
Metagenomic Approaches to Assess Bacteriophages in Various Environmental Niches



Abstract
:1. Introduction
2. Culture-Dependent vs. Culture-Independent Methods of Bacteriophage Study
Culture-Independent Methods of Bacteriophage Study
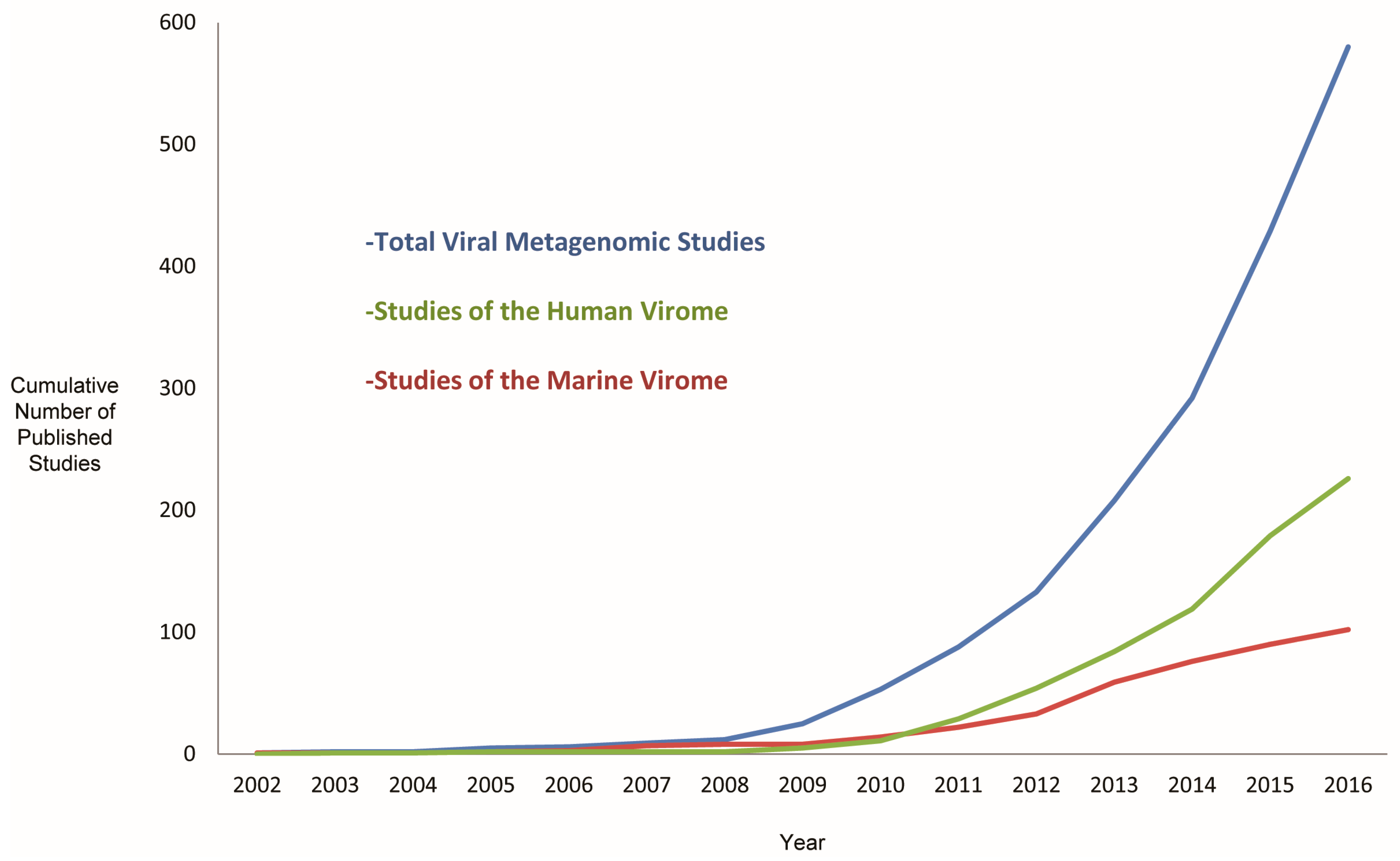
3. Metagenomics
4. Viral Metagenomic Sample to Sequence Pipeline
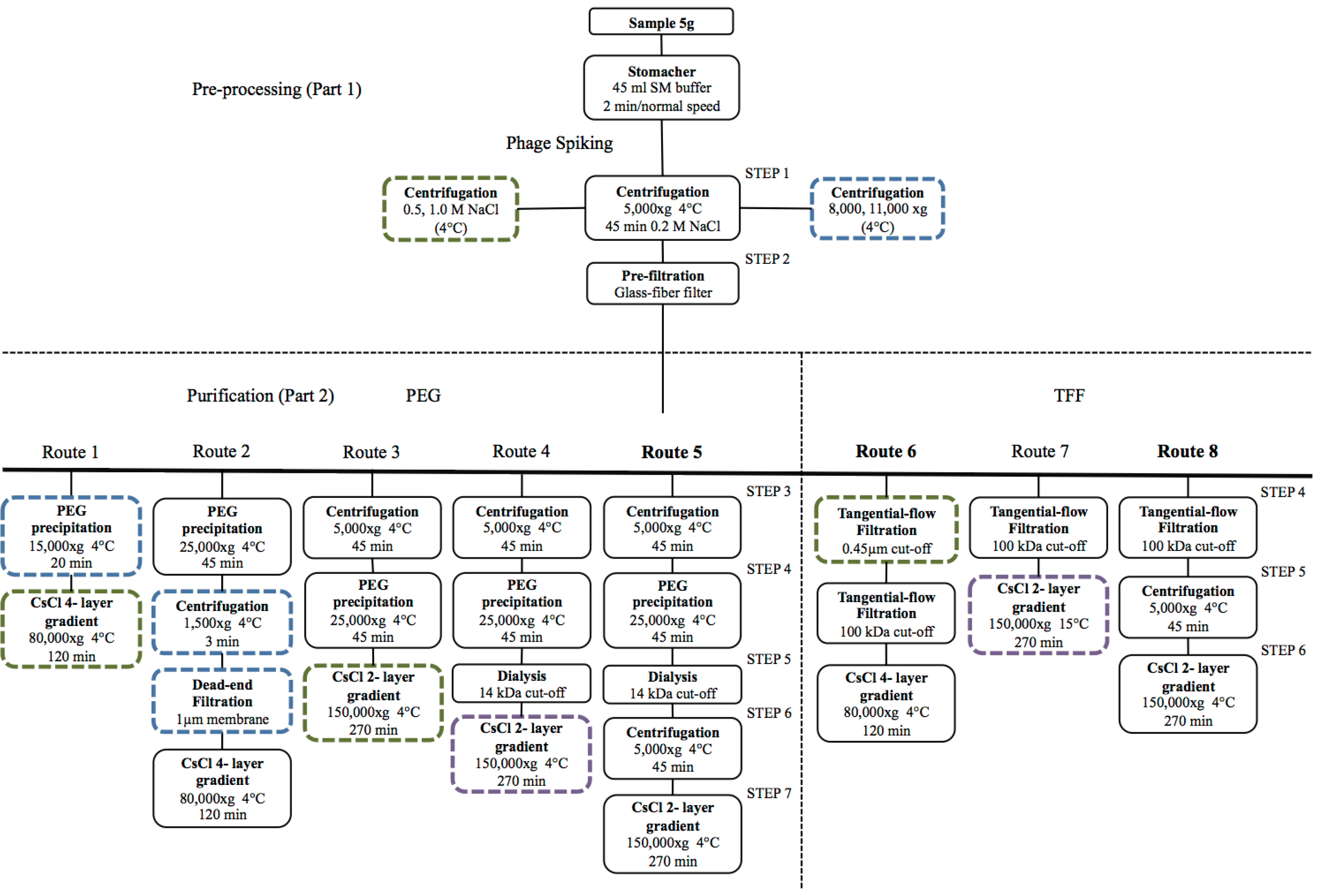
4.1. Viral Particle Extraction
4.2. Nucleic Acid Extraction
4.3. Library Preparation and Sequencing
4.4. Analysis
4.5. Bioinformatic Tools for Viral Metagenome Analysis
5. Current and Potential Areas of Interest for Viral Metagenomics
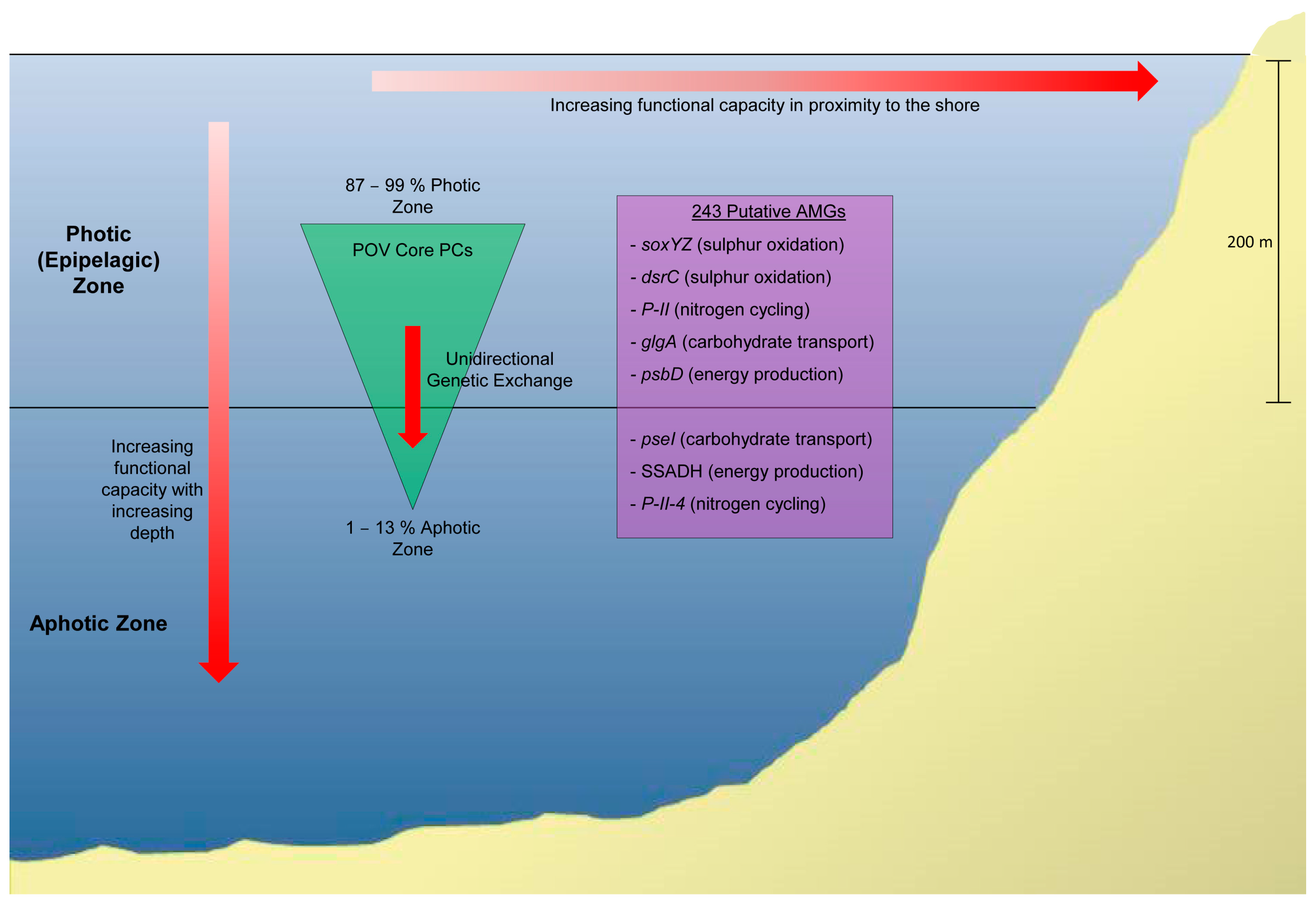
5.1. Marine Viral Metagenomics
5.2. Human Viral Metagenomics
5.2.1. Virome of the Skin and Oral Cavity
5.2.2. Virome of the Lungs
5.2.3. Virome of the Human Gut
5.3. Potential Applications
6. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rohwer, F. Global phage diversity. Cell 2003, 113, 141. [Google Scholar] [CrossRef]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Bergh, Ø.; Børsheim, K.Y.; Bratbak, G.; Heldal, M. High abundance of viruses found in aquatic environments. Nature 1989, 340, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Hoyles, L.; McCartney, A.L.; Neve, H.; Gibson, G.R.; Sanderson, J.D.; Heller, K.J.; van Sinderen, D. Characterization of virus-like particles associated with the human faecal and caecal microbiota. Res. Microbiol. 2014, 165, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Park, E.-J.; Roh, S.W.; Bae, J.-W. Diversity and abundance of single-stranded DNA viruses in human feces. Appl. Environ. Microbiol. 2011, 77, 8062–8070. [Google Scholar] [CrossRef] [PubMed]
- Clokie, M.R.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef] [PubMed]
- McGrath, S.; Fitzgerald, G.F.; van Sinderen, D. The impact of bacteriophage genomics. Curr. Opin. Biotechnol. 2004, 15, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Waldor, M.K.; Mekalanos, J.J. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 1996, 272, 1910–1914. [Google Scholar] [CrossRef] [PubMed]
- Waldor, M.K.; Friedman, D.I. Phage regulatory circuits and virulence gene expression. Curr. Opin. Microbiol. 2005, 8, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H.; Hendrix, R.W. Phage genomics: Small is beautiful. Cell 2002, 108, 13–16. [Google Scholar] [CrossRef]
- Gómez, P.; Buckling, A. Bacteria-phage antagonistic coevolution in soil. Science 2011, 332, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Pal, C.; Maciá, M.D.; Oliver, A.; Schachar, I.; Buckling, A. Coevolution with viruses drives the evolution of bacterial mutation rates. Nature 2007, 450, 1079–1081. [Google Scholar] [CrossRef] [PubMed]
- Canchaya, C.; Fournous, G.; Chibani-Chennoufi, S.; Dillmann, M.-L.; Brüssow, H. Phage as agents of lateral gene transfer. Curr. Opin. Microbiol. 2003, 6, 417–424. [Google Scholar] [CrossRef]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Valera, F.; Martin-Cuadrado, A.-B.; Rodriguez-Brito, B.; Pašić, L.; Thingstad, T.F.; Rohwer, F.; Mira, A. Explaining microbial population genomics through phage predation. Nat. Rev. Microbiol. 2009, 7, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Morris, J.J.; Décima, M.; Stukel, M.R. Mortality in the oceans: Causes and consequences. Assoc. Sci. Limnol. Oceanogr. 2014, 16–48. [Google Scholar] [CrossRef]
- Rohwer, F.; Youle, M. Consider something viral in your research. Nat. Rev. Microbiol. 2011, 9, 308–309. [Google Scholar] [CrossRef]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Mokili, J.L.; Rohwer, F.; Dutilh, B.E. Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2012, 2, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Amann, R.I.; Ludwig, W.; Schleifer, K.-H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 1995, 59, 143–169. [Google Scholar] [PubMed]
- Connon, S.A.; Giovannoni, S.J. High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Appl. Environ. Microbiol. 2002, 68, 3878–3885. [Google Scholar] [CrossRef] [PubMed]
- Rappe, M.S.; Giovannoni, S.J. The uncultured microbial majority. Annu. Rev. Microbiol. 2003, 57, 369–394. [Google Scholar] [CrossRef] [PubMed]
