
Viral Ubiquitin Ligase Stimulates Selective Host MicroRNA Expression by Targeting ZEB Transcriptional Repressors

 , , ,
, , ,  , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses
2.2. Cell Lines and Infections
2.3. Preparation and HSV-1 Infection of Superior Cervical Ganglia Neuron Cultures
2.4. Ethics Statement
2.5. RNA Extraction and Reverse Transcription Quantitative Polymerase Chain Reaction
2.6. High-Throughput Small RNA-Sequencing
2.7. Immunoblotting
2.8. Pharmacological Applications
2.9. Immunofluorescence Microscopy
2.10. RNAi Depletion
2.11. Accession Number
3. Results
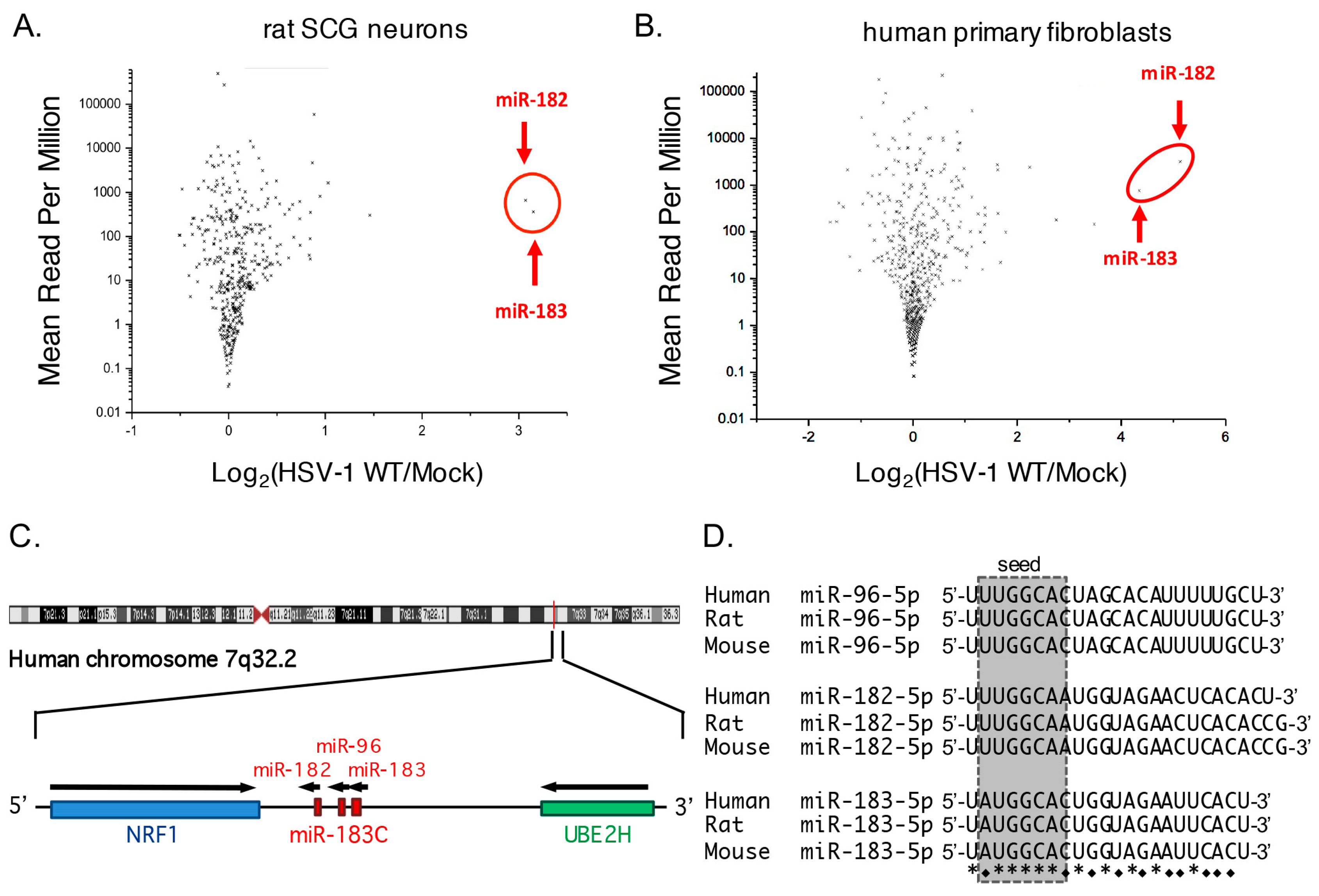
3.1. Increased Abundance of Host miR-182 and miR-183 during HSV-1 Infection
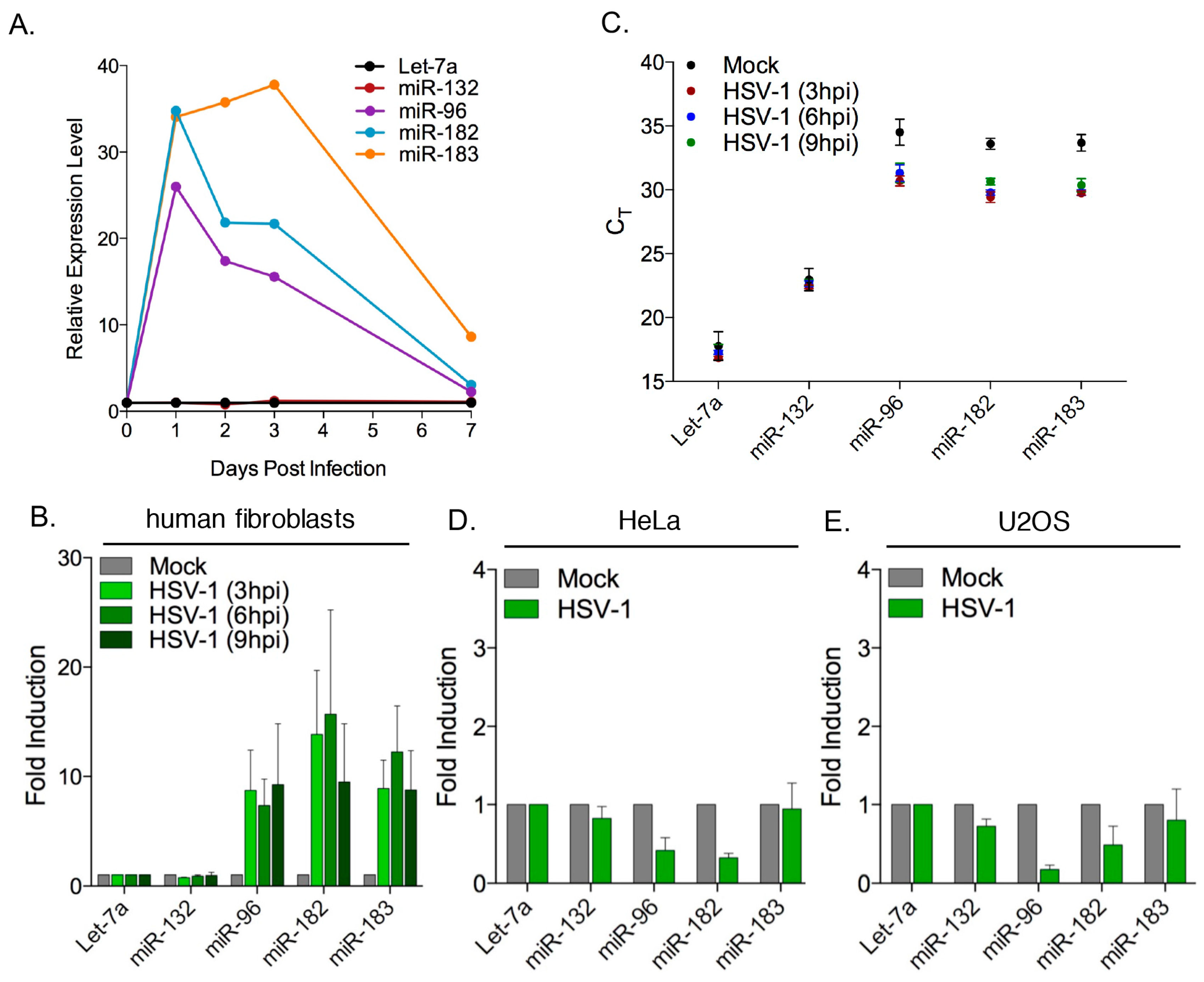
3.2. Robust Induction of the miR-183/96/182 Cluster during Acute HSV-1 Infection
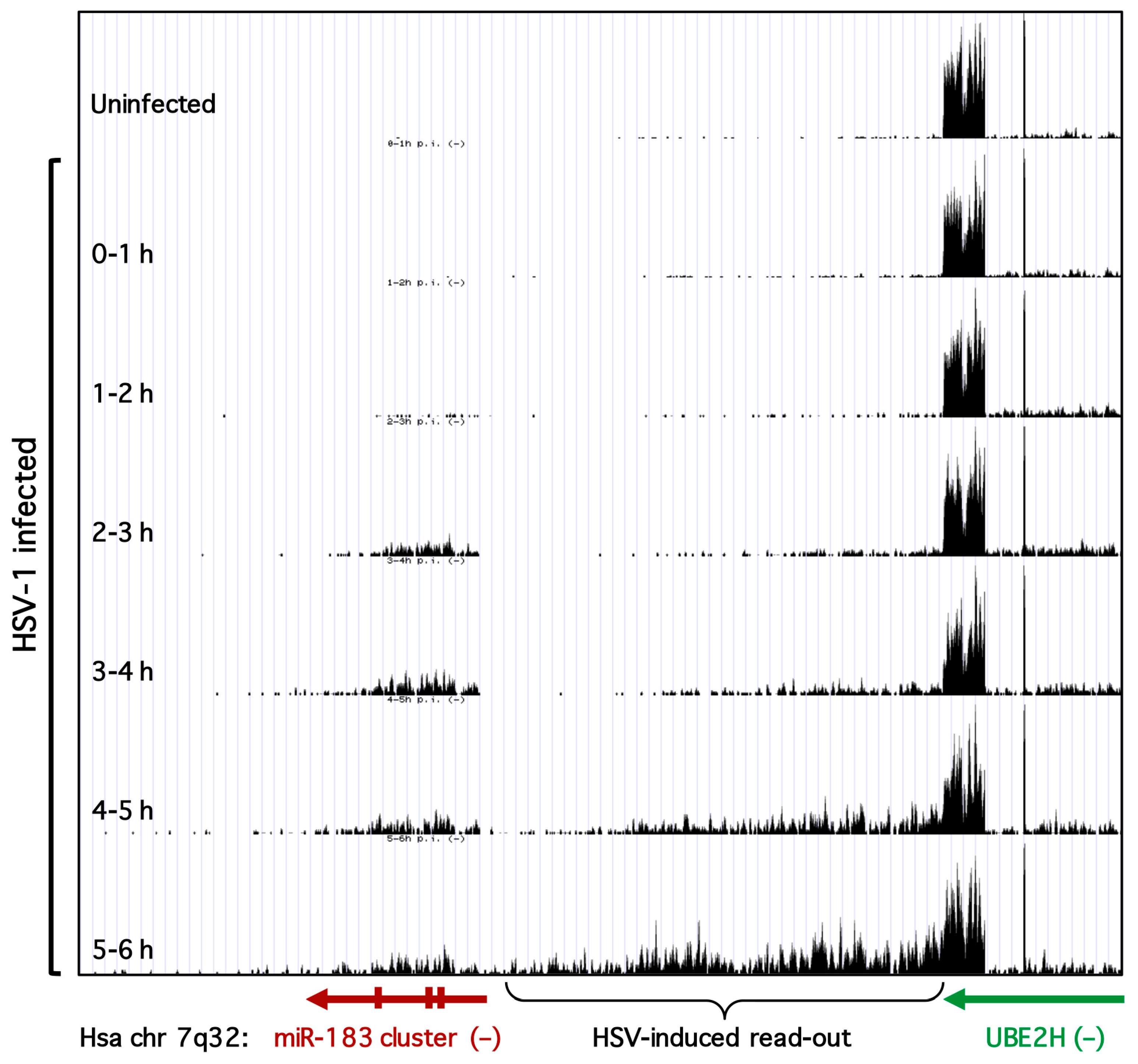
3.3. Transcription of the miR-183/96/182 Cluster Is Stimulated by HSV-1
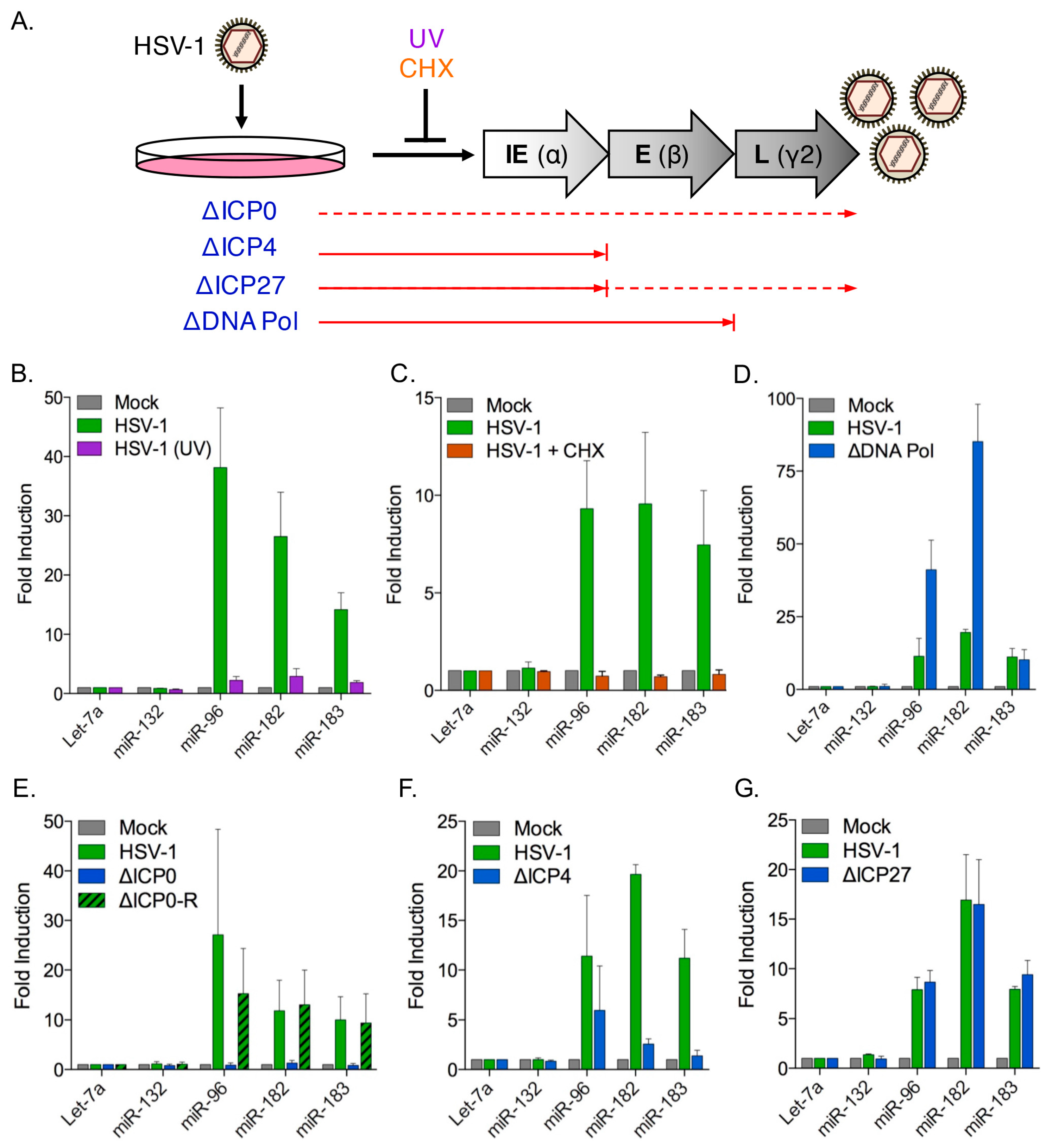
3.4. Induction of the miR-183/96/182 Cluster Requires Viral Gene Expression
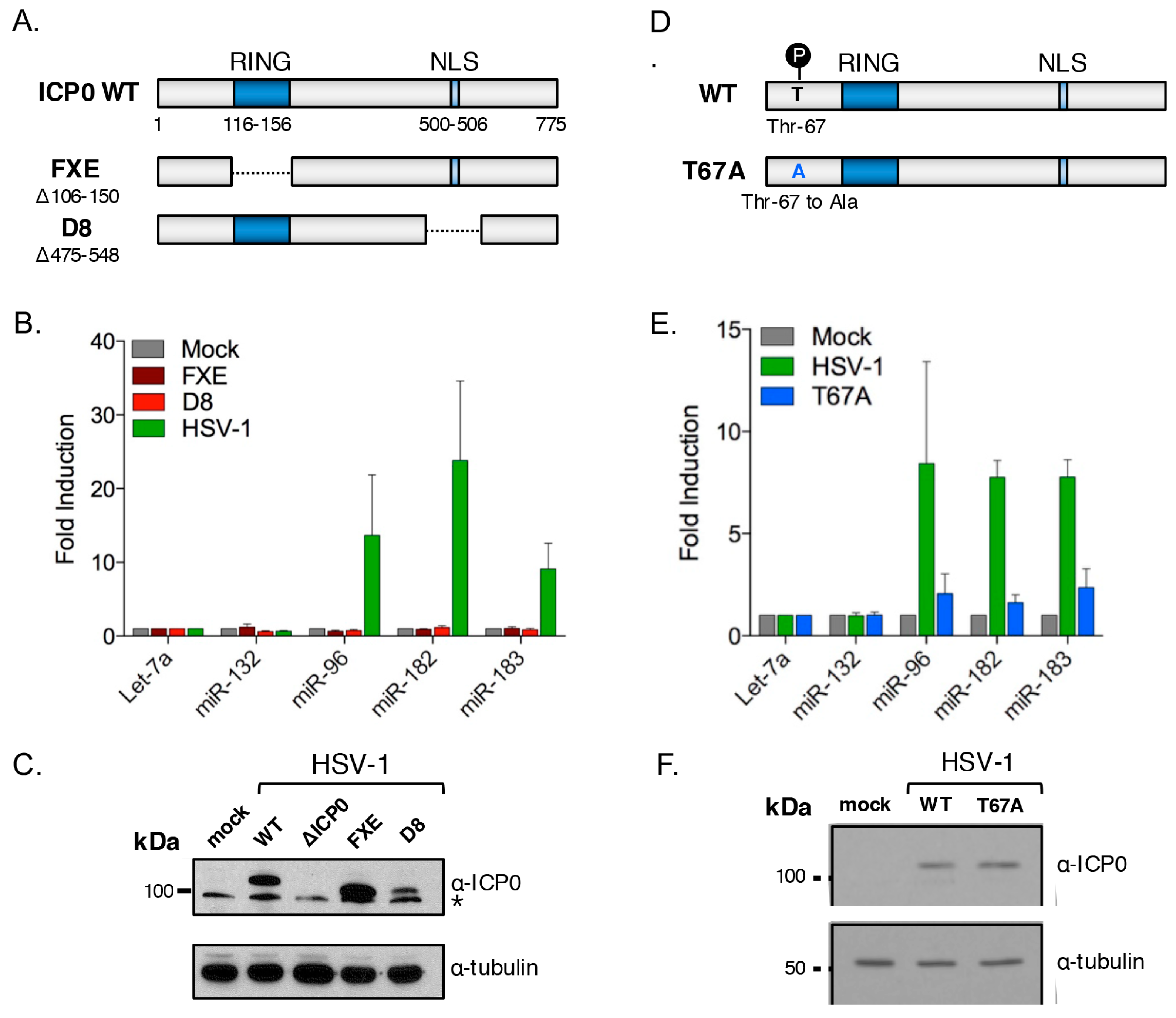
3.5. The HSV-1 Immediate-Early Proteins ICP0 and ICP4 Are Necessary for Maximal miR-183/96/182 Cluster Induction
3.6. ICP0 Is Sufficient to Induce the miR-183/96/182 Cluster
3.7. ICP0 Must Be Nuclear to Induce miR-183/96/182
3.8. Cluster Induction Requires the E3 Ligase Function of ICP0
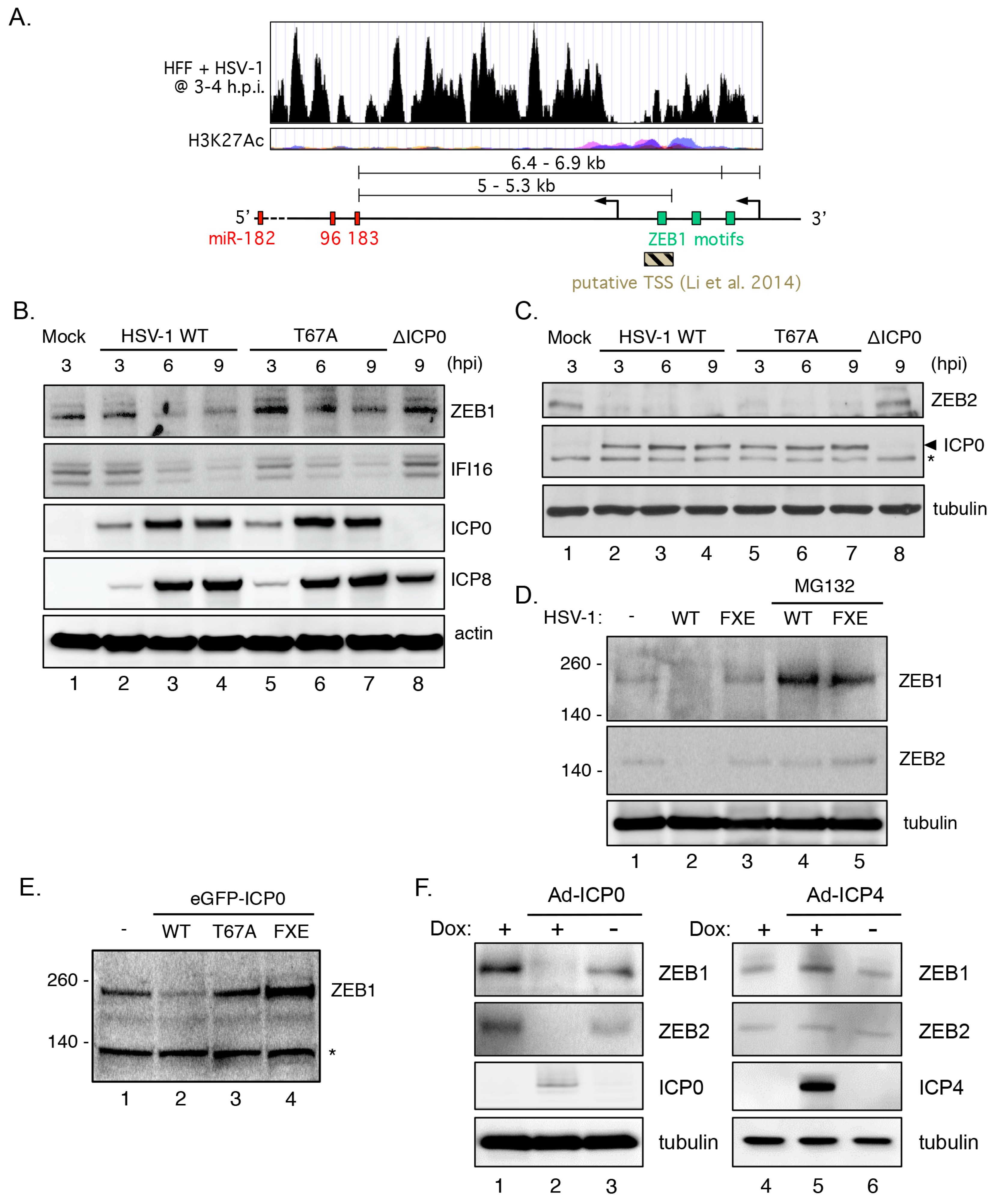
3.9. Transcriptional Repressor ZEB1 Is Targeted for Degradation by ICP0
3.10. ZEB1 Suppresses miR-183/96/182 Cluster Transcription in Primary Fibroblasts
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Vanícek, J.; Murphy, E.A. Host microRNA regulation of human cytomegalovirus immediate early protein translation promotes viral latency. J. Virol. 2014, 88, 5524–5532. [Google Scholar] [CrossRef] [PubMed]
- Powdrill, M.H.; Desrochers, G.F.; Singaravelu, R.; Pezacki, J.P. The role of microRNAs in metabolic interactions between viruses and their hosts. Curr. Opin. Virol. 2016, 19, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Song, Y.; He, L.; Wan, X.; Lai, L.; Dai, F.; Liu, Y.; Wang, Q. MicroRNA-223 promotes type I interferon production in antiviral innate immunity by targeting forkhead box protein O3 (FOXO3). J. Biol. Chem. 2016, 291, 14706–14716. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.A.; Hsieh, W.Y.; Forster, T.; Blanc, M.; Lu, H.; Crick, P.J.; Yutuc, E.; Watterson, S.; Martin, K.; Griffiths, S.J.; et al. An interferon regulated microrna provides broad cell-intrinsic antiviral immunity through multihit host-directed targeting of the sterol pathway. PLoS Biol. 2016, 14, e1002364. [Google Scholar] [CrossRef] [PubMed]
- Piedade, D.; Azevedo-Pereira, J.M. The role of microRNAs in the pathogenesis of herpesvirus infection. Viruses 2016, 8, 156. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Jurak, I.; Kramer, M.F.; Mellor, J.C.; van Lint, A.L.; Roth, F.P.; Knipe, D.M.; Coen, D.M. Numerous conserved and divergent microRNAs expressed by herpes simplex viruses 1 and 2. J. Virol. 2010, 84, 4659–4672. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Nagel, M.A.; Cohrs, R.J.; Gilden, D.H.; Cullen, B.R. Analysis of Human alphaherpesvirus microrna expression in latently infected human trigeminal ganglia. J. Virol. 2009, 83, 10677–10683. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.F.; Jurak, I.; Pesola, J.M.; Boissel, S.; Knipe, D.M.; Coen, D.M. Herpes simplex virus 1 microRNAs expressed abundantly during latent infection are not essential for latency in mouse trigeminal ganglia. Virology 2011, 417, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Flores, O.; Umbach, J.L.; Pesola, J.M.; Bentley, P.; Rosato, P.C.; Leib, D.A.; Cullen, B.R.; Coen, D.M. A neuron-specific host microRNA targets herpes simplex virus-1 ICP0 expression and promotes latency. Cell Host Microbe 2014, 15, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Flores, O.; Nakayama, S.; Whisnant, A.W.; Javanbakht, H.; Cullen, B.R.; Bloom, D.C. Mutational inactivation of hsv-1 micrornas identifies viral mRNA targets and reveals phenotypic effects in culture. J. Virol. 