
The Role of Caveolin 1 in HIV Infection and Pathogenesis

Abstract
:1. Introduction
2. Caveolin 1 and Its Function
3. Cav-1 Mediated HIV Inhibition
4. HIV Induces Cav-1 Up-Regulation
5. Cav-1 Mediated Cholesterol Balance and Role in HIV Macrophage Infection
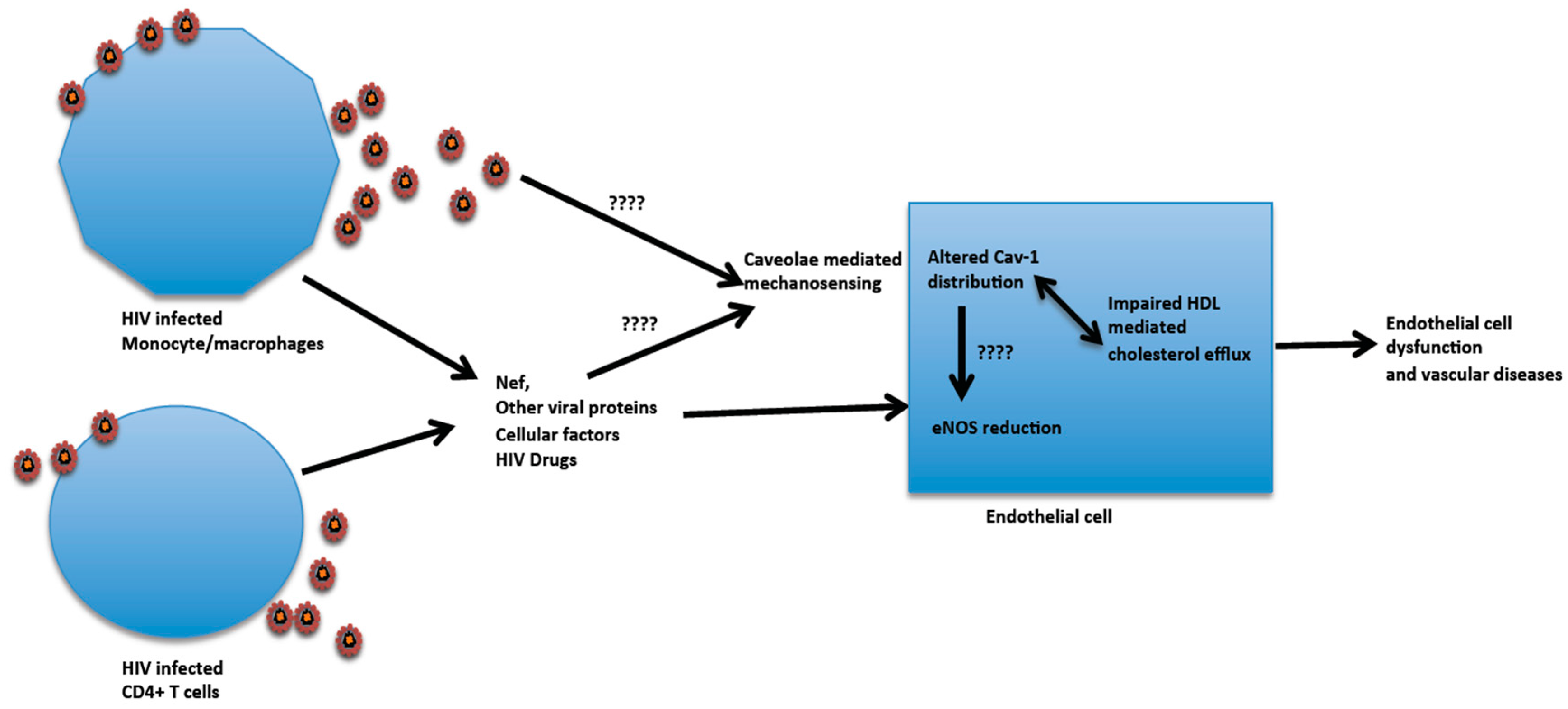
6. HIV Infection and Influence on Aortic Endothelial Cell Cav-1 Function
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Chun, T.W.; Moir, S.; Fauci, A.S. HIV reservoirs as obstacles and opportunities for an HIV cure. Nat. Immunol. 2015, 16, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Siliciano, R.F. Recent developments in the effort to cure HIV infection: Going beyond n = 1. J. Clin. Investig. 2016, 126, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Kline, C.; Ndjomou, J.; Franks, T.; Kiser, R.; Coalter, V.; Smedley, J.; Piatak, M., Jr.; Mellors, J.W.; Lifson, J.D.; Ambrose, Z. Persistence of viral reservoirs in multiple tissues after antiretroviral therapy suppression in a macaque RT-SHIV model. PLoS ONE 2013, 8, e84275. [Google Scholar] [CrossRef] [PubMed]
- Blankson, J.N.; Persaud, D.; Siliciano, R.F. The challenge of viral reservoirs in HIV-1 infection. Annu. Rev. Med. 2002, 53, 557–593. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–557. [Google Scholar] [PubMed]
- Archin, N.M.; Sung, J.M.; Garrido, C.; Soriano-Sarabia, N.; Margolis, D.M. Eradicating HIV-1 infection: Seeking to clear a persistent pathogen. Nat. Rev. Microbiol. 2014, 12, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Montagna, C.; Mazzuti, L.; Falasca, F.; Maida, P.; Bucci, M.; D’Ettorre, G.; Mezzaroma, I.; Fantauzzi, A.; Alvaro, N.; Vullo, V.; et al. Trends in drug resistance-associated mutations in a real-life cohort of italian patients infected with HIV-1. J. Glob. Antimicrob. Resist. 2015, 3, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lu, J.; Wang, J.; Yan, H.; Li, J.; Xu, X.; Zhang, Z.; Qiu, T.; Ding, P.; Fu, G.; et al. Prevalence of HIV antiretroviral drug resistance and its impacts on HIV-1 virological failures in Jiangsu, China: A cross-sectional study. Biomed Res. Int. 2016, 2016, 1752437. [Google Scholar] [CrossRef] [PubMed]
- Barral, M.F.; Sousa, A.K.; Santos, A.F.; Abreu, C.M.; Tanuri, A.; Soares, M.A. Identification of novel resistance-related polymorphisms in HIV-1 subtype C RT connection and RNAse H domains from patients under virological failure in brazil. AIDS Res. Hum. Retrovir. 2017, 33, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Little, S.J.; Holte, S.; Routy, J.P.; Daar, E.S.; Markowitz, M.; Collier, A.C.; Koup, R.A.; Mellors, J.W.; Connick, E.; Conway, B.; et al. Antiretroviral-drug resistance among patients recently infected with HIV. N. Engl. J. Med. 2002, 347, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Lucas, G.M. Antiretroviral adherence, drug resistance, viral fitness and HIV disease progression: A tangled web is woven. J. Antimicrob. Chemother. 2005, 55, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Lucas, G.M.; Chaisson, R.E.; Moore, R.D. Highly active antiretroviral therapy in a large urban clinic: Risk factors for virologic failure and adverse drug reactions. Ann. Intern. Med. 1999, 131, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Havlir, D.V.; Bassett, R.; Levitan, D.; Gilbert, P.; Tebas, P.; Collier, A.C.; Hirsch, M.S.; Ignacio, C.; Condra, J.; Gunthard, H.F.; et al. Prevalence and predictive value of intermittent viremia with combination HIV therapy. JAMA 2001, 286, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Havlir, D.V.; Koelsch, K.K.; Strain, M.C.; Margot, N.; Lu, B.; Ignacio, C.C.; Miller, M.D.; Wong, J.K. Predictors of residual viremia in HIV-infected patients successfully treated with efavirenz and lamivudine plus either tenofovir or stavudine. J. Infect. Dis. 2005, 191, 1164–1168. [Google Scholar] [CrossRef] [PubMed]
- Maldarelli, F.; Palmer, S.; King, M.S.; Wiegand, A.; Polis, M.A.; Mican, J.; Kovacs, J.A.; Davey, R.T.; Rock-Kress, D.; Dewar, R.; et al. ART suppresses plasma HIV-1 RNA to a stable set point predicted by pretherapy viremia. PLoS Pathog. 2007, 3, e46. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.; Wiegand, A.P.; Maldarelli, F.; Bazmi, H.; Mican, J.M.; Polis, M.; Dewar, R.L.; Planta, A.; Liu, S.; Metcalf, J.A.; et al. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J. Clin. Microbiol. 2003, 41, 4531–4536. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.; Josefsson, L.; Coffin, J.M. HIV reservoirs and the possibility of a cure for HIV infection. J. Intern. Med. 2011, 270, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.Z.; Wightman, F.; Lewin, S.R. HIV reservoirs and strategies for eradication. Curr. HIV/AIDS Rep. 2012, 9, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Nickle, D.C.; Justement, J.S.; Meyers, J.H.; Roby, G.; Hallahan, C.W.; Kottilil, S.; Moir, S.; Mican, J.M.; Mullins, J.I.; et al. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J. Infect. Dis. 2008, 197, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Cu-Uvin, S.; DeLong, A.K.; Venkatesh, K.K.; Hogan, J.W.; Ingersoll, J.; Kurpewski, J.; De Pasquale, M.P.; D'Aquila, R.; Caliendo, A.M. Genital tract HIV-1 RNA shedding among women with below detectable plasma viral load. AIDS 2010, 24, 2489–2497. [Google Scholar] [CrossRef] [PubMed]
- Thacker, T.C.; Zhou, X.; Estes, J.D.; Jiang, Y.; Keele, B.F.; Elton, T.S.; Burton, G.F. Follicular dendritic cells and human immunodeficiency virus type 1 transcription in CD4+ T cells. J. Virol. 2009, 83, 150–158. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, E.; Ter Heine, R.; van der Veen, F.; Repping, S.; Beijnen, J.H.; Prins, J.M. Penetration of atazanavir in seminal plasma of men infected with human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2007, 51, 335–337. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Gianella, S.; Sinclair, E.; Epling, L.; Li, Q.; Duan, L.; Choi, A.L.; Girling, V.; Ho, T.; Li, P.; et al. Differences in HIV burden and immune activation within the gut of HIV-positive patients receiving suppressive antiretroviral therapy. J. Infect. Dis. 2010, 202, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Shergill, A.K.; McQuaid, K.; Gianella, S.; Lampiris, H.; Hare, C.B.; Pandori, M.; Sinclair, E.; Gunthard, H.F.; Fischer, M.; et al. Effect of raltegravir-containing intensification on HIV burden and T-cell activation in multiple gut sites of HIV-positive adults on suppressive antiretroviral therapy. AIDS 2010, 24, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.C.; Onafuwa-Nuga, A.; McNamara, L.A.; Riddell, J.t.; Bixby, D.; Savona, M.R.; Collins, K.L. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat. Med. 2010, 16, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.C.; McNamara, L.A.; Onafuwa-Nuga, A.; Shackleton, M.; Riddell, J., 4th; Bixby, D.; Savona, M.R.; Morrison, S.J.; Collins, K.L. HIV-1 utilizes the CXCR4 chemokine receptor to infect multipotent hematopoietic stem and progenitor cells. Cell Host Microbe 2011, 9, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.R.; Sedaghat, A.R.; Kieffer, T.; Brennan, T.; Lee, P.K.; Wind-Rotolo, M.; Haggerty, C.M.; Kamireddi, A.R.; Liu, Y.; Lee, J.; et al. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J. Virol. 2006, 80, 6441–6457. [Google Scholar] [CrossRef] [PubMed]
- Keele, B.F.; Tazi, L.; Gartner, S.; Liu, Y.; Burgon, T.B.; Estes, J.D.; Thacker, T.C.; Crandall, K.A.; McArthur, J.C.; Burton, G.F. Characterization of the follicular dendritic cell reservoir of human immunodeficiency virus type 1. J. Virol. 2008, 82, 5548–5561. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Muthui, D.; Holte, S.; Nickle, D.; Feng, F.; Brodie, S.; Hwangbo, Y.; Mullins, J.I.; Corey, L. Evidence for human immunodeficiency virus type 1 replication in vivo in CD14(+) monocytes and its potential role as a source of virus in patients on highly active antiretroviral therapy. J. Virol. 2002, 76, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Alexaki, A.; Liu, Y.; Wigdahl, B. Cellular reservoirs of HIV-1 and their role in viral persistence. Curr. HIV Res. 2008, 6, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Alexaki, A.; Wigdahl, B. HIV-1 infection of bone marrow hematopoietic progenitor cells and their role in trafficking and viral dissemination. PLoS Pathog. 2008, 4, e1000215. [Google Scholar] [CrossRef] [PubMed]
- Coleman, C.M.; Wu, L. HIV interactions with monocytes and dendritic cells: Viral latency and reservoirs. Retrovirology 2009, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Fischer-Smith, T.; Croul, S.; Adeniyi, A.; Rybicka, K.; Morgello, S.; Khalili, K.; Rappaport, J. Macrophage/microglial accumulation and proliferating cell nuclear antigen expression in the central nervous system in human immunodeficiency virus encephalopathy. Am. J. Pathol. 2004, 164, 2089–2099. [Google Scholar] [CrossRef]