- Duhaime, M.B.; Sullivan, M.B. Ocean viruses: Rigorously evaluating the metagenomic sample-to-sequence pipeline. Virology 2012, 434, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Willner, D.; Hugenholtz, P. From deep sequencing to viral tagging: Recent advances in viral metagenomics. Bioessays 2013, 35, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Novick, R.P. Phage-mediated intergeneric transfer of toxin genes. Science 2009, 323, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Łoś, M.; Węgrzyn, G. Pseudolysogeny. Adv. Virus Res. 2011, 82, 339–349. [Google Scholar]
- Bryan, D.; El-Shibiny, A.; Hobbs, Z.; Porter, J.; Kutter, E.M. Bacteriophage T4 Infection of Stationary Phase E. coli: Life after Log from a Phage Perspective. Front. Microbiol. 2016, 7, 1391. [Google Scholar] [CrossRef] [PubMed]
- Chibani-Chennoufi, S.; Bruttin, A.; Dillmann, M.-L.; Brüssow, H. Phage-host interaction: An ecological perspective. J. Bacteriol. 2004, 186, 3677–3686. [Google Scholar] [CrossRef] [PubMed]
- Barns, S.M.; Fundyga, R.E.; Jeffries, M.W.; Pace, N.R. Remarkable archaeal diversity detected in a Yellowstone National Park hot spring environment. Proc. Natl. Acad. Sci. USA 1994, 91, 1609–1613. [Google Scholar] [CrossRef] [PubMed]
- Hugenholtz, P.; Pace, N.R. Identifying microbial diversity in the natural environment: A molecular phylogenetic approach. Trends Biotechnol. 1996, 14, 190–197. [Google Scholar] [CrossRef]
- Olsen, G.J.; Lane, D.J.; Giovannoni, S.J.; Pace, N.R.; Stahl, D.A. Microbial ecology and evolution: A ribosomal RNA approach. Annu. Rev. Microbiol. 1986, 40, 337–365. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-T.; Marsh, T.L.; Cheng, H.; Forney, L.J. Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl. Environ. Microbiol. 1997, 63, 4516–4522. [Google Scholar] [PubMed]
- Giovannoni, S.J.; Mullins, T.D.; Field, K.G. Microbial diversity in oceanic systems: rRNA approaches to the study of unculturable microbes. In Molecular Ecology of Aquatic Microbes; Springer: Berlin/Heidelberg, Germany, 1995; pp. 217–248. [Google Scholar]
- Schmidt, T.M.; DeLong, E.; Pace, N. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J. Bacteriol. 1991, 173, 4371–4378. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.A.; Rohwer, F. Viral metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Winget, D.M.; Wommack, K.E. Randomly amplified polymorphic DNA PCR as a tool for assessment of marine viral richness. Appl. Environ. Microbiol. 2008, 74, 2612–2618. [Google Scholar] [CrossRef] [PubMed]
- Brussaard, C.P. Optimization of procedures for counting viruses by flow cytometry. Appl. Environ. Microbiol. 2004, 70, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Børsheim, K.; Bratbak, G.; Heldal, M. Enumeration and biomass estimation of planktonic bacteria and viruses by transmission electron microscopy. Appl. Environ. Microbiol. 1990, 56, 352–356. [Google Scholar] [PubMed]
- Bratbak, G.; Heldal, M. Total count of viruses in aquatic environments. In Handbook of Methods in Aquatic Microbial Ecology; Lewis Publishers: Boca Raton, FL, USA, 1993; pp. 135–138. [Google Scholar]
- Allen, L.Z.; Ishoey, T.; Novotny, M.A.; McLean, J.S.; Lasken, R.S.; Williamson, S.J. Single virus genomics: A new tool for virus discovery. PLoS ONE 2011, 6, e17722. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Gregory, A.; Yilmaz, S.; Poulos, B.T.; Hugenholtz, P.; Sullivan, M.B. Contrasting life strategies of viruses that infect photo-and heterotrophic bacteria, as revealed by viral tagging. MBio 2012, 3, e00373-12. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Miyake, J.H.; Rohwer, F. Global distribution of nearly identical phage-encoded DNA sequences. FEMS Microbiol. Lett. 2004, 236, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.B.; Lindell, D.; Lee, J.A.; Thompson, L.R.; Bielawski, J.P.; Chisholm, S.W. Prevalence and evolution of core photosystem II genes in marine cyanobacterial viruses and their hosts. PLoS Biol. 2006, 4, e234. [Google Scholar] [CrossRef] [PubMed]