2013, 87, 6589–6603. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Brown, D.; Osorio, N.; Hsiang, C.; Li, L.; Chan, L.; BenMohamed, L.; Wechsler, S.L. A herpes simplex virus type 1 mutant disrupted for microRNA H2 with increased neurovirulence and rate of reactivation. J. Neurovirol. 2015, 21, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Pesola, J.M.; Li, G.; McCarron, S.; Coen, D.M. Mutations inactivating herpes simplex virus-1 miR-H2 do not detectably increase ICP0 gene expression in infected cultured cells or mouse trigeminal ganglia. J. Virol. 2016, 91, e02001–e02016. [Google Scholar] [CrossRef] [PubMed]
- Nachmani, D.; Lankry, D.; Wolf, D.G.; Mandelboim, O. The human cytomegalovirus microRNA miR-UL112 acts synergistically with a cellular microRNA to escape immune elimination. Nat. Immunol. 2010, 11, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; McBride, J.; Fewell, C.; Lacey, M.; Wang, X.; Lin, Z.; Cameron, J.; Flemington, E.K. MicroRNA-155 is an Epstein–Barr virus-induced gene that modulates Epstein–Barr virus-regulated gene expression pathways. J. Virol. 2008, 82, 5295–5306. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.J.; Rabinowitz, G.S.; Yario, T.A.; Luna, J.M.; Darnell, R.B.; Steitz, J.A. EBV and human microRNAs co-target oncogenic and apoptotic viral and human genes during latency. EMBO J. 2012, 31, 2207–2221. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Corcoran, D.L.; Gottwein, E.; Frank, C.L.; Kang, D.; Hafner, M.; Nusbaum, J.D.; Feederle, R.; Delecluse, H.-J.; Luftig, M.A.; et al. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog. 2012, 8, e1002484. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Boccellato, F.; Vincenti, S.; Rosato, P.; Bozzoni, I.; Frati, L.; Faggioni, A.; Presutti, C.; Trivedi, P. Epstein–Barr virus encoded LMP1 downregulates TCL1 oncogene through miR-29b. Oncogene 2010, 29, 1316–1328. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, R.P.; Burke, J.M.; Sullivan, C.S. RNA virus microRNA that mimics a B-cell oncomiR. Proc. Natl. Acad. Sci. USA 2012, 109, 3077–3082. [Google Scholar] [CrossRef] [PubMed]
- Cazalla, D.; Yario, T.; Steitz, J.A.; Steitz, J. Downregulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science 2010, 328, 1563–1566. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.-Z.; Weber, F.; Croce, C.; Liu, C.-G.; Liao, X.; Pellett, P.E. Human cytomegalovirus infection alters the expression of cellular microRNA species that affect its replication. J. Virol. 2008, 82, 9065–9074. [Google Scholar] [CrossRef] [PubMed]
- Kwong, A.D.; Frenkel, N. Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 1926–1930. [Google Scholar] [CrossRef] [PubMed]
- Read, G.S.; Frenkel, N. Herpes simplex virus mutants defective in the virion-associated shutoff of host polypeptide synthesis and exhibiting abnormal synthesis of alpha (immediate early) viral polypeptides. J. Virol. 1983, 46, 498–512. [Google Scholar] [PubMed]
- Abrisch, R.G.; Eidem, T.M.; Yakovchuk, P.; Kugel, J.F.; Goodrich, J.A. Infection by herpes simplex virus type-1 causes near-complete loss of RNA polymerase II occupancy on the host cell genome. J. Virol. 2015, 90, 2503–2513. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, A.J.; Erhard, F.; L’Hernault, A.; Bonfert, T.; Schilhabel, M.; Crump, C.; Rosenstiel, P.; Efstathiou, S.; Zimmer, R.; Friedel, C.C.; et al. Widespread disruption of host transcription termination in HSV-1 infection. Nat. Commun. 2015, 6, 7126. [Google Scholar] [CrossRef] [PubMed]
- Sandri-Goldin, R.M. The many roles of the regulatory protein ICP27 during herpes simplex virus infection. Front. Biosci. 2008, 13, 5241–5256. [Google Scholar] [CrossRef] [PubMed]