- Cosenza, M.A.; Zhao, M.L.; Si, Q.; Lee, S.C. Human brain parenchymal microglia express CD14 and CD45 and are productively infected by HIV-1 in HIV-1 encephalitis. Brain Pathol. 2002, 12, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Collman, R.G. CNS inflammation and macrophage/microglial biology associated with HIV-1 infection. J. Neuroimmune Pharmacol. 2009, 4, 430–447. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.; Nath, A. Where does HIV hide? A focus on the central nervous system. Curr. Opin. HIV AIDS 2013, 8, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Tornatore, C.; Meyers, K.; Atwood, W.; Conant, K.; Major, E. Temporal patterns of human immunodeficiency virus type 1 transcripts in human fetal astrocytes. J. Virol. 1994, 68, 93–102. [Google Scholar] [PubMed]
- Gorry, P.R.; Ong, C.; Thorpe, J.; Bannwarth, S.; Thompson, K.A.; Gatignol, A.; Vesselingh, S.L.; Purcell, D.F. Astrocyte infection by HIV-1: Mechanisms of restricted virus replication, and role in the pathogenesis of HIV-1-associated dementia. Curr. HIV Res. 2003, 1, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Castro, V.; Toborek, M. Infection of human pericytes by HIV-1 disrupts the integrity of the blood-brain barrier. J. Cell. Mol. Med. 2012, 16, 2950–2957. [Google Scholar] [CrossRef] [PubMed]
- Varatharajan, L.; Thomas, S.A. The transport of anti-HIV drugs across blood-CNS interfaces: Summary of current knowledge and recommendations for further research. Antivir. Res. 2009, 82, A99–109. [Google Scholar] [CrossRef] [PubMed]
- Ene, L.; Duiculescu, D.; Ruta, S.M. How much do antiretroviral drugs penetrate into the central nervous system? J. Med. Life 2011, 4, 432–439. [Google Scholar] [PubMed]
- Cribbs, S.K.; Lennox, J.; Caliendo, A.M.; Brown, L.A.; Guidot, D.M. Healthy HIV-1-infected individuals on highly active antiretroviral therapy harbor HIV-1 in their alveolar macrophages. AIDS Res. Hum. Retrovir. 2015, 31, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Zalar, A.; Figueroa, M.I.; Ruibal-Ares, B.; Bare, P.; Cahn, P.; de Bracco, M.M.; Belmonte, L. Macrophage HIV-1 infection in duodenal tissue of patients on long term haart. Antivir. Res. 2010, 87, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.; Mattapallil, J.J. Gastrointestinal tract and the mucosal macrophage reservoir in HIV infection. Clin. Vaccine Immunol. 2014, 21, 1469–1473. [Google Scholar] [CrossRef] [PubMed]
- Gougeon, M.-L. To kill or be killed: How HIV exhausts the immune system. Cell Death Differ. 2005, 12, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; Kashif, K.A. Is HIV infection a TNF receptor signalling-driven disease? Trends Immunol. 2008, 29, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Gartner, S.; Markovits, P.; Markovitz, D.M.; Kaplan, M.H.; Gallo, R.C.; Popovic, M. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science 1986, 233, 215–219. [Google Scholar] [CrossRef] [PubMed]
- McElrath, M.J.; Pruett, J.E.; Cohn, Z.A. Mononuclear phagocytes of blood and bone marrow: Comparative roles as viral reservoirs in human immunodeficiency virus type 1 infections. Proc. Natl. Acad. Sci. USA 1989, 86, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Orenstein, J.M.; Fox, C.; Wahl, S.M. Macrophages as a source of HIV during opportunistic infections. Science 1997, 276, 1857–1861. [Google Scholar] [CrossRef] [PubMed]
- Sharova, N.; Swingler, C.; Sharkey, M.; Stevenson, M. Macrophages archive HIV-1 virions for dissemination in trans. EMBO J. 2005, 24, 2481–2489. [Google Scholar] [CrossRef] [PubMed]
- Verani, A.; Gras, G.; Pancino, G. Macrophages and HIV-1: Dangerous liaisons. Mol. Immunol. 2005, 42, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Perno, C.F.; Svicher, V.; Schols, D.; Pollicita, M.; Balzarini, J.; Stefano, A. Therapeutic strategies towards HIV-1 infection in macrophages. Antivir. Res. 2006, 71, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Blom, J.; Nielsen, C.; Rhodes, J.M. An ultrastructural study of HIV-infected human dendritic cells and monocytes/macrophages. Apmis 1993, 101, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Gendelman, H.E.; Orenstein, J.M.; Martin, M.A.; Ferrua, C.; Mitra, R.; Phipps, T.; Wahl, L.A.; Lane, H.C.; Fauci, A.S.; Burke, D.S.; et al. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J. Exp. Med. 1998, 167, 1428–1441. [Google Scholar] [CrossRef]
- Orenstein, J.M.; Meltzer, M.S.; Phipps, T.; Gendelman, H.E. Cytoplasmic assembly and accumulation of human immunodeficiency virus types 1 and 2 in recombinant human colony-stimulating factor-1-treated human monocytes: An ultrastructural study. J. Virol. 1998, 62, 2578–2586. [Google Scholar]
- Orenstein, J.M.; Jannotta, F. Human immunodeficiency virus and papovavirus infections in acquired immunodeficiency syndrome: An ultrastructural study of three cases. Hum. Pathol. 1988, 19, 350–361. [Google Scholar] [CrossRef]
- Demirov, D.G.; Orenstein, J.M.; Freed, E.O. The late domain of human immunodeficiency virus type 1 p6 promotes virus release in a cell type-dependent manner. J. Virol. 2002, 76, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.G.; Booth, A.; Gould, S.J.; Hildreth, J.E. Evidence that HIV budding in primary macrophages occurs through the exosome release pathway. J. Biol. Chem. 2003, 278, 52347–52354. [Google Scholar] [CrossRef] [PubMed]
- Pelchen-Matthews, A.; Kramer, B.; Marsh, M. Infectious HIV-1 assembles in late endosomes in primary macrophages. J. Cell. Biol. 2003, 162, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Chertova, E.; Chertov, O.; Coren, L.V.; Roser, J.D.; Trubey, C.M.; Bess, J.W., Jr.; Sowder, R.C., II; Barsov, E.; Hood, B.L.; Fisher, R.J.; et al. Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J. Virol. 2006, 80, 9039–9052. [Google Scholar] [CrossRef] [PubMed]
- Jouve, M.; Sol-Foulon, N.; Watson, S.; Schwartz, O.; Benaroch, P. HIV-1 buds and accumulates in “nonacidic” endosomes in macrophages. Cell Host Microbe 2007, 2, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Neil, S.J.D.; Bess, C.; Johnson, M.C.; Virgen, C.A.; Simon, S.M.; Bieniasz, P.D. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006, 4, 2296–2310. [Google Scholar] [CrossRef] [PubMed]
- Deneka, M.; Pelchen-Matthews, A.; Byland, R.; Ruiz-Mateos, E.; Marsh, M. Macrophages, HIV-1 assembles into an intracellular plasma membrane domain containing the tetraspanins CD81, CD9, and CD53. J. Cell Biol. 2007, 177, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Keppler, O.T.; Habermann, A.; Allespach, I.; Krijnse-Locker, J.; Usslich, H.-G.K. HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog. 2007, 3, 0001–0011. [Google Scholar] [CrossRef] [PubMed]
- Nkwe, D.O.; Pelchen-Matthews, A.; Burden, J.J.; Collinson, L.M.; Marsh, M. The intracellular plasma membrane-connected compartment in the assembly of HIV-1 in human macrophages. BMC Biol. 2016, 14, 50. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Sattentau, Q.J. The HIV-1-containing macrophage compartment: A perfect cellular niche? Trends Microbiol. 2013, 21, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Salahuddin, S.Z.; Rose, R.M.; Groopman, J.E.; Markham, P.D.; Gallo, R.C. Human T lymphotropic virus typeIII infection of human alveolar macrophages. Blood 1986, 68, 281–284. [Google Scholar] [PubMed]
- Guillemard, E.; Jacquemot, C.; Aillet, F.; Schmitt, N.; Barre-Sinoussi, F.; Israel, N. Human immunodeficiency virus 1 favors the persistence of infection by activating macrophages through TNF. Virology 2004, 329, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Giri, M.S.; Nebozyhn, M.; Raymond, A.; Gekonge, B.; Hancock, A.; Creer, S.; Nicols, C.; Yousef, M.; Foulkes, A.S.; Mounzer, K.; et al. Circulating monocytes in HIV-1-infected viremic subjects exhibit an antiapoptosis gene signature and virus- and host-mediated apoptosis resistance1. J. Immunol. 2009, 182, 4459–4470. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O. HIV-1 and the host cell: An intimate association. Trends Microbiol. 2004, 12, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Emerman, M. HIV-1 accessory proteins. Ensuring viral survival in a hostile environment. Cell Host Microbe 2008, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Goff, S.P. Host restriction factors blocking retroviral replication. Annu. Rev. Genet. 2008, 42, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.; Bieniasz, P. Human immunodeficiency virus, restriction factors, and interferon. J. Interferon Cytokine Res. 2009, 29, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Kluge, S.F.; Sauter, D.; Kirchhoff, F. Snapshot: Antiviral restriction factors. Cell 2015, 163, 774–774.e1. [Google Scholar] [CrossRef] [PubMed]