- Sharon, I.; Tzahor, S.; Williamson, S.; Shmoish, M.; Man-Aharonovich, D.; Rusch, D.B.; Yooseph, S.; Zeidner, G.; Golden, S.S.; Mackey, S.R.; et al. Viral photosynthetic reaction center genes and transcripts in the marine environment. ISME J. 2007, 1, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Chenard, C.; Suttle, C. Phylogenetic diversity of sequences of cyanophage photosynthetic gene psbA in marine and freshwaters. Appl. Environ. Microbiol. 2008, 74, 5317–5324. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Krisch, H.M. The capsid of the T4 phage superfamily: The evolution, diversity, and structure of some of the most prevalent proteins in the biosphere. Mol. Biol. Evol. 2008, 25, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.B. Viromes, not gene markers, for studying double-stranded DNA virus communities. J. Virol. 2015, 89, 2459–2461. [Google Scholar] [CrossRef] [PubMed]
- Hadrys, H.; Balick, M.; Schierwater, B. Applications of random amplified polymorphic DNA (RAPD) in molecular ecology. Mol. Ecol. 1992, 1, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Hara, S.; Terauchi, K.; Koike, I. Abundance of viruses in marine waters: Assessment by epifluorescence and transmission electron microscopy. Appl. Environ. Microbiol. 1991, 57, 2731–2734. [Google Scholar] [PubMed]
- Weinbauer, M.; Suttle, C. Comparison of epifluorescence and transmission electron microscopy for counting viruses in natural marine waters. Aquat. Microb. Ecol. 1997, 13, 225–232. [Google Scholar] [CrossRef]
- Noble, R.T.; Fuhrman, J.A. Use of SYBR Green I for rapid epifluorescence counts of marine viruses and bacteria. Aquat. Microb. Ecol. 1998, 14, 113–118. [Google Scholar] [CrossRef]
- Wen, K.; Ortmann, A.C.; Suttle, C.A. Accurate estimation of viral abundance by epifluorescence microscopy. Appl. Environ. Microbiol. 2004, 70, 3862–3867. [Google Scholar] [CrossRef] [PubMed]
- Marie, D.; Brussaard, C.P.; Thyrhaug, R.; Bratbak, G.; Vaulot, D. Enumeration of marine viruses in culture and natural samples by flow cytometry. Appl. Environ. Microbiol. 1999, 65, 45–52. [Google Scholar] [PubMed]
- Brussaard, C.P.; Marie, D.; Bratbak, G. Flow cytometric detection of viruses. J. Virol. Methods 2000, 85, 175–182. [Google Scholar] [CrossRef]
- Ohno, S.; Okano, H.; Tanji, Y.; Ohashi, A.; Watanabe, K.; Takai, K.; Imachi, H. A method for evaluating the host range of bacteriophages using phages fluorescently labeled with 5-ethynyl-2′-deoxyuridine (EdU). Appl. Microbiol. Biotechnol. 2012, 95, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Sullivan, M.B. Rising to the challenge: Accelerated pace of discovery transforms marine virology. Nat. Rev. Microbiol. 2015, 13, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J.; Goodman, R.M. Molecular biological access to the chemistry of unknown soil microbes: A new frontier for natural products. Chem. Biol. 1998, 5, R245–R249. [Google Scholar] [CrossRef]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics-a guide from sampling to data analysis. Microb. Inform. Exp. 2012, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.L.; Madsen, K.L.; et al. Characterization of the gut microbiome using 16S or shotgun metagenomics. Front. Microbiol. 2016, 7, 459. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, J. Metagenetics: Spending our inheritance on the future. Microb. Biotechnol. 2009, 2, 138–139. [Google Scholar] [CrossRef] [PubMed]
- Oulas, A.; Pavloudi, C.; Polymenakou, P.; Pavlopoulos, G.A.; Papanikolaou, N.; Kotoulas, G.; Arvanitidis, C.; Iliopoulos, I. Metagenomics: Tools and insights for analyzing next-generation sequencing data derived from biodiversity studies. Bioinform. Biol. Insights 2015, 9, 75–88. [Google Scholar] [PubMed]
- De Filippis, F.; Parente, E.; Ercolini, D. Metagenomics insights into food fermentations. Microb. Biotechnol. 2016, 10, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, V.M.; Chen, I.-M.A.; Palaniappan, K.; Chu, K.; Szeto, E.; Grechkin, Y.; Ratner, A.; Jacob, B.; Huang, J.; Williams, P.; et al. IMG: The integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012, 40, D115–D122. [Google Scholar] [CrossRef] [PubMed]