- Gaglia, M.M.; Covarrubias, S.; Wong, W.; Glaunsinger, B.A. A common strategy for host RNA degradation by divergent viruses. J. Virol. 2012, 86, 9527–9530. [Google Scholar] [CrossRef] [PubMed]
- Elgadi, M.M.; Hayes, C.E.; Smiley, J.R. The herpes simplex virus vhs protein induces endoribonucleolytic cleavage of target RNAs in cell extracts. J. Virol. 1999, 73, 7153–7164. [Google Scholar] [PubMed]
- Jan, E.; Mohr, I.; Walsh, D. A cap-to-tail guide to mRNA translation strategies in virus-infected cells. Annu. Rev. Virol. 2016, 3, 283–307. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.E.; Steitz, J.A. Virus meets host microRNA: The destroyer, the booster, the hijacker. Mol. Cell Biol. 2014, 34, 3780–3787. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-L.; Hara, T.; Choi, Y.; Subramanian, M.; Francis, P.; Bilke, S.; Walker, R.L.; Pineda, M.; Zhu, Y.; Yang, Y.; et al. A p21-ZEB1 complex inhibits epithelial-mesenchymal transition through the miR-183–96–182 cluster. Mol. Cell Biol. 2013, 34, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Lodrini, M.; Oehme, I.; Schroeder, C.; Milde, T.; Schier, M.C.; Kopp-Schneider, A.; Schulte, J.H.; Fischer, M.; De Preter, K.; Pattyn, F.; et al. MYCN and HDAC2 cooperate to repress miR-183 signaling in neuroblastoma. Nucleic Acids Res. 2013, 41, 6018–6033. [Google Scholar] [CrossRef] [PubMed]
- Benboudjema, L.; Mulvey, M.; Gao, Y.; Pimplikar, S.W.; Mohr, I. Association of the herpes simplex virus type 1 Us11 gene product with the cellular kinesin light-chain-related protein PAT1 results in the redistribution of both polypeptides. J. Virol. 2003, 77, 9192–9203. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Schaffer, P.A. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J. Virol. 1992, 66, 2904–2915. [Google Scholar] [PubMed]
- Deluca, N.A.; Schaffer, P.A. Physical and functional domains of the herpes simplex virus transcriptional regulatory protein ICP4. J. Virol. 1988, 62, 732–743. [Google Scholar] [PubMed]
- Rice, S.A.; Knipe, D.M. Genetic evidence for two distinct transactivation functions of the herpes simplex virus alpha protein ICP27. J. Virol. 1990, 64, 1704–1715. [Google Scholar] [PubMed]
- Marcy, A.I.; Yager, D.R.; Coen, D.M. Isolation and characterization of herpes simplex virus mutants containing engineered mutations at the DNA polymerase locus. J. Virol. 1990, 64, 2208–2216. [Google Scholar] [PubMed]
- Halford, W.P.; Kemp, C.D.; Isler, J.A.; Davido, D.J.; Schaffer, P.A. ICP0, ICP4, or VP16 expressed from adenovirus vectors induces reactivation of latent herpes simplex virus type 1 in primary cultures of latently infected trigeminal ganglion cells. J. Virol. 2001, 75, 6143–6153. [Google Scholar] [CrossRef] [PubMed]
- Stoller, M.L.; Chang, H.C.; Fekete, D.M. Bicistronic gene transfer tools for delivery of miRNAs and protein coding sequences. Int. J. Mol. Sci. 2013, 14, 18239–18255. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Kim, J.Y.; Camarena, V.; Roehm, P.C.; Chao, M.V.; Wilson, A.C.; Mohr, I. A primary neuron culture system for the study of herpes simplex virus latency and reactivation. J. Vis. Exp. 2012, 62, e3823. [Google Scholar] [CrossRef] [PubMed]
- Camarena, V.; Kobayashi, M.; Kim, J.Y.; Roehm, P.C.; Perez, R.; Gardner, J.; Wilson, A.C.; Mohr, I.; Chao, M.V. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe 2010, 8, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Shiflett, L.A.; Linderman, J.A.; Mohr, I.; Wilson, A.C. Using homogeneous primary neuron cultures to study fundamental aspects of HSV-1 latency and reactivation. Methods Mol. Biol. 2014, 1144, 167–179. [Google Scholar] [PubMed]
- Barturen, G.; Rueda, A.; Hamberg, M.; Alganza, A.; Lebron, R.; Kotsyfakis, M.; Shi, B.-J.; Koppers-Lalic, D.; Hackenberg, M. sRNAbench: Profiling of small RNAs and its sequence variants in single or multi-species high-throughput experiments. Methods Next Gener. Seq. 2014, 1, 21–31. [Google Scholar] [CrossRef]