- Kirchhoff, F. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 2010, 8, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Bieniasz, P.D. HIV restriction factors and mechanisms of evasion. Cold Spring Harb. Perspect. Med. 2012, 2, a006940. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, A.; Pancino, G. Host hindrance to HIV-1 replication in monocytes and macrophages. Retrovirology 2010, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Wissing, S.; Galloway, N.L.; Greene, W.C. HIV-1 Vif versus the APOBEC3 cytidine deaminases: An intracellular duel between pathogen and host restriction factors. Mol. Asp. Med. 2010, 31, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in old world monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, N.; Greenwell-Wild, T.; Marinos, N.J.; Swaim, W.D.; Nares, S.; Ott, D.E.; Schubert, U.; Henklein, P.; Orenstein, J.M.; Sporn, M.B.; et al. Human Immunodeficiency Virus Type 1-induced macrophage gene expression includes p21, a target for viral regulation. J. Virol. 2005, 79, 4479–4491. [Google Scholar] [CrossRef] [PubMed]
- Giri, M.S.; Nebozhyn, M.; Showe, L.; Montaner, L.J. Microarray data on gene modulation by HIV-1 in immune cells: 2000–2006. J. Leukoc. Biol. 2006, 80, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, K.G.; Heuser, J.E.; Donzell, W.C.; Ying, Y.S.; Glenney, J.R.; Anderson, R.G. Caveolin, a protein component of caveolae membrane coats. Cell 1992, 68, 673–682. [Google Scholar] [CrossRef]
- Palade, G.E. Fine structure of blood capillaries. J. Appl. Phys. 1953, 24, 1424. [Google Scholar]
- Yamada, E. The fine structures of the gall bladder epithelium of the mouse. J. Biophys. Biochem. Cytol. 1955, 1, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Stan, R.V. Structure of caveolae. Biochim. Biophys. 2005, 1746, 334–348. [Google Scholar] [CrossRef] [PubMed]
- Stan, R.V.; Tkachenko, E.; Niesman, I.R. PV1 is a key structural component for the formation of the stomatal and fenestral diaphragms. Mol. Biol. Cell 2004, 15, 3615–3630. [Google Scholar] [CrossRef] [PubMed]
- Parton, R.G.; Simons, K. The multiple faces of caveolae. Nat. Rev. 2007, 8, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Gargalovic, P.; Dory, L. Caveolins and macrophage lipid metabolism. J. Lipid Res. 2003, 44, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Werling, D.; Koss, M.; Monaghan, P.; Taylor, G.; Howard, C.J. Expression of caveolin by bovine lymphocytes and antigen-presenting cells. Immunology 2002, 105, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Quest, A.F.G.; Leyton, L.; Párraga, M. Caveolins, caveolae, and lipid rafts in cellular transport, signaling, and disease. Biochem. Cell. Biol. 2004, 82, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Mercier, I.; Jasmin, J.-F.; Pavlides, S.; Minetti, C.; Flomenberg, N.; Pestell, R.G.; Frank, P.G.; Sotgia, F.; Lisanti1, M.P. Clinical and translational implications of the caveolin gene family: Lessons from mouse models and human genetic disorders. Lab. Investig. 2009, 89, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Fra, A.M.; Williamson, E.; Simons, K.; Parton, R.G. De novo formation of caveolae in lymphocytes by expression of VIP21-caveolin. Proc. Natl. Acad. Sci. USA 1995, 92, 8655–8659. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, M.; Maeda, T.; Ikemoto, T.; Mori, H.; Seya, T.; Shimizu, A. Expression of caveolin-1 in human T cell leukemia cell lines. Biochem. Biophys. Res. Commun. 1998, 253, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, J.; Hardin, C.D. Expression of caveolin-1 in lymphocytes induces caveolae formation and recruitment of phosphofructokinase to the plasma membrane. FASEB J. 2005, 16, 586–587. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.M.; Bastiani, M.; Luetterforst, R.; Kirkham, M.; Kirkham, A.; Nixon, S.J.; Walser, P.; Abankwa, D.; Oorschot, V.M.; Martin, S.; et al. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 2008, 132, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Ariotti, N.; Parton, R.G. Snapshot: Caveolae, caveolins, and cavins. Cell 2013, 154, 704–704.e1. [Google Scholar] [CrossRef] [PubMed]
- Chidlow, J.H., Jr.; Sessa, W.C. Caveolae, caveolins, and cavins: Complex control of cellular signalling and inflammation. Cardiovasc. Res. 2010, 86, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Way, M.; Parton, R.G. M-caveolin, a muscle-specific caveolin-related protein. FEBS Lett. 1995, 376, 108–112. [Google Scholar] [CrossRef]
- Tang, Z.; Scherer, P.E.; Okamoto, T.; Song, K.; Chu, C.; Kohtz, D.S.; Nishimoto, I.; Lodish, H.F.; Lisanti, M.P. Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J. Biol. Chem. 1996, 271, 2255–2261. [Google Scholar] [PubMed]
- Sowa, G.; Pypaert, M.; Fulton, D.; Sessa, W.C. The phosphorylation of caveolin-2 on serines 23 and 36 modulates caveolin-1-dependent caveolae formation. Proc. Natl. Acad. Sci. USA 2003, 100, 6511–6516. [Google Scholar] [CrossRef] [PubMed]
- Lahtinen, U.; Honsho, M.; Parton, R.G.; Simons, K.; Verkade, P. Involvement of caveolin-2 in caveolar biogenesis in MDCK cells. FEBS Lett. 2003, 538, 85–88. [Google Scholar] [CrossRef]
- Drab, M.; Verkade, P.; Elger, M.; Kasper, M.; Lohn, M.; Lauterbach, B.; Menne, J.; Lindschau, C.; Mende, F.; Luft, F.C.; et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 2001, 293, 2449–2452. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, F.; Engelman, J.A.; Volonte, D.; Zhang, X.L.; Minetti, C.; Li, M.; Hou, H., Jr.; Kneitz, B.; Edelmann, W.; Lisanti, M.P. Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin–glycoprotein complex, and T-tubule abnormalities. J. Biol. Chem. 2001, 276, 21425–21433. [Google Scholar] [CrossRef] [PubMed]
- Stoeber, M.; Schellenberger, P.; Siebert, C.A.; Leyrat, C.; Helenius, A.; Grunewald, K. Model for the architecture of caveolae based on a flexible, net-like assembly of cavin1 and caveolin discs. Proc. Natl. Acad. Sci. USA 2016, 113, E8069–E8078. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.P.; Nichols, B.J. Caveolae: One function or many? Trends Cell Biol. 2016, 26, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Lisanti, M.P.; Tang, Z.; Scherer, P.E.; Kübler, E.; Koleske, A.J.; Sargiacomo, M. Caveolae, transmembrane signalling and cellular transformation. Mol. Membr. Biol. 1995, 12, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Schlegel, A.; Scherer, P.E.; Lisanti, M.P. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J. Biol. Chem. 1998, 273, 5419–5422. [Google Scholar] [CrossRef] [PubMed]
- Kurzchalia, T.V.; Parton, R.G. Membrane microdomains and caveolae. Curr. Opin. Cell Biol. 1999, 11, 424–431. [Google Scholar] [CrossRef]
- Liu, P.; Rudick, M.; Anderson, R.G. Multiple functions of caveolin-1. J. Biol. Chem. 2002, 277, 41295–41298. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.S.; Gao, Z.; Abraham, S.N. Involvement of cellular caveolae in bacterial entry into mast cells. Science 2000, 289, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Pelkmans, L.; Kartenbeck, J.; Helenius, A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 2001, 3, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.M.; Lisanti, M.P. Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am. J. Physiol. Cell Physiol. 2005, 288, C494–C506. [Google Scholar] [CrossRef] [PubMed]
- Frank, P.G.; Pavlides, S.; Cheung, M.W.; Daumer, K.; Lisanti, M.P. Role of caveolin-1 in the regulation of lipoprotein metabolism. Am. J. Physiol Cell Physiol. 2008, 295, C242–C248. [Google Scholar] [CrossRef] [PubMed]
- Frank, P.G.; Pavlides, S.; Lisanti, M.P. Caveolae and transcytosis in endothelial cells: Role in atherosclerosis. Cell Tissue Res. 2009, 335, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Spisni, E.; Bianco, M.C.; Griffoni, C.; Toni, M.; D'Angelo, R.; Santi, S.; Riccio, M.; Tomasi, V. Mechanosensing role of caveolae and caveolar constituents in human endothelial cells. J. Cell. Physiol. 2003, 197, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Spisni, E.; Toni, M.; Strillacci, A.; Galleri, G.; Santi, S.; Griffoni, C.; Tomasi, V. Caveolae and caveolae constituents in mechanosensing: Effect of modeled microgravity on cultured human endothelial cells. Cell Biochem. Biophys. 2006, 46, 155–164. [Google Scholar] [CrossRef]