- Culligan, E.P.; Sleator, R.D.; Marchesi, J.R.; Hill, C. Metagenomics and novel gene discovery: Promise and potential for novel therapeutics. Virulence 2014, 5, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Thies, S.; Rausch, S.C.; Kovacic, F.; Schmidt-Thaler, A.; Wilhelm, S.; Rosenau, F.; Daniel, R.; Streit, W.; Pietruszka, J.; Jaeger, K.-E. Metagenomic discovery of novel enzymes and biosurfactants in a slaughterhouse biofilm microbial community. Sci. Rep. 2016, 6, 27035. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, T.; Miyazaki, K. Functional metagenomics for enzyme discovery: Challenges to efficient screening. Curr. Opin. Biotechnol. 2009, 20, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, J. Metagenomics: Application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 2004, 68, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Krupovic, M.; Debroas, D.; Forterre, P.; Enault, F. Assessment of viral community functional potential from viral metagenomes may be hampered by contamination with cellular sequences. Open Biol. 2013, 3, 130160. [Google Scholar] [CrossRef] [PubMed]
- Labonté, J.M.; Suttle, C.A. Previously unknown and highly divergent ssDNA viruses populate the oceans. ISME J. 2013, 7, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Van Regenmortel, M.H.; Fauquet, C.M.; Bishop, D.H.; Carstens, E.; Estes, M.; Lemon, S.; Maniloff, J.; Mayo, M.; McGeoch, D.; Pringle, C. Virus taxonomy: Classification and nomenclature of viruses. In Seventh Report of the International Committee on Taxonomy of Viruses; Academic Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Kleiner, M.; Hooper, L.V.; Duerkop, B.A. Evaluation of methods to purify virus-like particles for metagenomic sequencing of intestinal viromes. BMC Genom. 2015, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Salamon, P.; Andresen, B.; Mahaffy, J.M.; Segall, A.M.; Mead, D.; Azam, F.; Rohwer, F. Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. USA 2002, 99, 14250–14255. [Google Scholar] [CrossRef] [PubMed]
- Castro-Mejía, J.L.; Muhammed, M.K.; Kot, W.; Neve, H.; Franz, C.M.; Hansen, L.H.; Vogensen, F.K.; Nielsen, D.S. Optimizing protocols for extraction of bacteriophages prior to metagenomic analyses of phage communities in the human gut. Microbiome 2015, 3, 64. [Google Scholar] [CrossRef] [PubMed]
- Thurber, R.V.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009, 4, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Iker, B.C.; Bright, K.R.; Pepper, I.L.; Gerba, C.P.; Kitajima, M. Evaluation of commercial kits for the extraction and purification of viral nucleic acids from environmental and fecal samples. J. Virol. Methods 2013, 191, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.J.; Wang, J.; Todd, A.K.; Bissielo, A.B.; Yen, S.; Strydom, H.; Moore, N.E.; Ren, X.; Huang, Q.S.; Carter, P.E.; et al. Evaluation of rapid and simple techniques for the enrichment of viruses prior to metagenomic virus discovery. J. Virol. Methods 2014, 195, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Pelzek, A.J.; Schuch, R.; Schmitz, J.E.; Fischetti, V.A. Isolation of bacteriophages from environmental sources, and creation and functional screening of phage DNA libraries. Curr. Protoc. Essent. Lab. Tech. 2013, 7, 13.3.1–13.3.35. [Google Scholar]
- John, S.G.; Mendez, C.B.; Deng, L.; Poulos, B.; Kauffman, A.K.M.; Kern, S.; Brum, J.; Polz, M.F.; Boyle, E.A.; Sullivan, M.B. A simple and efficient method for concentration of ocean viruses by chemical flocculation. Environ. Microbiol. Rep. 2011, 3, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, B.L.; Deng, L.; Poulos, B.T.; Sullivan, M.B. Evaluation of methods to concentrate and purify ocean virus communities through comparative, replicated metagenomics. Environ. Microbiol. 2013, 15, 1428–1440. [Google Scholar] [CrossRef] [PubMed]
- Wesolowska-Andersen, A.; Bahl, M.I.; Carvalho, V.; Kristiansen, K.; Sicheritz-Pontén, T.; Gupta, R.; Licht, T.R. Choice of bacterial DNA extraction method from fecal material influences community structure as evaluated by metagenomic analysis. Microbiome 2014, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Sachsenröder, J.; Twardziok, S.; Hammerl, J.A.; Janczyk, P.; Wrede, P.; Hertwig, S.; Johne, R. Simultaneous identification of DNA and RNA viruses present in pig faeces using process-controlled deep sequencing. PLoS ONE 2012, 7, e34631. [Google Scholar] [CrossRef] [PubMed]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, B.L.; U’ren, J.M.; Youens-Clark, K. Computational prospecting the great viral unknown. FEMS Microbiol. Lett. 2016, 363, fnw077. [Google Scholar] [CrossRef] [PubMed]