- Jurak, I.; Hackenberg, M.; Kim, J.Y.; Pesola, J.M.; Everett, R.D.; Preston, C.M.; Wilson, A.C.; Coen, D.M. Expression of herpes simplex virus 1 miRNAs in cell culture models of quiescent and latent infection. J. Virol. 2014, 88, 2337–2339. [Google Scholar] [CrossRef] [PubMed]
- Hackenberg, M.; Sturm, M.; Langenberger, D.; Falcón-Pérez, J.M.; Aransay, A.M. miRanalyzer: A microRNA detection and analysis tool for next-generation sequencing experiments. Nucleic Acids Res. 2009, 37, W68–W76. [Google Scholar] [CrossRef] [PubMed]
- McMahon, R.; Walsh, D. Efficient quiescent infection of normal human diploid fibroblasts with wild-type herpes simplex virus type 1. J. Virol. 2008, 82, 10218–10230. [Google Scholar] [CrossRef] [PubMed]
- Dambal, S.; Shah, M.; Mihelich, B.L.; Nonn, L. The microRNA-183 cluster: The family that plays together stays together. Nucleic Acids Res. 2015, 43, 7173–7188. [Google Scholar] [CrossRef] [PubMed]
- Pierce, M.L.; Weston, M.D.; Fritzsch, B.; Gabel, H.W.; Ruvkun, G.; Soukup, G.A. MicroRNA-183 family conservation and ciliated neurosensory organ expression. Evol. Dev. 2008, 10, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Lumayag, S.; Haldin, C.E.; Corbett, N.J.; Wahlin, K.J.; Cowan, C.; Turturro, S.; Larsen, P.E.; Kovacs, B.; Witmer, P.D.; Valle, D.; et al. Inactivation of the microRNA-183/96/182 cluster results in syndromic retinal degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, E507–E516. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Witmer, P.D.; Lumayag, S.; Kovacs, B.; Valle, D. MicroRNA (miRNA) transcriptome of mouse retina and identification of a sensory organ-specific miRNA cluster. J. Biol. Chem. 2007, 282, 25053–25066. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Sheng, C.; Huang, L.; Zhang, H.; Huang, L.; Cheng, Z.; Zhu, Q. MiR-183/-96/-182 cluster is upregulated in most breast cancers and increases cell proliferation and migration. Breast Cancer Res. 2014, 16, 473. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.; Zhang, Y.; Khan, D.; Heid, B.; Caudell, D.; Crasta, O.; Ahmed, S.A. Identification of a common lupus disease-associated microRNA expression pattern in three different murine models of lupus. PLoS ONE 2010, 5, e14302. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Liu, Y.; Bai, L.; Kijlstra, A.; Yang, P. Predisposition to Behçet’s disease and VKH syndrome by genetic variants of miR-182. J. Mol. Med. 2014, 92, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Kye, M.J.; Niederst, E.D.; Wertz, M.H.; Gonçalves, I.D.C.G.; Akten, B.; Dover, K.Z.; Peters, M.; Riessland, M.; Neveu, P.; Wirth, B.; et al. SMN regulates axonal local translation via miR-183/mTOR pathway. Hum. Mol. Genet. 2014, 23, 6318–6331. [Google Scholar] [CrossRef] [PubMed]
- Ubhi, K.; Rockenstein, E.; Kragh, C.; Inglis, C.; Spencer, B.; Michael, S.; Mante, M.; Adame, A.; Galasko, D.; Masliah, E. Widespread microRNA dysregulation in multiple system atrophy—Disease-related alteration in miR-96. Eur. J. Neurosci. 2014, 39, 1026–1041. [Google Scholar] [CrossRef] [PubMed]
- Davari, M.; Soheili, Z.-S.; Samiei, S.; Sharifi, Z.; Pirmardan, E.R. Overexpression of miR-183/-96/-182 triggers neuronal cell fate in human retinal pigment epithelial (hRPE) cells in culture. Biochem. Biophys. Res. Commun. 2016, 483, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Woldemichael, B.T.; Jawaid, A.; Kremer, E.A.; Gaur, N.; Krol, J.; Marchais, A.; Mansuy, I.M. The microRNA cluster miR-183/96/182 contributes to long-term memory in a protein phosphatase 1-dependent manner. Nat. Commun. 2016, 7, 12594. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Li, L.; Zhang, M.