- Parton, R.G.; del Pozo, M.A. Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell Biol. 2013, 14, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Echarri, A.; Del Pozo, M.A. Caveolae - mechanosensitive membrane invaginations linked to actin filaments. J. Cell Sci 2015, 128, 2747–2758. [Google Scholar] [CrossRef] [PubMed]
- Nassoy, P.; Lamaze, C. Stressing caveolae new role in cell mechanics. Trends Cell Biol. 2012, 22, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, F.; Volonte, D.; Liu, J.; Capozza, F.; Frank, P.G.; Zhu, L.; Pestell, R.G.; Lisanti, M.P. Caveolin-1 expression negatively regulates cell cycle progression by inducing G0/G1 arrest via a p53/p21WAF1/Cip1-dependent mechanism. Mol. Biol. Cell 2001, 12, 2229–2244. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Werling, D.; Hope, J.C.; Taylor, G.; Howard, C.J. Caveolae and caveolin in immune cells: Distribution and functions. Trends Immunol. 2002, 23, 158–164. [Google Scholar] [CrossRef]
- Lee, S.W.; Reimer, C.L.; Oh, P.; Campbell, D.B.; Schnitzer, J.E. Tumor cell growth inhibition by caveolin re-expression in human breast cancer cells. Oncogene 1998, 16, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Uittenbogaard, A.; Everson, W.V.; Matveev, S.V.; Smart, E.J. Cholesteryl ester is transported from caveolae to internal membranes as part of a caveolin-annexin II lipid-protein complex. J. Biol. Chem. 2002, 277, 4925–4931. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Stoppani, E.; Volonte, D.; Galbiati, F. Caveolin-1, cellular senescence and age-related diseases. Mech. Ageing Dev. 2011, 132, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Lamaze, C.; Torrino, S. Caveolae and cancer: A new mechanical perspective. Biomed. J. 2015, 38, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.H.; Murray, F.; Insel, P.A. Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 359–391. [Google Scholar] [CrossRef] [PubMed]
- Fridolfsson, H.N.; Roth, D.M.; Insel, P.A.; Patel, H.H. Regulation of intracellular signaling and function by caveolin. FASEB J. 2014, 28, 3823–3831. [Google Scholar] [CrossRef] [PubMed]
- Llano, M.; Kelly, T.; Vanegas, M.; Peretz, M.; Peterson, T.E.; Simari, R.D.; Poeschla, E.M. Blockade of human immunodeficiency virus type 1 expression by caveolin-1. J. Virol. 2002, 76, 9152–9164. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-M.; Nadeau, P.E.; Lin, S.; Abbott, J.E.; Mergia, A. Caveolin 1 inhibits HIV replication by transcriptional repression mediated through NF-κB. J. Virol. 2011, 85, 5483–5493. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.E., Jr.; Taylor, H.E.; Hildreth, J.E. Caveolin-1 suppresses human immunodeficiency virus-1 replication by inhibiting acetylation of NF-κB. Virology 2012, 432, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.A.; Lempicki, R.A.; Sidorov, I.A.; Adelsberger, J.W.; Herpin, B.; Metcalf, J.A.; Sereti, I.; Polis, M.A.; Davey, R.T.; Tavel, J.; et al. Identification of dynamically distinct subpopulations of T lymphocytes that are differentially affected by HIV. J. Exp. Med. 2001, 194, 1731–1741. [Google Scholar] [CrossRef] [PubMed]
- McCune, J.M.; Hanley, M.B.; Cesar, D.; Halvorsen, R.; Hoh, R.; Schmidt, D.; Wieder, E.; Deeks, S.; Siler, S.; Neese, R.; et al. Factors influencing T-cell turnover in HIV-1-seropositive patients. J. Clin. Investig. 2000, 105, R1–R8. [Google Scholar] [CrossRef] [PubMed]
- Hellerstein, M.; McCune, J. T cell turnover in HIV-1 disease. Immunity 1997, 7, 583–589. [Google Scholar] [CrossRef]
- Hellerstein, M.; Hanley, M.B.; Cesar, D.; Siler, S.; Papageorgopoulos, C.; Wieder, E.; Schmidt, D.; Hoh, R.; Neese, R.; Macallan, D.; et al. Directly measured kinetics of circulating T lymphocytes in normal and HIV-1-infected humans. Nat. Med. 1999, 5, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Valdez, H.; Lederman, M.M. Cytokines and cytokine therapies in HIV infection. AIDS Clin. Rev. 1998, 1997–1998, 187–228. [Google Scholar]
- Epple, H.J.; Schneider, T.; Troeger, H.; Kunkel, D.; Allers, K.; Moos, V.; Amasheh, M.; Loddenkemper, C.; Fromm, M.; Zeitz, M.; et al. Impairment of the intestinal barrier is evident in untreated but absent in suppressively treated HIV-infected patients. Gut 2009, 58, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Nazli, A.; Chan, O.; Dobson-Belaire, W.N.; Ouellet, M.; Tremblay, M.J.; Gray-Owen, S.D.; Arsenault, A.L.; Kaushic, C. Exposure to HIV-1 directly impairs mucosal epithelial barrier integrity allowing microbial translocation. PLoS Pathog. 2010, 6, e1000852. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Ancuta, P.; Kamat, A.; Kunstman, K.J.; Kim, E.Y.; Autissier, P.; Wurcel, A.; Zaman, T.; Stone, D.; Mefford, M.; Morgello, S.; et al. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS ONE 2008, 3, e2516. [Google Scholar] [CrossRef] [PubMed]
- Cassol, E.; Malfeld, S.; Mahasha, P.; van der Merwe, S.; Cassol, S.; Seebregts, C.; Alfano, M.; Poli, G.; Rossouw, T. Persistent microbial translocation and immune activation in HIV-1-infected south africans receiving combination antiretroviral therapy. J. Infect. Dis. 2010, 202, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.G.; Morrison, D.C. Differential expression of caveolin-1 in lipopolysaccharide-activated murine macrophages. Infect. Immun. 2000, 68, 5084–5089. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.G.; Tan, X.; Qureshi, N.; Morrison, D.C. Regulation of cellular caveolin-1 protein expression in murine macrophages by microbial products. Infect. Immun. 2005, 73, 8136–8143. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Kim, H.P.; Nakahira, K.; Ryter, S.W.; Choi, A.M. The heme oxygenase-1/carbon monoxide pathway suppresses tlr4 signaling by regulating the interaction of TLR4 with caveolin-1. J. Immunol. 2009, 182, 3809–3818. [Google Scholar] [CrossRef] [PubMed]
- Lang, T.; Mansell, A. The negative regulation of Toll-like receptor and associated pathways. Immunol. Cell Biol. 2007, 85, 425–434. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A. Tampering with Toll-like receptor signaling. Cell 2007, 131, 1039–1041. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A. ‘Fine tuning’ TLR signaling. Nat. Immunol. 2008, 9, 459–461. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G.; Sher, A. Cooperation of Toll-like receptor signals in innate immune defence. Nat. Rev. Immunol. 2007, 7, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Effros, R.B.; Allsopp, R.; Chiu, C.P.; Hausner, M.A.; Hirji, K.; Wang, L.; Harley, C.B.; Villeponteau, B.; West, M.D.; Giorgi, J.V. Shortened telomeres in the expanded CD28-CD8+ cell subset in HIV disease implicate replicative senescence in HIV pathogenesis. AIDS 1996, 10, F17–22. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Karandikar, N.J.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Crotty, L.E.; Casazza, J.P.; Kuruppu, J.; Migueles, S.A.; Connors, M.; et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood 2003, 101, 2711–2720. [Google Scholar] [CrossRef] [PubMed]
- Grossman, Z.; Meier-Schellersheim, M.; Sousa, A.E.; Victorino, R.M.; Paul, W.E. CD4+ T-cell depletion in HIV infection: Are we closer to understanding the cause? Nat. Med. 2002, 8, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Dyck, L.; Mills, K.H.G. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur. J. Immunol. 2017, 47, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, L.; Janbazian, L.; Chomont, N.; Said, E.A.; Gimmig, S.; Bessette, B.; Boulassel, M.R.; Delwart, E.; Sepulveda, H.; Balderas, R.S.; et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006, 12, 1198–1202. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, D.E.; Walker, B.D. Programmed death-1 as a factor in immune exhaustion and activation in HIV infection. Curr. Opin. HIV AIDS 2008, 3, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, M.; Porichis, F.; de Jong, O.G.; Levi, K.; Diefenbach, T.J.; Lifson, J.D.; Freeman, G.J.; Walker, B.D.; Kaufmann, D.E.; Kavanagh, D.G. Expression of PD-l1 and PD-l2 on human macrophages is up-regulated by HIV-1 and differentially modulated by IL-10. J. Leukoc. Biol. 2011, 89, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Petrovas, C.; Casazza, J.P.; Brenchley, J.M.; Price, D.A.; Gostick, E.; Adams, W.C.; Precopio, M.L.; Schacker, T.; Roederer, M.; Douek, D.C.; et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J. Exp. Med. 2006, 203, 2281–2292. [Google Scholar] [CrossRef] [PubMed]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Wen, Q.; Zhao, Y.; Gao, Q.; Bai, Y. NF-κB plays a key role in inducing CD274 expression in human monocytes after lipopolysaccharide treatment. PLoS ONE 2013, 8, e61602. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Akiba, H.; Iwai, H.; Matsuda, H.; Aoki, M.; Tanno, Y.; Shin, T.; Tsuchiya, H.; Pardoll, D.M.; Okumura, K.; et al. Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol. 2002, 169, 5538–5545. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; X-M, W.; Nadeau, P.E.; Mergia, A. HIV infection upregulates caveolin 1 (Cav-1) expression to restrict virus production. J. Virol. 2010, 84, 9487–9496. [Google Scholar] [CrossRef] [PubMed]