- Solonenko, S.A.; Sullivan, M.B. Preparation of metagenomic libraries from naturally occurring marine viruses. Methods Enzyml. 2013, 531, 143–160. [Google Scholar]
- Kim, K.-H.; Bae, J.-W. Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl. Environ. Microbiol. 2011, 77, 7663–7668. [Google Scholar] [CrossRef] [PubMed]
- Woyke, T.; Xie, G.; Copeland, A.; Gonzalez, J.M.; Han, C.; Kiss, H.; Saw, J.H.; Senin, P.; Yang, C.; Chatterji, S.; et al. Assembling the marine metagenome, one cell at a time. PLoS ONE 2009, 4, e5299. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, S.; Allgaier, M.; Hugenholtz, P. Multiple displacement amplification compromises quantitative analysis of metagenomes. Nat. Methods 2010, 7, 943–944. [Google Scholar] [CrossRef] [PubMed]
- Bikel, S.; Valdez-Lara, A.; Cornejo-Granados, F.; Rico, K.; Canizales-Quinteros, S.; Soberón, X.; Del Pozo-Yauner, L.; Ochoa-Leyva, A. Combining metagenomics, metatranscriptomics and viromics to explore novel microbial interactions: Towards a systems-level understanding of human microbiome. Comput. Struct. Biotechnol. J. 2015, 13, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, W.A.; Bártfai, R.; Françoijs, K.-J.; Stunnenberg, H.G. Linear amplification for deep sequencing. Nat. Protoc. 2011, 6, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Marine, R.; Polson, S.W.; Ravel, J.; Hatfull, G.; Russell, D.; Sullivan, M.; Syed, F.; Dumas, M.; Wommack, K.E. Evaluation of a transposase protocol for rapid generation of shotgun high-throughput sequencing libraries from nanogram quantities of DNA. Appl. Environ. Microbiol. 2011, 77, 8071–8079. [Google Scholar] [CrossRef] [PubMed]
- Chafee, M.; Maignien, L.; Simmons, S.L. The effects of variable sample biomass on comparative metagenomics. Environ. Microbiol. 2015, 17, 2239–2253. [Google Scholar] [CrossRef] [PubMed]
- Henn, M.R.; Sullivan, M.B.; Stange-Thomann, N.; Osburne, M.S.; Berlin, A.M.; Kelly, L.; Yandava, C.; Kodira, C.; Zeng, Q.; Weiand, M.; et al. Analysis of high-throughput sequencing and annotation strategies for phage genomes. PLoS ONE 2010, 5, e9083. [Google Scholar] [CrossRef] [PubMed]
- Duhaime, M.B.; Deng, L.; Poulos, B.T.; Sullivan, M.B. Towards quantitative metagenomics of wild viruses and other ultra-low concentration DNA samples: A rigorous assessment and optimization of the linker amplification method. Environ. Microbiol. 2012, 14, 2526–2537. [Google Scholar] [CrossRef] [PubMed]
- Dean, F.B.; Hosono, S.; Fang, L.; Wu, X.; Faruqi, A.F.; Bray-Ward, P.; Sun, Z.; Zong, Q.; Du, Y.; Du, J.; et al. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. USA 2002, 99, 5261–5266. [Google Scholar] [CrossRef] [PubMed]
- Zong, C.; Lu, S.; Chapman, A.R.; Xie, X.S. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 2012, 338, 1622–1626. [Google Scholar] [CrossRef] [PubMed]
- Bowers, R.M.; Clum, A.; Tice, H.; Lim, J.; Singh, K.; Ciobanu, D.; Ngan, C.Y.; Cheng, J.-F.; Tringe, S.G.; Woyke, T. Impact of library preparation protocols and template quantity on the metagenomic reconstruction of a mock microbial community. BMC Genom. 2015, 16, 856. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, P.; Murphy, J.; Mahony, J.; Van Sinderen, D. Next-generation sequencing as an approach to dairy starter selection. Dairy Sci. Technol. 2015, 95, 545–568. [Google Scholar] [CrossRef] [PubMed]
- Bibby, K. Improved bacteriophage genome data is necessary for integrating viral and bacterial ecology. Microb. Ecol. 2014, 67, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Glass, E.M.; Wilkening, J.; Wilke, A.; Antonopoulos, D.; Meyer, F. Using the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5368. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Gibbons, T.; Ghodsi, M.; Treangen, T.; Pop, M. Accurate and fast estimation of taxonomic profiles from metagenomic shotgun sequences. BMC Genom. 2011, 12, S4. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, W.; Jünemann, S.; Tille, F.; Goesmann, A.; Stoye, J. WebCARMA: A web application for the functional and taxonomic classification of unassembled metagenomic reads. BMC Bioinform. 2009, 10, 430. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, B.L.; Westveld, A.H.; Brum, J.R.; Sullivan, M.B. Modeling ecological drivers in marine viral communities using comparative metagenomics and network analyses. Proc. Natl. Acad. Sci. USA 2014, 111, 10714–10719. [Google Scholar] [CrossRef] [PubMed]