-D.; Bengtsson Gonzales, C.; Parisien, M.; Belfer, I.; Usoskin, D.; Abdo, H.; Furlan, A.; Häring, M.; et al. miR-183 cluster scales mechanical pain sensitivity by regulating basal and neuropathic pain genes. Science 2017, 356, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Weston, M.D.; Pierce, M.L.; Rocha-Sanchez, S.; Beisel, K.W.; Soukup, G.A. MicroRNA gene expression in the mouse inner ear. Brain Res. 2006, 1111, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, M.; Mito, J.K.; Lee, C.-L.; Zhang, M.; Li, Z.; Dodd, R.D.; Cason, D.; Luo, L.; Ma, Y.; Van Mater, D.; et al. MicroRNA-182 drives metastasis of primary sarcomas by targeting multiple genes. J. Clin. Investig. 2016, 126, 1606. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-B.; Chen, P.-H.; Hsu, T.; Fu, T.-F.; Su, W.-C.; Liaw, H.; Chang, W.-C.; Hung, J.-J. Sp1-mediated microRNA-182 expression regulates lung cancer progression. Oncotarget 2014, 5, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Howell, P.M.; Riker, A.I. Upregulation of miR-182 expression after epigenetic modulation of human melanoma cells. Ann. Surg. Oncol. 2013, 20, 1745–1752. [Google Scholar] [CrossRef] [PubMed]
- Dodd, R.D.; Sachdeva, M.; Mito, J.K.; Eward, W.C.; Brigman, B.E.; Ma, Y.; Dodd, L.; Kim, Y.; Lev, D.; Kirsch, D.G. Myogenic transcription factors regulate pro-metastatic miR-182. Oncogene 2015, 35, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, D.; Wang, X.; Yao, X.; Ye, C.; Zhang, S.; Wang, H.; Chang, C.; Xia, H.; Wang, Y.-C.; et al. Hypoxia-inducible miR-182 enhances HIF1α signaling via targeting PHD2 and FIH1 in prostate cancer. Sci. Rep. 2015, 5, 12495. [Google Scholar] [CrossRef] [PubMed]
- Lagos, D.; Pollara, G.; Henderson, S.; Gratrix, F.; Fabani, M.; Milne, R.S.B.; Gotch, F.; Boshoff, C. miR-132 regulates antiviral innate immunity through suppression of the p300 transcriptional co-activator. Nat. Cell Biol. 2010, 12, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Honess, R.W.; Roizman, B. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 1974, 14, 8–19. [Google Scholar] [PubMed]
- Boutell, C.; Everett, R.D. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J. Gen. Virol. 2013, 94, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Dixon, R.A.; Schaffer, P.A. Fine-structure mapping and functional analysis of temperature-sensitive mutants in the gene encoding the herpes simplex virus type 1 immediate early protein VP175. J. Virol. 1980, 36, 189–203. [Google Scholar] [PubMed]
- O’Hare, P.; Hayward, G.S. Three trans-acting regulatory proteins of herpes simplex virus modulate immediate-early gene expression in a pathway involving positive and negative feedback regulation. J. Virol. 1985, 56, 723–733. [Google Scholar] [PubMed]
- Sandri-Goldin, R.M. The many roles of the highly interactive HSV protein ICP27, a key regulator of infection. Future Microbiol. 2011, 6, 1261–1277. [Google Scholar] [CrossRef] [PubMed]
- Mullen, M.A.; Ciufo, D.M.; Hayward, G.S. Mapping of intracellular localization domains and evidence for colocalization interactions between the IE110 and IE175 nuclear transactivator proteins of herpes simplex virus. J. Virol. 1994, 68, 3250–3266. [Google Scholar] [PubMed]
- Lopez, P.; Van Sant, C.; Roizman, B. Requirements for the nuclear-cytoplasmic translocation of infected-cell protein 0 of herpes simplex virus 1. J. Virol. 2001, 75, 3832–3840. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D. Analysis of the functional domains of herpes simplex virus type 1 immediate-early polypeptide Vmw110. J. Mol. Biol. 1988, 202, 87–96. [Google Scholar] [CrossRef]
- Knipe, D.M.; Smith, J.L. A mutant herpesvirus protein leads to a block in nuclear localization of other viral proteins. Mol. Cell Biol. 1986, 6, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; DeLuca, N.A.; Schaffer, P.A. Overexpression of the herpes simplex virus type 1 immediate-early regulatory protein, ICP27, is responsible for the aberrant localization of ICP0 and mutant forms of ICP4 in ICP4 mutant virus-infected cells. J. Virol. 1996, 70, 5346–5356. [Google Scholar] [PubMed]