- Ito, J.; Nagayasu, Y.; Miura, Y.; Yokoyama, S.; Michikawa, M. Astrocytes endogenous apoE generates HDL-like lipoproteins using previously synthesized cholesterol through interaction with ABCA1. Brain Res. 2014, 1570, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; He, X.; Zhang, Q.; Zhang, N.; Li, L.; Hui, Y.; Li, W.; Wang, D.; Wu, Z. Alpha-synuclein induces microglial cell migration through stimulating HIF-1α accumulation. J. Neurosci. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tiruppathi, C.; Shimizu, J.; Miyawaki-Shimizu, K.; Vogel, S.M.; Bair, A.M.; Minshall, R.D.; Predescu, D.; Malik, A.B. Role of NF-κB-dependent caveolin-1 expression in the mechanism of increased endothelial permeability induced by lipopolysaccharide. J. Biol. Chem. 2008, 283, 4210–4218. [Google Scholar] [CrossRef] [PubMed]
- Gargalovic, P.; Dory, L. Cellular apoptosis is associated with increased caveolin-1 expression in macrophages. J. Lipid Res. 2003, 44, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Wang, S.H.; Kuan, I.I.; Tseng, W.K.; Chen, M.F.; Wu, J.C.; Chen, Y.L. OXLDL upregulates caveolin-1 expression in macrophages: Role for caveolin-1 in the adhesion of OXLDL-treated macrophages to endothelium. J. Cell. Biochem. 2009, 107, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Lee, R.J.; Karnezis, A.; Bearss, D.J.; Webster, M.; Siegel, P.; Muller, W.J.; Windle, J.J.; Pestell, R.G.; Lisanti, M.P. Reciprocal regulation of Neu tyrosine kinase activity and caveolin-1 protein expression in vitro and in vivo. Implications for human breast cancer. J. Biol. Chem. 1998, 273, 20448–20455. [Google Scholar] [CrossRef] [PubMed]
- Koleske, A.J.; Baltimore, D.; Lisanti, M.P. Reduction of caveolin and caveolae in oncogenically transformed cells. Proc. Natl. Acad. Sci. USA 1995, 92, 1381–1385. [Google Scholar] [CrossRef] [PubMed]
- Park, D.S.; Razani, B.; Lasorella, A.; Schreiber-Agus, N.; Pestell, R.G.; Iavarone, A.; Lisanti, M.P. Evidence that Myc isoforms transcriptionally repress caveolin-1 gene expression via an INR-dependent mechanism. Biochemistry 2001, 40, 3354–3362. [Google Scholar] [CrossRef] [PubMed]
- Timme, T.L.; Goltsov, A.; Tahir, S.; Li, L.; Wang, J.; Ren, C.; Johnston, R.N.; Thompson, T.C. Caveolin-1 is regulated by c-myc and suppresses c-myc-induced apoptosis. Oncogene 2000, 19, 3256–3265. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.T.; Nadeau, P.E.; Lin, S.; Mergia, A. Establishing restricted expression of caveolin-1 in HIV infected cells and inhibition of virus replication. Open Microbiol. J. 2014, 8, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Kim, H.P.; Song, R.; Choi, A.M. Caveolin-1 confers antiinflammatory effects in murine macrophages via the MKK3/p38 MAPK pathway. Am. J. Respir. Cell Mol. Biol. 2006, 34, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Bellyei, S.; Schally, A.V.; Zarandi, M.; Varga, J.L.; Vidaurre, I.; Pozsgai, E. GHRH antagonists reduce the invasive and metastatic potential of human cancer cell lines in vitro. Cancer Lett. 2010, 293, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Bist, A.; Fielding, C.J.; Fielding, P.E. P53 regulates caveolin gene transcription, cell cholesterol, and growth by a novel mechanism. Biochemistry 2000, 39, 1966–1972. [Google Scholar] [CrossRef] [PubMed]
- Bist, A.; Fielding, P.E.; Fielding, C.J. Two sterol regulatory element-like sequences mediate up-regulation of caveolin gene transcription in response to low density lipoprotein free cholesterol. Proc. Natl. Acad. Sci. USA 1997, 94, 10693–10698. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Fernandez-Zapico, M.E.; Jin, D.; Puri, V.; Cook, T.A.; Lerman, L.O.; Zhu, X.Y.; Urrutia, R.; Shah, V. KLF11-mediated repression antagonizes Sp1/sterol-responsive element-binding protein-induced transcriptional activation of caveolin-1 in response to cholesterol signaling. J. Biol. Chem. 2005, 280, 1901–1910. [Google Scholar] [CrossRef] [PubMed]
- Fielding, C.J.; Bist, A.; Fielding, P.E. Intracellular cholesterol transport in synchronized human skin fibroblasts. Biochemistry 1999, 38, 2506–2513. [Google Scholar] [CrossRef] [PubMed]
- Costales, P.; Castellano, J.; Revuelta-Lopez, E.; Cal, R.; Aledo, R.; Llampayas, O.; Nasarre, L.; Juarez, C.; Badimon, L.; Llorente-Cortes, V. Lipopolysaccharide downregulates CD91/low-density lipoprotein receptor-related protein 1 expression through SREBP-1 overexpression in human macrophages. Atherosclerosis 2013, 227, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Feig, J.E.; Shang, Y.; Rotllan, N.; Vengrenyuk, Y.; Wu, C.; Shamir, R.; Torra, I.P.; Fernandez-Hernando, C.; Fisher, E.A.; Garabedian, M.J. Statins promote the regression of atherosclerosis via activation of the CCR7-dependent emigration pathway in macrophages. PLoS ONE 2011, 6, e28534. [Google Scholar] [CrossRef] [PubMed]
- Dasari, A.; Bartholomew, J.N.; Volonte, D.; Galbiati, F. Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements. Cancer Res. 2006, 66, 10805–10814. [Google Scholar] [CrossRef] [PubMed]
- Chretien, A.; Piront, N.; Delaive, E.; Demazy, C.; Ninane, N.; Toussaint, O. Increased abundance of cytoplasmic and nuclear caveolin 1 in human diploid fibroblasts in H2O2-induced premature senescence and interplay with p38αMAPK. FEBS Lett. 2008, 582, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Lin, J.C.; Yang, S.C.; Lin, C.W.; Chen, J.J.; Shih, J.Y.; Hong, T.M.; Yang, P.C. Modulation of the expression of the invasion-suppressor CRMP-1 by cyclooxygenase-2 inhibition via reciprocal regulation of Sp1 and C/EBPα. Mol. Cancer Ther. 2008, 7, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, N.; Huang, J.; Xin, J.; Peng, F.; Ren, Y.; Zhang, S.; Miao, J. Inhibition of phosphatidylcholine-specific phospholipase C prevents bone marrow stromal cell senescence in vitro. J. Cell. Biochem. 2009, 108, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.M.; Sutliff, R.L. HIV-1, reactive oxygen species, and vascular complications. Free Radic. Biol. Med. 2012, 53, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Smart, E.J.; Weksler, B.; Couraud, P.O.; Hennig, B.; Toborek, M. Caveolin-1 regulates human immunodeficiency virus-1 TAT-induced alterations of tight junction protein expression via modulation of the Ras signaling. J. Neurosci. 2008, 28, 7788–7796. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Andras, I.E.; Rha, G.B.; Hennig, B.; Toborek, M. PPARα and PPARγ protect against HIV-1-induced MMP-9 overexpression via caveolae-associated ERK and Akt signaling. FASEB J. 2011, 25, 3979–3988. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Feng, X.; Xie, X.; Zhang, J.; Wu, D.; Xu, L. HIV-1 TAT protein increases the permeability of brain endothelial cells by both inhibiting occludin expression and cleaving occludin via matrix metalloproteinase-9. Brain Res. 2012, 1436, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.L.; Song, J.N.; Zhang, M. Role of caveolin-1 in the biology of the blood-brain barrier. Rev. Neurosci. 2014, 25, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Klucken, J.; Buchler, C.; Orso, E.; Kaminski, W.E.; Porsch-Ozcurumez, M.; Liebisch, G.; Kapinsky, M.; Diederich, W.; Drobnik, W.; Dean, M.; et al. ABCG1 (ABC8), the human homolog of the Drosophila white gene, is a regulator of macrophage cholesterol and phospholipid transport. Proc. Natl. Acad. Sci. USA 2000, 97, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Bodzioch, M.; Orso, E.; Klucken, J.; Langmann, T.; Bottcher, A.; Diederich, W.; Drobnik, W.; Barlage, S.; Buchler, C.; Porsch-Ozcurumez, M.; et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat. Genet. 1999, 22, 347–351. [Google Scholar] [PubMed]
- De La Llera-Moya, M.; Connelly, M.A.; Drazul, D.; Klein, S.M.; Favari, E.; Yancey, P.G.; Williams, D.L.; Rothblat, G.H. Scavenger receptor class B type I affects cholesterol homeostasis by magnifying cholesterol flux between cells and HDL. J. Lipid Res. 2001, 42, 1969–1978. [Google Scholar] [PubMed]
- Arakawa, R.; Abe-Dohmae, S.; Asai, M.; Ito, J.I.; Yokoyama, S. Involvement of caveolin-1 in cholesterol enrichment of high density lipoprotein during its assembly by apolipoprotein and THP-1 cells. J. Lipid Res. 2000, 41, 1952–1962. [Google Scholar] [PubMed]
