie, R. Zinc folds the N-terminal domain of HIV-1 integrase, promotes multimerization, and enhances catalytic activity. Proc. Natl. Acad. Sci. USA 1996, 93, 13659–13664. [Google Scholar] [CrossRef] [PubMed]
- Woerner, A.M.; Marcus-Sekura, C.J. Characterization of a DNA binding domain in the C-terminus of HIV-1 integrase by deletion mutagenesis. Nucleic Acids Res. 1993, 21, 3507–3511. [Google Scholar] [CrossRef] [PubMed]
- Malik, H.S.; Henikoff, S.; Eickbush, T.H. Poised for contagion, evolutionary origins of the infectious abilities of invertebrate retroviruses. Genome Res. 2000, 10, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.R. SIRE-1, a putative plant retrovirus is closely related to a legume Ty1-copia retrotransposon family. Cell. Mol. Biol. Lett. 2007, 12, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.; Veith, A.M.; Volff, J.N. A family of neofunctionalized Ty3/gypsy retrotransposon genes in mammalian genomes. Cytogenet. Genome Res. 2005, 110, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Casacuberta, E.; González, J. The impact of transposable elements in environmental adaptation. J. Mol. Ecol. 2013, 22, 1503–1517. [Google Scholar] [CrossRef] [PubMed]
- Elbarbary, R.A.; Lucas, B.A.; Maquat, L.E. Retrotransposons as regulators of gene expression. Science 2016, 351, aac7247. [Google Scholar] [CrossRef] [PubMed]
- Nefedova, L.; Kuzmin, I.; Makhnovskii, P.; Kim, A. Domesticated retroviral gag gene in Drosophila, new functions for an old gene. Virology 2014, 450–451, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Malik, H.S.; Henikoff, S. Positive selection of Iris, a retroviral envelope-derived host gene in Drosophila melanogaster. PLoS Genet. 2005, 1, e44. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nefedova, L.; Kim, A. Mechanisms of LTR‐Retroelement Transposition: Lessons from Drosophila melanogaster. Viruses 2017, 9, 81. https://doi.org/10.3390/v9040081
Nefedova L, Kim A. Mechanisms of LTR‐Retroelement Transposition: Lessons from Drosophila melanogaster. Viruses. 2017; 9(4):81. https://doi.org/10.3390/v9040081
Chicago/Turabian StyleNefedova, Lidia, and Alexander Kim. 2017. "Mechanisms of LTR‐Retroelement Transposition: Lessons from Drosophila melanogaster" Viruses 9, no. 4: 81. https://doi.org/10.3390/v9040081
APA StyleNefedova, L., & Kim, A. (2017). Mechanisms of LTR‐Retroelement Transposition: Lessons from Drosophila melanogaster. Viruses, 9(4), 81. https://doi.org/10.3390/v9040081