- Zhu, Z.; Cai, W.; Schaffer, P.A. Cooperativity among herpes simplex virus type 1 immediate-early regulatory proteins: ICP4 and ICP27 affect the intracellular localization of ICP0. J. Virol. 1994, 68, 3027–3040. [Google Scholar] [PubMed]
- Everett, R.D. Construction and characterization of herpes simplex virus type 1 mutants with defined lesions in immediate early gene 1. J. Gen. Virol. 1989, 70 Pt 5, 1185–1202. [Google Scholar] [CrossRef] [PubMed]
- Boutell, C.; Sadis, S.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 2002, 76, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Vanni, E.; Gatherer, D.; Tong, L.; Everett, R.D.; Boutell, C. Functional characterization of residues required for the herpes simplex virus 1 E3 ubiquitin ligase ICP0 to interact with the cellular E2 ubiquitin-conjugating enzyme UBE2D1 (UbcH5a). J. Virol. 2012, 86, 6323–6333. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Meredith, M.; Orr, A. The ability of herpes simplex virus type 1 immediate-early protein Vmw110 to bind to a ubiquitin-specific protease contributes to its roles in the activation of gene expression and stimulation of virus replication. J. Virol. 1999, 73, 417–426. [Google Scholar] [PubMed]
- Kummer, M.; Turza, N.M.; Mühl-Zürbes, P.; Lechmann, M.; Boutell, C.; Coffin, R.S.; Everett, R.D.; Steinkasserer, A.; Prechtel, A.T. Herpes simplex virus type 1 induces CD83 degradation in mature dendritic cells with immediate-early kinetics via the cellular proteasome. J. Virol. 2007, 81, 6326–6338. [Google Scholar] [CrossRef] [PubMed]
- Lomonte, P.; Sullivan, K.F.; Everett, R.D. Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J. Biol. Chem. 2001, 276, 5829–5835. [Google Scholar] [CrossRef] [PubMed]
- Cuchet-Lourenço, D.; Vanni, E.; Glass, M.; Orr, A.; Everett, R.D. Herpes simplex virus 1 ubiquitin ligase ICP0 interacts with PML isoform I and induces its SUMO-independent degradation. J. Virol. 2012, 86, 11209–11222. [Google Scholar] [CrossRef] [PubMed]
- Chaurushiya, M.S.; Lilley, C.E.; Aslanian, A.; Meisenhelder, J.; Scott, D.C.; Landry, S.; Ticau, S.; Boutell, C.; Yates, J.R.; Schulman, B.A.; et al. Viral E3 ubiquitin ligase-mediated degradation of a cellular E3: Viral mimicry of a cellular phosphorylation mark targets the RNF8 FHA domain. Mol. Cell 2012, 46, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Tie, F.; Banerjee, R.; Saiakhova, A.R.; Howard, B.; Monteith, K.E.; Scacheri, P.C.; Cosgrove, M.S.; Harte, P.J. Trithorax monomethylates histone H3K4 and interacts directly with CBP to promote H3K27 acetylation and antagonize Polycomb silencing. Development 2014, 141, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.-H.; Sun, Y.-M.; Chang, W.-C.; Chiang-Hsieh, P.-Y.; Lee, T.-Y.; Tsai, W.-C.; Horng, J.-T.; Tsou, A.-P.; Huang, H.-D. Identifying transcriptional start sites of human microRNAs based on high-throughput sequencing data. Nucleic Acids Res. 2011, 39, 9345–9356. [Google Scholar] [CrossRef] [PubMed]
- Ozsolak, F.; Poling, L.L.; Wang, Z.; Liu, H.; Liu, X.S.; Roeder, R.G.; Zhang, X.; Song, J.S.; Fisher, D.E. Chromatin structure analyses identify miRNA promoters. Genes Dev. 2008, 22, 3172–3183. [Google Scholar] [CrossRef] [PubMed]
- Sloan, E.; Tatham, M.H.; Groslambert, M.; Glass, M.; Orr, A.; Hay, R.T.; Everett, R.D. Analysis of the SUMO2 Proteome during HSV-1 Infection. PLoS Pathog. 2015, 11, e1005059. [Google Scholar] [CrossRef] [PubMed]
- Rivas, H.G.; Schmaling, S.K.; Gaglia, M.M. Shutoff of host gene expression in influenza a virus and herpesviruses: Similar mechanisms and common themes. Viruses 2016, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Mohr, I.; Sonenberg, N. Host translation at the nexus of infection and immunity. Cell Host Microbe 2012, 12, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Kim, E.T.; Vladimirova, O.; Dheekollu, J.; Wang, Z.; Newhart, A.; Liu, D.; Myers, J.L.; Hensley, S.E.; Moffat, J.; et al. HSV-1 remodels host telomeres to facilitate viral replication. Cell Rep. 2014, 9, 2263–2278. [Google Scholar] [CrossRef] [PubMed]