- Escher, G.; Krozowski, Z.; Croft, K.D.; Sviridov, D. Expression of sterol 27-hydroxylase (CYP27A1) enhances cholesterol efflux. J. Biol. Chem. 2003, 278, 11015–11019. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.X.; Cao, D.L.; Xiong, Y.; Peng, X.H.; Liao, D.F. A novel model of cholesterol efflux from lipid-loaded cells. Acta Pharmacol. Sin. 2010, 31, 1243–1257. [Google Scholar] [CrossRef] [PubMed]
- Frank, P.G.; Cheung, M.W.; Pavlides, S.; Llaverias, G.; Park, D.S.; Lisanti, M.P. Caveolin-1 and the regulation of cellular cholesterol homeostasis. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H677–H686. [Google Scholar] [CrossRef] [PubMed]
- Fielding, C.J.; Fielding, P.E. Caveolae and intracellular trafficking of cholesterol. Adv. Drug Deliv. Rev. 2001, 49, 251–264. [Google Scholar] [CrossRef]
- Razani, B.; Woodman, S.E.; Lisanti, M.P. Caveolae: From cell biology to animal physiology. Pharmacol. Rev. 2002, 54, 431–467. [Google Scholar] [CrossRef] [PubMed]
- Kabouridis, P.S.; Janzen, J.; Magee, A.L.; Ley, S.C. Cholesterol depletion disrupts lipid rafts and modulates the activity of multiple signaling pathways in T lymphocytes. Eur. J. Immunol. 2000, 30, 954–963. [Google Scholar] [CrossRef]
- Parpal, S.; Karlsson, M.; Thorn, H.; Stralfors, P. Cholesterol depletion disrupts caveolae and insulin receptor signaling for metabolic control via insulin receptor substrate-1, but not for mitogen-activated protein kinase control. J. Biol. Chem. 2001, 276, 9670–9678. [Google Scholar] [CrossRef] [PubMed]
- Mahammad, S.; Dinic, J.; Adler, J.; Parmryd, I. Limited cholesterol depletion causes aggregation of plasma membrane lipid rafts inducing T cell activation. Biochim. Biophys. Acta 2010, 1801, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.Z.; McIntosh, V.J.; Abou Samra, A.B.; Mohammad, R.M.; Lasley, R.D. Cholesterol depletion alters cardiomyocyte subcellular signaling and increases contractility. PLoS ONE 2016, 11, e0154151. [Google Scholar] [CrossRef] [PubMed]
- Le Lay, S.; Hajduch, E.; Lindsay, M.R.; Le Liepvre, X.; Thiele, C.; Ferre, P.; Parton, R.G.; Kurzchalia, T.; Simons, K.; Dugail, I. Cholesterol-induced caveolin targeting to lipid droplets in adipocytes: A role for caveolar endocytosis. Traffic 2006, 7, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G. Caveolin, cholesterol, and lipid droplets? J. Cell Biol. 2001, 152, F29–34. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Signore, S.C.; Zerr, I.; Althaus, H.H. Oligodendroglial process formation is differentially affected by modulating the intra- and extracellular cholesterol content. J. Mol. Neurosci. 2013, 49, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Zhu, N.; Ao, B.X.; Liu, C.; Shi, Y.N.; Du, K.; Chen, J.X.; Zheng, X.L.; Liao, D.F. Caveolae and caveolin-1 integrate reverse cholesterol transport and inflammation in atherosclerosis. Int. J. Mol. Sci. 2016, 17, 429. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Peranen, J.; Schreiner, R.; Wieland, F.; Kurzchalia, T.V.; Simons, K. VIP21/caveolin is a cholesterol-binding protein. Proc. Natl. Acad. Sci. USA 1995, 92, 10339–10343. [Google Scholar] [CrossRef] [PubMed]
- Smart, E.J.; Ying, Y.; Donzell, W.C.; Anderson, R.G.W. A role for caveolin in transport of cholesterol from endoplasmic reticulum to plasma membrane. J. Biol. Chem. 1996, 271, 29427–29435. [Google Scholar] [PubMed]
- Uittenbogaard, A.; Ying, Y.; Smart, E.J. Characterization of a cytosolic heat-shock protein-caveolin chaperone complex. Involvement in cholesterol trafficking. J. Biol. Chem. 1998, 273, 6525–6532. [Google Scholar] [CrossRef] [PubMed]
- Pol, A.; Luetterforst, R.; Lindsay, S.; Heino, S.; Ikonen, E.; Parton, R.G. A caveolin dominant negative mutant associates with lipid bodies and induces intracellular cholesterol imbalance. J. Cell Biol. 2001, 152, 1057–1070. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Hoang, A.; Escher, G.; Parton, R.G.; Krozowski, Z.; Sviridov, D. Expression of caveolin-1 enhances cholesterol efflux in hepatic cells. J. Biol. Chem. 2004, 279, 14140–14146. [Google Scholar] [CrossRef] [PubMed]
- Pol, A.; Martin, S.; Fernandez, M.A.; Ferguson, C.; Carozzi, A.; Luetterforst, R.; Enrich, C.; Parton, R.G. Dynamic and regulated association of caveolin with lipid bodies: Modulation of lipid body motility and function by a dominant negative mutant. Mol. Biol. Cell 2004, 15, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Meshulam, T.; Simard, J.R.; Wharton, J.; Hamilton, J.A.; Pilch, P.F. Role of caveolin-1 and cholesterol in transmembrane fatty acid movement. Biochemistry 2006, 45, 2882–2893. [Google Scholar] [CrossRef] [PubMed]
- Puglielli, L.; Rigotti, A.; Greco, A.V.; Santos, M.J.; Nervi, F. Sterol carrier protein-2 is involved in cholesterol transfer from the endoplasmic reticulum to the plasma membrane in human fibroblasts. J. Biol. Chem. 1995, 270, 18723–18726. [Google Scholar] [PubMed]
- Zhou, M.; Parr, R.D.; Petrescu, A.D.; Payne, H.R.; Atshaves, B.P.; Kier, A.B.; Ball, J.M.; Schroeder, F. Sterol carrier protein-2 directly interacts with caveolin-1 in vitro and in vivo. Biochemistry 2004, 43, 7288–7306. [Google Scholar] [CrossRef] [PubMed]
- Jessup, W.; Gelissen, I.C.; Gaus, K.; Kritharides, L. Roles of ATP binding cassette transporters A1 and B1, scavenger receptor BI and membrane lipid domains in cholesterol export from macrophages. Curr. Opin. Lipidol. 2006, 17, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Oram, J.F.; Lawn, R.M. ABCA1. The gatekeeper for eliminating excess tissue cholesterol. J. Lipid Res. 2001, 42, 1173–1179. [Google Scholar] [PubMed]
- Argmann, C.A.; Edwards, J.Y.; Sawyez, C.G.; O’Neil, C.H.; Hegele, R.A.; Pickering, J.G.; Huff, M.W. Regulation of macrophage cholesterol efflux through hydroxymethylglutaryl-CoA reductase inhibition: A role for RhoA in ABCA1-mediated cholesterol efflux. J. Biol. Chem. 2005, 280, 22212–22221. [Google Scholar] [CrossRef] [PubMed]
- Landry, Y.D.; Denis, M.; Nandi, S.; Bell, S.; Vaughan, A.M.; Zha, X. ATP-binding cassette transporter A1 expression disrupts raft membrane microdomains through its ATPase-related functions. J. Biol. Chem. 2006, 281, 36091–36101. [Google Scholar] [CrossRef] [PubMed]
- Aloia, R.C.; Tian, H.; Jensen, F.C. Lipid composition and fluidity of the human immunodeficiency virus envelope and host cell plasma membranes. Proc. Natl. Acad. Sci. USA 1993, 90, 5181–5185. [Google Scholar] [CrossRef] [PubMed]
- Maziere, J.C.; Landureau, J.C.; Giral, P.; Auclair, M.; Fall, L.; Lachgar, A.; Achour, A.; Zagury, D. Lovastatin inhibits HIV-1 expression in H9 human T lymphocytes cultured in cholesterol-poor medium. Biomed. Pharmacother. 1994, 48, 63–67. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Hildreth, J.E. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J. Virol. 2000, 74, 3264–3272. [Google Scholar] [CrossRef] [PubMed]
- Espeseth, A.S.; Felock, P.; Wolfe, A.; Witmer, M.; Grobler, J.; Anthony, N.; Egbertson, M.; Melamed, J.Y.; Young, S.; Hamill, T.; et al. HIV-1 integrase inhibitors that compete with the target DNA substrate define a unique strand transfer conformation for integrase. Proc. Natl. Acad. Sci. USA 2000, 97, 11244–11249. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Derdowski, A.; Wang, J.J.; Spearman, P. Independent segregation of human immunodeficiency virus type 1 Gag protein complexes and lipid rafts. J. Virol. 2003, 77, 1916–1926. [Google Scholar] [CrossRef] [PubMed]
- Holm, K.; Weclewicz, K.; Hewson, R.; Suomalainen, M. Human immunodeficiency virus type 1 assembly and lipid rafts: Pr55gag associates with membrane domains that are largely resistant to Brij98 but sensitive to Triton X-100. J. Virol. 2003, 77, 4805–4817. [Google Scholar] [CrossRef] [PubMed]
- Bukrinsky, M.; Sviridov, D. Human immunodeficiency virus infection and macrophage cholesterol metabolism. J. Leukoc. Biol. 2006, 80, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.H.; Plemenitas, A.; Linnemann, T.; Fackler, O.T.; Peterlin, B.M. Nef increases infectivity of HIV via lipid rafts. Curr. Biol. 2001, 11, 875–879. [Google Scholar] [CrossRef]
- Lin, S.; Nadeau, P.E.; Wang, X.; Mergia, A. Caveolin-1 reduces HIV-1 infectivity by restoration of HIV Nef mediated impairment of cholesterol efflux by apoA-I. Retrovirology 2012, 9, 85. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Graham, D.R.; Hildreth, J.E. Lipid rafts and HIV pathogenesis: Virion-associated cholesterol is required for fusion and infection of susceptible cells. AIDS Res. Hum. Retrovir. 2003, 19, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Carter, G.C.; Bernstone, L.; Sangani, D.; Bee, J.W.; Harder, T.; James, W. HIV entry in macrophages is dependent on intact lipid rafts. Virology 2009, 386, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.H.; Plemenitas, A.; Fielding, C.J.; Peterlin, B.M. Nef increases the synthesis of and transports cholesterol to lipid rafts and HIV-1 progeny virions. Proc. Natl. Acad. Sci. USA 2003, 100, 8460–8465. [Google Scholar] [CrossRef] [PubMed]