- Ounit, R.; Wanamaker, S.; Close, T.J.; Lonardi, S. CLARK: Fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genom. 2015, 16, 236. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, B.L.; Sullivan, M.B. The Pacific Ocean Virome (POV): A marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PLoS ONE 2013, 8, e57355. [Google Scholar] [CrossRef] [PubMed]
- Wommack, K.E.; Bhavsar, J.; Polson, S.W.; Chen, J.; Dumas, M.; Srinivasiah, S.; Furman, M.; Jamindar, S.; Nasko, D.J. VIROME: A standard operating procedure for analysis of viral metagenome sequences. Stand. Genom. Sci. 2012, 6, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Tournayre, J.; Mahul, A.; Debroas, D.; Enault, F. Metavir 2: New tools for viral metagenome comparison and assembled virome analysis. BMC Bioinform. 2014, 15, 76. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, H.A.; Hoover, J.; Inman, J.; Safford, T.; Murphy, S.; Kagan, L.; Williamson, S.J. The Viral MetaGenome Annotation Pipeline (VMGAP): An automated tool for the functional annotation of viral Metagenomic shotgun sequencing data. Stand. Genom. Sci. 2011, 4, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Tangherlini, M.; Dell’Anno, A.; Allen, L.Z.; Riccioni, G.; Corinaldesi, C. Assessing viral taxonomic composition in benthic marine ecosystems: Reliability and efficiency of different bioinformatic tools for viral metagenomic analyses. Sci. Rep. 2016, 6, 28428. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Semenkovich, N.P.; Whiteson, K.; Rohwer, F.; Gordon, J.I. Going viral: Next-generation sequencing applied to phage populations in the human gut. Nat. Rev. Microbiol. 2012, 10, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Wommack, K.E.; Nasko, D.J.; Chopyk, J.; Sakowski, E.G. Counts and sequences, observations that continue to change our understanding of viruses in nature. J. Microbiol. 2015, 53, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Angly, F.E.; Felts, B.; Breitbart, M.; Salamon, P.; Edwards, R.A.; Carlson, C.; Chan, A.M.; Haynes, M.; Kelley, S.; Liu, H.; et al. The marine viromes of four oceanic regions. PLoS Biol. 2006, 4, e368. [Google Scholar] [CrossRef] [PubMed]
- Dutilh, B.E.; Schmieder, R.; Nulton, J.; Felts, B.; Salamon, P.; Edwards, R.A.; Mokili, J.L. Reference-independent comparative metagenomics using cross-assembly: CrAss. Bioinformatics 2012, 28, 3225–3231. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.; Constantinides, B.; Tapinos, A.; Robertson, D.L.; Prosperi, M. Challenges in the analysis of viral metagenomes. Virus Evol. 2016, 2, vew022. [Google Scholar] [CrossRef]
- Sharma, D.; Priyadarshini, P.; Vrati, S. Unraveling the web of viroinformatics: Computational tools and databases in virus research. J. Virol. 2015, 89, 1489–1501. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef] [PubMed]
- Jurtz, V.I.; Villarroel, J.; Lund, O.; Larsen, M.V.; Nielsen, M. MetaPhinder—Identifying Bacteriophage Sequences in Metagenomic Data Sets. PLoS ONE 2016, 11, e0163111. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Wu, G.; Lim, E.S.; Droit, L.; Krishnamurthy, S.; Barouch, D.H.; Virgin, H.W.; Wang, D. VirusSeeker, a computational pipeline for virus discovery and virome composition analysis. Virology 2017, 503, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, B.; Youens-Clark, K.; Roux, S.; Hurwitz, B.L.; Sullivan, M.B. iVirus: Facilitating new insights in viral ecology with software and community data sets imbedded in a cyberinfrastructure. ISME J. 2016, 11, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Yooseph, S.; Sutton, G.; Rusch, D.B.; Halpern, A.L.; Williamson, S.J.; Remington, K.; Eisen, J.A.; Heidelberg, K.B.; Manning, G.; Li, W.; et al. The Sorcerer II Global Ocean Sampling expedition: Expanding the universe of protein families. PLoS Biol. 2007, 5, e16. [Google Scholar] [CrossRef] [PubMed]
- ONeill, K.; Klimke, W.; Tatusova, T. Protein Clusters: A Collection of Proteins Grouped by Sequence Similarity and Function; Protein Clusters Help; National Center for Biotechnology Information: Bethesda, MD, USA, 2007. Available online: https://www.ncbi.nlm.nih.gov/books/NBK3797/ (accessed on 30 March 2017).