- Majer, A.; Caligiuri, K.A.; Gale, K.K.; Niu, Y.; Phillipson, C.S.; Booth, T.F.; Booth, S.A. Induction of multiple miR-200/182 members in the brains of mice are associated with acute herpes simplex virus 1 encephalitis. PLoS ONE 2017, 12, e0169081. [Google Scholar]
- Cai, W.; Astor, T.L.; Liptak, L.M.; Cho, C.; Coen, D.M.; Schaffer, P.A. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J. Virol. 1993, 67, 7501–7512. [Google Scholar] [PubMed]
- Halford, W.P.; Schaffer, P.A. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J. Virol. 2001, 75, 3240–3249. [Google Scholar] [CrossRef] [PubMed]
- Leib, D.A.; Coen, D.M.; Bogard, C.L.; Hicks, K.A.; Yager, D.R.; Knipe, D.M.; Tyler, K.L.; Schaffer, P.A. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J. Virol. 1989, 63, 759–768. [Google Scholar] [PubMed]
- Thompson, R.L.; Sawtell, N.M. Evidence that the herpes simplex virus type 1 ICP0 protein does not initiate reactivation from latency in vivo. J. Virol. 2006, 80, 10919–10930. [Google Scholar] [CrossRef] [PubMed]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. USA 2012, 109, E3008–E3017. [Google Scholar] [CrossRef] [PubMed]
- Paladino, P.; Collins, S.E.; Mossman, K.L. Cellular localization of the herpes simplex virus ICP0 protein dictates its ability to block IRF3-mediated innate immune responses. PLoS ONE 2010, 5, e10428. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Raja, P.; Knipe, D.M. Herpesviral ICP0 protein promotes two waves of heterochromatin removal on an early viral promoter during lytic infection. MBio 2016, 7, e02007–e02015. [Google Scholar] [CrossRef] [PubMed]
- Raja, P.; Lee, J.S.; Pan, D.; Pesola, J.M.; Coen, D.M.; Knipe, D.M. A Herpesviral lytic protein regulates the structure of latent viral chromatin. MBio 2016, 7, e00633-16. [Google Scholar] [CrossRef] [PubMed]
- Boutell, C.; Cuchet-Lourenço, D.; Vanni, E.; Orr, A.; Glass, M.; McFarlane, S.; Everett, R.D. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog. 2011, 7, e1002245. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Freemont, P.; Saitoh, H.; Dasso, M.; Orr, A.; Kathoria, M.; Parkinson, J. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 1998, 72, 6581–6591. [Google Scholar] [PubMed]
- Chen, S.-H.; Lee, L.Y.; Garber, D.A.; Schaffer, P.A.; Knipe, D.M.; Coen, D.M. Neither LAT nor open reading frame P mutations increase expression of spliced or intron-containing ICP0 transcripts in mouse ganglia latently infected with herpes simplex virus. J. Virol. 2002, 76, 4764–4772. [Google Scholar] [CrossRef] [PubMed]
- Maillet, S.; Naas, T.; Crepin, S.; Roque-Afonso, A.-M.; Lafay, F.; Efstathiou, S.; Labetoulle, M. Herpes simplex virus type 1 latently infected neurons differentially express latency-associated and ICP0 transcripts. J. Virol. 2006, 80, 9310–9321. [Google Scholar] [CrossRef] [PubMed]
- Proença, J.T.; Coleman, H.M.; Connor, V.; Winton, D.J.; Efstathiou, S. A historical analysis of herpes simplex virus promoter activation in vivo reveals distinct populations of latently infected neurones. J. Gen. Virol. 2008, 89, 2965–2974. [Google Scholar] [CrossRef] [PubMed]
- Proença, J.T.; Nelson, D.; Nicoll, M.P.; Connor, V.; Efstathiou, S. Analyses of HSV-1 latency and reactivation at the single cell level using fluorescent reporter mice. J. Gen. Virol. 2015, 97, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Oba, S.; Mizutani, T.; Suzuki, E.; Nishimatsu, H.; Takahashi, M.; Ogawa, Y.; Kimura, K.; Hirata, Y.; Fujita, T. A useful method of identifying of miRNAs which can downregulate Zeb-2. BMC Res. Notes 2013, 6, 470. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Seo, D.; Kim, D.; Hong, Y.; Chang, H.; Baek, D.; Kim, V.N.; Lee, S.; Ahn, K. Temporal landscape of microRNA-mediated host-virus crosstalk during productive human cytomegalovirus infection. Cell Host Microbe 2015, 17, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Stark, T.J.; Arnold, J.D.; Spector, D.H.; Yeo, G.W. High-resolution profiling and analysis of viral and host small RNAs during human cytomegalovirus infection. J. Virol. 2012, 86, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Everett, R.D. Analysis of the functional interchange between the IE1 and pp71 proteins of HCMV and ICP0 of HSV-1. J. Virol. 2014, 89, 3062–3075. [Google Scholar] [CrossRef] [PubMed]