- Van‘t Wout, A.B.; Swain, J.V.; Schindler, M.; Rao, U.; Pathmajeyan, M.S.; Mullins, J.I.; Kirchhoff, F. Nef induces multiple genes involved in cholesterol synthesis and uptake in human immunodeficiency virus type 1-infected T cells. J. Virol. 2005, 79, 10053–10058. [Google Scholar] [CrossRef] [PubMed]
- Mujawar, Z.; Rose, H.; Morrow, M.P.; Pushkarsky, T.; Dubrovsky, L.; Mukhamedova, N.; Fu, Y.; Dart, A.; Orenstein, J.M.; Bobryshev, Y.V.; et al. Human immunodeficiency virus impairs reverse cholesterol transport from macrophages. PLoS Biol. 2006, 4, 1970–1983. [Google Scholar] [CrossRef] [PubMed]
- Mujawar, Z.; Tamehiro, N.; Grant, A.; Sviridov, D.; Bukrinsky, M.; Fitzgerald, M.L. Mutation of the ATP cassette binding transporter A1 (ABCA1) c-terminus disrupts HIV-1 Nef binding but does not block the Nef enhancement of ABCA1 protein degradation. Biochemistry 2010, 49, 8338–8349. [Google Scholar] [CrossRef] [PubMed]
- Jennelle, L.; Hunegnaw, R.; Dubrovsky, L.; Pushkarsky, T.; Fitzgerald, M.L.; Sviridov, D.; Popratiloff, A.; Brichacek, B.; Bukrinsky, M. HIV-1 protein Nef inhibits activity of ATP-binding cassette transporter A1 by targeting endoplasmic reticulum chaperone calnexin. J. Biol. Chem. 2014, 289, 28870–28884. [Google Scholar] [CrossRef] [PubMed]
- Kalyana Sundaram, R.V.; Li, H.; Bailey, L.; Rashad, A.A.; Aneja, R.; Weiss, K.; Huynh, J.; Bastian, A.R.; Papazoglou, E.; Abrams, C.; et al. Impact of HIV-1 membrane cholesterol on cell-independent lytic inactivation and cellular infectivity. Biochemistry 2016, 55, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.H.; Lu, L.; Hong, L.; Chen, X.; Jiang, S.; Chen, Y.-H. Identification of the HIV-1 gp41 core-binding motif in the scaffolding domain of caveolin-1. J. Biol. Chem. 2007, 282, 6143–6152. [Google Scholar] [CrossRef] [PubMed]
- Hovanessian, A.G.; Briand, J.P.; Said, E.A.; Svab, J.; Ferris, S.; Dali, H.; Muller, S.; Desgranges, C.; Krust, B. The caveolin-1 binding domain of HIV-1 glycoprotein gp41 is an efficient B cell epitope vaccine candidate against virus infection. Immunity 2004, 21, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Nadeau, P.E.; Lo, Y.-T.; Mergia, A. Caveolin-1 modulates HIV-1 envelope induced bystander apoptosis through gp41. J. Virol. 2010, 84, 6515–6526. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.K.; Yuan, J.; Gao, X.P.; Garrean, S.; Brovkovych, V.; Malik, A.B.; Tiruppathi, C.; Zhao, Y.Y. Caveolin-1 deficiency dampens Toll-like receptor 4 signaling through eNOS activation. Am. J. Pathol. 2010, 176, 2344–2351. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.H.; Chen, S.F.; Huang, T.Y.; Tzeng, C.F.; Chiang, A.S.; Kou, Y.R.; Lee, T.S.; Shyue, S.K. Impaired CD14 and CD36 expression, bacterial clearance, and Toll-like receptor 4-Myd88 signaling in caveolin-1-deleted macrophages and mice. Shock 2011, 35, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Yi, F.; Dai, Z.; Huang, X.; Zhao, Y.D.; Mirza, M.K.; Xu, J.; Vogel, S.M.; Zhao, Y.Y. Loss of caveolin-1 and adiponectin induces severe inflammatory lung injury following LPS challenge through excessive oxidative/nitrative stress. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L566–L573. [Google Scholar] [CrossRef] [PubMed]
- Medina, F.A.; de Almeida, C.J.; Dew, E.; Li, J.; Bonuccelli, G.; Williams, T.M.; Cohen, A.W.; Pestell, R.G.; Frank, P.G.; Tanowitz, H.B.; et al. Caveolin-1-deficient mice show defects in innate immunity and inflammatory immune response during Salmonella enterica serovar typhimurium infection. Infect. Immun. 2006, 74, 6665–6674. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Guo, L.; Song, Z.; Gao, H.; Wang, D.; Fu, W.; Han, J.; Li, Z.; Huang, B.; Li, X.A. Caveolin-1 protects against sepsis by modulating inflammatory response, alleviating bacterial burden, and suppressing thymocyte apoptosis. J. Biol. Chem. 2010, 285, 25154–25160. [Google Scholar] [CrossRef] [PubMed]
- Sathish, V.; Thompson, M.A.; Sinha, S.; Sieck, G.C.; Prakash, Y.S.; Pabelick, C.M. Inflammation, caveolae and CD38-mediated calcium regulation in human airway smooth muscle. Biochim. Biophys. Acta 2014, 1843, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Zhang, Y.; Yan, Z.; Wang, Z.G.; Liu, G.; Minshall, R.D.; Malik, A.B.; Hu, G. Caveolin-1 Tyr14 phosphorylation induces interaction with TLR4 in endothelial cells and mediates MyD88-dependent signaling and sepsis-induced lung inflammation. J. Immunol. 2013, 191, 6191–6199. [Google Scholar] [CrossRef] [PubMed]
- Hazenberg, M.D.; Otto, S.A.; van Benthem, B.H.; Roos, M.T.; Coutinho, R.A.; Lange, J.M.; Hamann, D.; Prins, M.; Miedema, F. Persistent immune activation in HIV-1 infection is associated with progression to aids. AIDS 2003, 17, 1881–1888. [Google Scholar] [CrossRef] [PubMed]
- Appay, V.; Sauce, D. Immune activation and inflammation in HIV-1 infection: Causes and consequences. J. Pathol. 2008, 214, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, J.; Jacobs, D.R., Jr.; Baker, J.V.; Calmy, A.; Duprez, D.; La Rosa, A.; Kuller, L.H.; Pett, S.L.; Ristola, M.; Ross, M.J.; et al. Markers of inflammation, coagulation, and renal function are elevated in adults with HIV infection. J. Infect. Dis. 2010, 201, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Ipp, H.; Zemlin, A. The paradox of the immune response in HIV infection: When inflammation becomes harmful. Clin. Chim. Acta 2013, 416, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, N.F.; Cool, C.D.; Flores, S. From viral infection to pulmonary arterial hypertension: A role for viral proteins? AIDS 2008, 22 (Suppl. 3), S49–S53. [Google Scholar] [CrossRef] [PubMed]
- Petitpretz, P.; Brenot, F.; Azarian, R.; Parent, F.; Rain, B.; Herve, P.; Simonneau, G. Pulmonary hypertension in patients with human immunodeficiency virus infection. Comparison with primary pulmonary hypertension. Circulation 1994, 89, 2722–2727. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.J.; Khan, I.A.; Mehta, R.N.; Sepkowitz, D.A. HIV-related pulmonary hypertension: Analytic review of 131 cases. Chest 2000, 118, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Limsukon, A.; Saeed, A.I.; Ramasamy, V.; Nalamati, J.; Dhuper, S. HIV-related pulmonary hypertension. Mt. Sinai J. Med. 2006, 73, 1037–1044. [Google Scholar] [PubMed]
- Marecki, J.; Cool, C.; Voelkel, N.; Luciw, P.; Flores, S. Evidence for vascular remodeling in the lungs of macaques infected with simian immunodeficiency virus/HIV Nef recombinant virus. Chest 2005, 128, 621S–622S. [Google Scholar] [CrossRef] [PubMed]
- Duffy, P.; Wang, X.; Lin, P.H.; Yao, Q.; Chen, C. HIV Nef protein causes endothelial dysfunction in porcine pulmonary arteries and human pulmonary artery endothelial cells. J. Surg. Res. 2009, 156, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, R.M.; Draznin, M.B.; Murad, F. Endothelium-dependent vasodilator-and nitrovasodilator-induced relaxation may be mediated through cyclic GMP formation and cyclic GMP-dependent protein phosphorylation. Trans. Assoc. Am. Physicians 1983, 96, 19–30. [Google Scholar] [PubMed]
- Palmer, R.M.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar] [PubMed]
- Cooke, J.P. The pivotal role of nitric oxide for vascular health. Can. J. Cardiol. 2004, 20 (Suppl. B), 7B–15B. [Google Scholar] [PubMed]
- Garcia-Cardena, G.; Martasek, P.; Masters, B.S.; Skidd, P.M.; Couet, J.; Li, S.; Lisanti, M.P.; Sessa, W.C. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the NOS caveolin binding domain in vivo. J. Biol. Chem. 1997, 272, 25437–25440. [Google Scholar] [CrossRef] [PubMed]
- Sowa, G.; Pypaert, M.; Sessa, W.C. Distinction between signaling mechanisms in lipid rafts vs. Caveolae. Proc. Natl. Acad. Sci. USA 2001, 98, 14072–14077. [Google Scholar] [CrossRef] [PubMed]