- Hurwitz, B.L.; Brum, J.R.; Sullivan, M.B. Depth-stratified functional and taxonomic niche specialization in the ‘core’and ‘flexible’ Pacific Ocean Virome. ISME J. 2015, 9, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Ignacio-Espinoza, J.C.; Solonenko, S.A.; Sullivan, M.B. The global virome: Not as big as we thought? Curr. Opin. Virol. 2013, 3, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 2016, 537, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Thompson, L.R.; Suttle, C.A.; Sullivan, M. Exploring the vast diversity of marine viruses. Oceanography 2007, 20, 135–139. [Google Scholar] [CrossRef]
- Hurwitz, B.L.; Hallam, S.J.; Sullivan, M.B. Metabolic reprogramming by viruses in the sunlit and dark ocean. Genome Biol. 2013, 14, R123. [Google Scholar] [CrossRef] [PubMed]
- Karsenti, E.; Acinas, S.G.; Bork, P.; Bowler, C.; De Vargas, C.; Raes, J.; Sullivan, M.; Arendt, D.; Benzoni, F.; Claverie, J.-M.; et al. A holistic approach to marine eco-systems biology. PLoS Biol. 2011, 9, e1001177. [Google Scholar] [CrossRef] [PubMed]
- Duarte, C.M. Seafaring in the 21st century: The Malaspina 2010 Circumnavigation Expedition. Limnol. Oceanogr. Bull. 2015, 24, 11–14. [Google Scholar] [CrossRef]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral dark matter and virus–host interactions resolved from publicly available microbial genomes. eLife 2015, 4, e08490. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, C.M.; Rodriguez-Valera, F.; Kimes, N.E.; Ghai, R. Expanding the marine virosphere using metagenomics. PLoS Genet. 2013, 9, e1003987. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.A.; McNair, K.; Faust, K.; Raes, J.; Dutilh, B.E. Computational approaches to predict bacteriophage-host relationships. FEMS Microbiol. Rev. 2016, 40, 258–272. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.F.; Banfield, J.F. Virus population dynamics and acquired virus resistance in natural microbial communities. Science 2008, 320, 1047–1050. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Solonenko, N.E.; Dang, V.T.; Poulos, B.T.; Schwenck, S.M.; Goldsmith, D.B.; Coleman, M.L.; Breitbart, M.; Sullivan, M.B. Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ 2016, 4, e2777. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.L.; Xu, S.Y.; Ren, Z.G.; Tao, L.; Jiang, J.W.; Zheng, S.S. Application of metagenomics in the human gut microbiome. World J. Gastroenterol. 2015, 21, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Hannigan, G.D.; Meisel, J.S.; Tyldsley, A.S.; Zheng, Q.; Hodkinson, B.P.; SanMiguel, A.J.; Minot, S.; Bushman, F.D.; Grice, E.A. The human skin double-stranded DNA virome: Topographical and temporal diversity, genetic enrichment, and dynamic associations with the host microbiome. MBio 2015, 6, e01578-15. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Conlan, S.; Deming, C.; Segre, J.A.; Kong, H.H.; Korlach, J.; Oh, J. Resolving the complexity of human skin metagenomes using single-molecule sequencing. MBio 2016, 7, e01948-15. [Google Scholar] [CrossRef] [PubMed]
- Edlund, A.; Santiago-Rodriguez, T.M.; Boehm, T.K.; Pride, D.T. Bacteriophage and their potential roles in the human oral cavity. J. Oral Microbiol. 2015, 7, 27423. [Google Scholar] [CrossRef] [PubMed]
- Pride, D.T.; Salzman, J.; Haynes, M.; Rohwer, F.; Davis-Long, C.; White, R.A., III; Loomer, P.; Armitage, G.C.; Relman, D.A. Evidence of a robust resident bacteriophage population revealed through analysis of the human salivary virome. ISME J. 2012, 6, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Boutin, S.; Graeber, S.Y.; Weitnauer, M.; Panitz, J.; Stahl, M.; Clausznitzer, D.; Kaderali, L.; Einarsson, G.; Tunney, M.M.; Elborn, J.S.; et al. Comparison of microbiomes from different niches of upper and lower airways in children and adolescents with cystic fibrosis. PLoS ONE 2015, 10, e0116029. [Google Scholar] [CrossRef] [PubMed]
- Moran Losada, P.; Chouvarine, P.; Dorda, M.; Hedtfeld, S.; Mielke, S.; Schulz, A.; Wiehlmann, L.; Tummler, B. The cystic fibrosis lower airways microbial metagenome. ERJ Open Res. 2016, 2, 00096-2015. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Lucht, L.; Tipton, L.; Rogers, M.B.; Fitch, A.; Kessinger, C.; Camp, D.; Kingsley, L.; Leo, N.; Greenblatt, R.M.; et al. Topographic diversity of the respiratory tract mycobiome and alteration in HIV and lung disease. Am. J. Respir. Crit. Care Med. 2015, 191, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.B.; Oliver, B.G.; Glanville, A.R. Translational aspects of the human respiratory virome. Am. J. Respir. Crit. Care Med. 2016, 194, 1458–1464. [Google Scholar] [CrossRef] [PubMed]
- Young, J.C.; Chehoud, C.; Bittinger, K.; Bailey, A.; Diamond, J.M.; Cantu, E.; Haas, A.R.; Abbas, A.; Frye, L.; Christie, J.D.; et al. Viral metagenomics reveal blooms of anelloviruses in the respiratory tract of lung transplant recipients. Am. J. Transplant. 2015, 15, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.W.; Schmieder, R.; Haynes, M.; Willner, D.; Furlan, M.; Youle, M.; Abbott, K.; Edwards, R.; Evangelista, J.; Conrad, D.; et al. Metagenomics and metatranscriptomics: Windows on CF-associated viral and microbial communities. J. Cyst. Fibros. 2013, 12, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Duerkop, B.A.; Hooper, L.V. Resident viruses and their interactions with the immune system. Nat. Immunol. 2013, 14, 654–659. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, P.W.; Jeffery, I.B. Gut microbiota and aging. Science 2015, 350, 1214–1215. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, I.B.; Lynch, D.B.; O’Toole, P.W. Composition and temporal stability of the gut microbiota in older persons. ISME J. 2016, 10, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.B.; Jeffery, I.B.; O’Toole, P.W. The role of the microbiota in ageing: Current state and perspectives. Wiley Interdiscip. Rev. Syst. Biol. Med. 2015, 7, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Mancabelli, L.; Milani, C.; Lugli, G.A.; Turroni, F.; Ferrario, C.; van Sinderen, D.; Ventura, M. Meta-analysis of the human gut microbiome from urbanized and pre-agricultural populations. Environ. Microbiol. 2017, 19, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Ferrario, C.; Turroni, F.; Duranti, S.; Mangifesta, M.; van Sinderen, D.; Ventura, M. The human gut microbiota and its interactive connections to diet. J. Hum. Nutr. Diet. 2016, 29, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.B.; Jeffery, I.B.; Cusack, S.; O’Connor, E.M.; O’Toole, P.W. Diet-microbiota-health interactions in older subjects: Implications for healthy aging. Interdiscip. Top. Gerontol. 2015, 40, 141–154. [Google Scholar] [PubMed]