- Oussaief, L.; Fendri, A.; Chane-Woon-Ming, B.; Poirey, R.; Delecluse, H.-J.; Joab, I.; Pfeffer, S. Modulation of the microRNA cluster miR-183-96-182 expression by the Epstein–Barr virus latent membrane protein 1. J. Virol. 2015, 89, 12178–12188. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Gaya, A.; Martinez, A.; Urbano-Ispizua, A.; Pons, A.; Balagué, O.; Gel, B.; Abrisqueta, P.; Lopez-Guillermo, A.; Artells, R.; et al. MicroRNA expression profiling in classic Hodgkin lymphoma. Blood 2008, 111, 2825–2832. [Google Scholar] [CrossRef] [PubMed]
- Hutzinger, R.; Mrázek, J.; Vorwerk, S.; Hüttenhofer, A. NcRNA-microchip analysis: A novel approach to identify differential expression of noncoding RNAs. RNA Biol. 2010, 7, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.-M.; Lyu, X.-M.; Luo, W.-R.; Cui, X.-F.; Ye, Y.-F.; Yuan, C.-C.; Peng, Q.-X.; Wu, D.-H.; Liu, T.-F.; Wang, E.; et al. EBV-miR-BART7-3p promotes the EMT and metastasis of nasopharyngeal carcinoma cells by suppressing the tumor suppressor PTEN. Oncogene 2015, 34, 2156–2166. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tian, W.-D.; Xu, X.; Nie, B.; Lu, J.; Liu, X.; Zhang, B.; Dong, Q.; Sunwoo, J.B.; Li, G.; et al. Epstein–Barr virus nuclear antigen 1 (EBNA1) protein induction of epithelial-mesenchymal transition in nasopharyngeal carcinoma cells. Cancer 2014, 120, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Ellis, A.L.; Wang, Z.; Yu, X.; Mertz, J.E. Either ZEB1 or ZEB2/SIP1 can play a central role in regulating the Epstein–Barr virus latent-lytic switch in a cell-type-specific manner. J. Virol. 2010, 84, 6139–6152. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, Z.; Mertz, J.E. ZEB1 regulates the latent-lytic switch in infection by Epstein–Barr virus. PLoS Pathog. 2007, 3, e194. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.-H.; Kraus, R.J.; Dickerson, S.J.; Lim, H.J.; Jones, R.J.; Yu, X.; Mertz, J.E.; Kenney, S.C. ZEB1 and c-Jun levels contribute to the establishment of highly lytic Epstein–Barr virus infection in gastric AGS cells. J. Virol. 2007, 81, 10113–10122. [Google Scholar] [CrossRef] [PubMed]









© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lutz, G.; Jurak, I.; Kim, E.T.; Kim, J.Y.; Hackenberg, M.; Leader, A.; Stoller, M.L.; Fekete, D.M.; Weitzman, M.D.; Coen, D.M.; et al. Viral Ubiquitin Ligase Stimulates Selective Host MicroRNA Expression by Targeting ZEB Transcriptional Repressors. Viruses 2017, 9, 210. https://doi.org/10.3390/v9080210
Lutz G, Jurak I, Kim ET, Kim JY, Hackenberg M, Leader A, Stoller ML, Fekete DM, Weitzman MD, Coen DM, et al. Viral Ubiquitin Ligase Stimulates Selective Host MicroRNA Expression by Targeting ZEB Transcriptional Repressors. Viruses. 2017; 9(8):210. https://doi.org/10.3390/v9080210
Chicago/Turabian StyleLutz, Gabriel, Igor Jurak, Eui Tae Kim, Ju Youn Kim, Michael Hackenberg, Andrew Leader, Michelle L. Stoller, Donna M. Fekete, Matthew D. Weitzman, Donald M. Coen, and et al. 2017. "Viral Ubiquitin Ligase Stimulates Selective Host MicroRNA Expression by Targeting ZEB Transcriptional Repressors" Viruses 9, no. 8: 210. https://doi.org/10.3390/v9080210
APA StyleLutz, G., Jurak, I., Kim, E. T., Kim, J. Y., Hackenberg, M., Leader, A., Stoller, M. L., Fekete, D. M., Weitzman, M. D., Coen, D. M., & Wilson, A. C. (2017). Viral Ubiquitin Ligase Stimulates Selective Host MicroRNA Expression by Targeting ZEB Transcriptional Repressors. Viruses, 9(8), 210. https://doi.org/10.3390/v9080210