- Bucci, M.; Gratton, J.P.; Rudic, R.D.; Acevedo, L.; Roviezzo, F.; Cirino, G.; Sessa, W.C. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat. Med. 2000, 6, 1362–1367. [Google Scholar] [PubMed]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Y.; Liu, Y.; Stan, R.V.; Fan, L.; Gu, Y.; Dalton, N.; Chu, P.H.; Peterson, K.; Ross, J., Jr.; Chien, K.R. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc. Natl. Acad. Sci. USA 2002, 99, 11375–11380. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Ma, L.; LeDuc, C.; Berman Rosenzweig, E.; Borczuk, A.; Phillips, J.A., III; Palomero, T.; Sumazin, P.; Kim, H.R.; Talati, M.H.; et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ. Cardiovasc. Genet. 2012, 5, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Kircher, M.; Del Campo, M.; Amato, R.S.; Agarwal, A.K.; University of Washington Center for Mendelian Genomics. Whole exome sequencing identifies de novo heterozygous CAV1 mutations associated with a novel neonatal onset lipodystrophy syndrome. Am. J. Med. Genet. A 2015, 167A, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Copeland, C.A.; Kawano, Y.; Rosenzweig, E.B.; Austin, E.D.; Shahmirzadi, L.; Tang, S.; Raghunathan, K.; Chung, W.K.; Kenworthy, A.K. Characterization of a caveolin-1 mutation associated with both pulmonary arterial hypertension and congenital generalized lipodystrophy. Traffic 2016, 17, 1297–1312. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Nadeau, P.E.; Mergia, A. HIV inhibits endothelial reverse cholesterol transport through impacting subcellular caveolin-1 trafficking. Retrovirology 2015, 12, 62. [Google Scholar] [CrossRef] [PubMed]
- Corrotte, M.; Almeida, P.E.; Tam, C.; Castro-Gomes, T.; Fernandes, M.C.; Millis, B.A.; Cortez, M.; Miller, H.; Song, W.; Maugel, T.K.; et al. Caveolae internalization repairs wounded cells and muscle fibers. eLife 2013, 2, e00926. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.; Samaras, K.; Chisholm, D.J.; Cooper, D.A. Pathogenesis of HIV-1-protease inhibitor-associated peripheral lipodystrophy, hyperlipidaemia, and insulin resistance. Lancet 1998, 1881–1883. [Google Scholar] [CrossRef]
- Barbaro, G. Cardiovascular manifestations of HIV infection. Circulation 2002, 106, 1420–1425. [Google Scholar] [CrossRef] [PubMed]
- Raulin, J. Human immunodeficiency virus and host cell lipids. Interesting pathways in research for a new HIV therapy. Prog. Lipid Res. 2002, 41, 27–65. [Google Scholar] [CrossRef]
- Ben-Romano, R.; Rudich, A.; Tirosh, A.; Potashnik, R.; Sasaoka, T.; Riesenberg, K.; Schlaeffer, F.; Bashan, N. Nelfinavir-induced insulin resistance is associated with impaired plasma membrane recruitment of the PI 3-kinase effectors Akt/PKB and PKC-zeta. Diabetologia 2004, 47, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.; Hurley, L.B.; Quesenberry, C.P., Jr.; Sidney, S. Do protease inhibitors increase the risk for coronary heart disease in patients with HIV-1 infection? J. Acquir. Immune Defic. Syndr. 2002, 30, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Charakida, M.; Donald, A.E.; Green, H.; Storry, C.; Clapson, M.; Caslake, M.; Dunn, D.T.; Halcox, J.P.; Gibb, D.M.; Klein, N.J.; et al. Early structural and functional changes of the vasculature in HIV-infected children: Impact of disease and antiretroviral therapy. Circulation 2005, 112, 103–109. [Google Scholar] [CrossRef] [PubMed]
- El-Sadr, W.M.; Mullin, C.M.; Carr, A.; Gibert, C.; Rappoport, C.; Visnegarwala, F.; Grunfeld, C.; Raghavan, S.S. Effects of HIV disease on lipid, glucose and insulin levels: Results from a large antiretroviral-naive cohort. HIV Med. 2005, 6, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Umpleby, A.M.; Das, S.; Stolinski, M.; Shojaee-Moradie, F.; Jackson, N.C.; Jefferson, W.; Crabtree, N.; Nightingale, P.; Shahmanesh, M. Low density lipoprotein apolipoprotein B metabolism in treatment-naive HIV patients and patients on antiretroviral therapy. Antivir. Ther. 2005, 10, 663–670. [Google Scholar] [PubMed]
- Barbaro, G. Visceral fat as target of highly active antiretroviral therapy associated metabolic syndrome. Curr. Pharm. Des. 2007, 13, 2208–2213. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.W.; Stephan, C.; Harmjanz, A.; Staszewski, S.; Buehler, A.; Bickel, M.; von Kegler, S.; Ruhkamp, D.; Steinmetz, H.; Sitzer, M. Both long-term HIV infection and highly active antiretroviral therapy are independent risk factors for early carotid atherosclerosis. Atherosclerosis 2007, 196, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Gagnon, A.; Angel, J.B.; Sorisky, A. Ritonavir increases the level of active ADD-1/SREBP-1 protein during adipogenesis. AIDS 2000, 14, 2467–2473. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.S.; Distler, O.; Cooper, D.A.; Jamil, H.; Deckelbaum, R.J.; Ginsberg, H.N.; Sturley, S.L. HIV protease inhibitors protect apolipoprotein B from degradation by the proteasome: A potential mechanism for protease inhibitor-induced hyperlipidemia. Nat. Med. 2001, 7, 1327–1331. [Google Scholar] [CrossRef] [PubMed]
- Dressman, J.; Kincer, J.; Matveev, S.V.; Guo, L.; Greenberg, R.N. HIV protease inhibitors promote atherosclerotic lesion formation independent of dyslipidemia by increasing CD36-dependent cholesteryl ester accumulation in macrophages. J. Clin. Investig. 2003, 111, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mu, H.; Chai, H.; Liao, D.; Yao, Q.; Chen, C. Human immunodeficiency virus protease inhibitor ritonavir inhibits cholesterol efflux from human macrophage-derived foam cells. Am. J. Pathol. 2007, 171, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Mergia, A. Nelfinavir also reduces Cav-1 expression in macrophages. University of Florida: Gainesville Florida, 2015; unpublished results. [Google Scholar]
- Hsue, P.Y.; Lo, J.C.; Franklin, A.; Bolger, A.F.; Martin, J.N.; Deeks, S.G.; Waters, D.D. Progression of atherosclerosis as assessed by carotid intima-media thickness in patients with HIV infection. Circulation 2004, 109, 1603–1608. [Google Scholar] [CrossRef] [PubMed]
- Guaraldi, G.; Zona, S.; Orlando, G.; Carli, F.; Ligabue, G.; Fiocchi, F.; Menozzi, M.; Rossi, R.; Modena, M.G.; Raggi, P. Human immunodeficiency virus infection is associated with accelerated atherosclerosis. J. Antimicrob. Chemother. 2011, 66, 1857–1860. [Google Scholar] [CrossRef] [PubMed]
- Coll, B.; Parra, S.; Aloonso-Villaverde, C.; de Groot, E.; Aragones, G.; Montero, M.; Tous, M.; Camps, J.; Joven, J.; Masana, L. HIV infected patients with lipodystrophy have higher rates of carotid atherosclerosis: The role of monocyte chemoattractant protein-1. Cytokine 2006, 34, 51–55. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Functions | Activities |
|---|---|
| Mechanosensing | Protection of cells from mechanical stress damage and other distress signals. |
| Lipid Homeostasis | Regulation of membrane lipid composition, cholesterol transport, and efflux. |
| Endocytosis | Proposed endocytosed cargos include glycosylphosphatidyl inositol (GPI)-anchored proteins, the insulin receptor, shiga and cholera toxins, cholesterol, albumin, and simian virus 40 (SV40). The specificity of caveolae and mechanisms for these cargos are still in question. |
| Transcytosis | Caveolae transport proteins and lipids such as low density lipoproteins (LDLs) albumin, and insulin from the blood across the interior of the endothelial cells to the opposite face and vice versa. |
| Signal transduction | Signaling platforms to diverse signaling molecules such as the insulin receptor, endothelial nitric oxide synthase( eNOS,) the epidermal growth factor receptor, and mitogen-activated protein kinase. |
| Inflammation | Cav-1 modulation of eNOS regulates inflammatory signaling through local control of NO production. |
| Ca2+ regulation and signaling can also be involved in inflammatory responses. | |
| Cav-1 serves as a protective protein against cellular stress and inflammation. | |
| Cancer | Serves as a tumor suppresser and at times as an oncogenic factor. Loss of Cav-1 in the stroma is linked to the clinical outcome of breast cancer. |
| Apoptosis | Involved in supporting multiple anti-apoptotic and survival pathways. |
| Angiogenesis | Involved in cancer angiogenesis. The Caveolin 1 pathway is also involved in the regulation of vascular endothelial growth factor (VEGF) in association with induced angiogenesis in brain ischemic. |
| Cellular senescence | Caveolin 1 plays a major role in controlling cellular senescence through its up-regulation by oxidative stress. |
| Cell cycle regulation | Caveolin 1 is important in the regulation of cyclin D1. |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mergia, A. The Role of Caveolin 1 in HIV Infection and Pathogenesis. Viruses 2017, 9, 129. https://doi.org/10.3390/v9060129
Mergia A. The Role of Caveolin 1 in HIV Infection and Pathogenesis. Viruses. 2017; 9(6):129. https://doi.org/10.3390/v9060129
Chicago/Turabian StyleMergia, Ayalew. 2017. "The Role of Caveolin 1 in HIV Infection and Pathogenesis" Viruses 9, no. 6: 129. https://doi.org/10.3390/v9060129
APA StyleMergia, A. (2017). The Role of Caveolin 1 in HIV Infection and Pathogenesis. Viruses, 9(6), 129. https://doi.org/10.3390/v9060129