- Joyce, S.A.; Gahan, C.G. Disease-associated changes in bile acid profiles and links to altered gut microbiota. Dig. Dis. 2017, 35, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Ticinesi, A.; Gerritsen, J.; Nouvenne, A.; Lugli, G.A.; Mancabelli, L.; Turroni, F.; Duranti, S.; Mangifesta, M.; Viappiani, A.; et al. Gut microbiota composition and Clostridium difficile infection in hospitalized elderly individuals: A metagenomic study. Sci. Rep. 2016, 6, 25945. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.A.; Milani, C.; Turroni, F.; Tremblay, D.; Ferrario, C.; Mancabelli, L.; Duranti, S.; Ward, D.V.; Ossiprandi, M.C.; Moineau, S.; et al. Prophages of the genus Bifidobacterium as modulating agents of the infant gut microbiota. Environ. Microbiol. 2016, 18, 2196–2213. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef] [PubMed]
- Manrique, P.; Bolduc, B.; Walk, S.T.; van der Oost, J.; de Vos, W.M.; Young, M.J. Healthy human gut phageome. Proc. Natl. Acad. Sci. USA 2016, 113, 10400–10405. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, P.; Eck, J. Metagenomics and industrial applications. Nat. Rev. Microbiol. 2005, 3, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, L.M.; Cotter, P.D.; Hill, C.; Alvarez-Ordóñez, A. Biotechnological applications of functional metagenomics in the food and pharmaceutical industries. Front. Microbiol. 2015, 6, 672. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, T.; Liles, M.; Wommack, K.E.; Polson, S.W.; Godiska, R.; Mead, D. Functional viral metagenomics and the next generation of molecular tools. Trends Microbiol. 2010, 18, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, T.W.; Moser, M.J.; Mead, D. Functional Viral Metagenomics and the Development of New Enzymes for DNA and RNA Amplification and Sequencing. In Encyclopedia of Metagenomics: Genes, Genomes and Metagenomes: Basics, Methods, Databases and Tools; Springer International Publishing: New York, NY, USA, 2015; pp. 198–218. [Google Scholar]
- Moser, M.J.; DiFrancesco, R.A.; Gowda, K.; Klingele, A.J.; Sugar, D.R.; Stocki, S.; Mead, D.A.; Schoenfeld, T.W. Thermostable DNA polymerase from a viral metagenome is a potent RT-PCR enzyme. PLoS ONE 2012, 7, e38371. [Google Scholar] [CrossRef] [PubMed]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage endolysins as novel antimicrobials. Future Microbiol. 2012, 7, 1147–1171. [Google Scholar] [CrossRef] [PubMed]
- Fischetti, V.A. Bacteriophage lysins as effective antibacterials. Curr. Opin. Microbiol. 2008, 11, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.E.; Schuch, R.; Fischetti, V.A. Identifying active phage lysins through functional viral metagenomics. Appl. Environ. Microbiol. 2010, 76, 7181–7187. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Method | Description | Limitations |
|---|---|---|
| Gene marker-based studies [43,44,45,46,47] | Utilise marker genes, ranging from major capsid proteins to photosynthesis related genes, to study the diversity of viruses in a sample. | Lack of universal viral gene limits the focus of studies to particular phage genera [48]; cannot provide quantitative analysis [24]. |
| Randomly Amplified Polymorphic DNA (RAPD) PCR [37] | Uses short, random primers to amplify fragments of environmental DNA of assorted sizes. Provides a rapid, rudimentary comparison of viral diversity. | Limited inferences possible; difficult to reproduce results due to high sensitivity of the technique to reaction conditions [49]. |
| Electron microscopy [3,39,40] | Allows enumeration of uncultured viruses, particularly in marine samples. Accuracy and speed improved by epifluorescent microscopy [50,51,52,53]. | Limited to observation of morphologies and rough estimates of quantity of viral particles; no sequence data generated. |
| Flow Cytometry [38,54,55] | Rapid enumeration of viral particles in a sample via their staining with highly fluorescent nucleic acid dyes followed by counting via flow cytometry. | Limited to estimations of quantity; no sequence data generated or morphology information. |
| Single virus genomics [41] | Enables isolation and complete genome sequencing of single viral particles. Involves sorting of single viruses by flow cytometry, followed by genome amplification via multiple displacement amplification (MDA) and whole genome sequencing. | Does not provide community-wide view of viral population. |
| Viral Tagging [42,56] | Allows study of phage–host interactions by fluorescently labelling phages and using them to ‘tag’ their host. Phages inject labelled genomes into their host, rendering the bacteria fluorescent. Potential hosts are then sorted via fluorescence-activated cell sorting (FACS). | Requires a culturable host, extensive optimisation required for each new host [57]. |
| Method | Nucleic Acid Quantity | Advantages | Drawbacks |
|---|---|---|---|
| Multiple displacement amplification (MDA) [95] | 1–100 ng | Rapid and high-throughput | Introduces both predictable and stochastic biases |
| Linear amplification for deep sequencing (LADS) [90] | 3–40 ng | Low levels of bias introduced, resulting in near-quantitative metagenomes | Low throughput, requires significant expertise |
| Linker amplified library construction [94] | >10 pg | Remains the most quantitatively accurate method, requires minimal nucleic acid input | Low throughput, requires significant expertise |
| Nextera XT (Illumina) | 50 pg | Rapid, combines fragmentation and tagging of DNA into single 5 min ‘tagmentation’ step | Slight sequence-dependent biases at low nucleic acid input levels [92] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayes, S.; Mahony, J.; Nauta, A.; Van Sinderen, D. Metagenomic Approaches to Assess Bacteriophages in Various Environmental Niches. Viruses 2017, 9, 127. https://doi.org/10.3390/v9060127
Hayes S, Mahony J, Nauta A, Van Sinderen D. Metagenomic Approaches to Assess Bacteriophages in Various Environmental Niches. Viruses. 2017; 9(6):127. https://doi.org/10.3390/v9060127
Chicago/Turabian StyleHayes, Stephen, Jennifer Mahony, Arjen Nauta, and Douwe Van Sinderen. 2017. "Metagenomic Approaches to Assess Bacteriophages in Various Environmental Niches" Viruses 9, no. 6: 127. https://doi.org/10.3390/v9060127
APA StyleHayes, S., Mahony, J., Nauta, A., & Van Sinderen, D. (2017). Metagenomic Approaches to Assess Bacteriophages in Various Environmental Niches. Viruses, 9(6), 127. https://doi.org/10.3390/v9060127