
Influenza Virus Infection, Interferon Response, Viral Counter-Response, and Apoptosis



Abstract
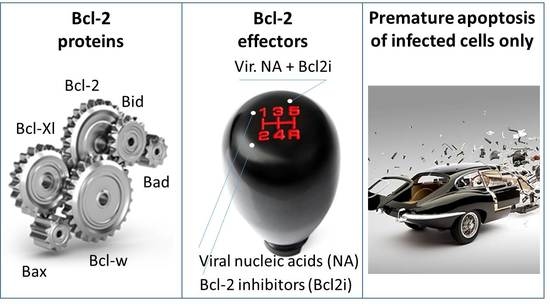
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
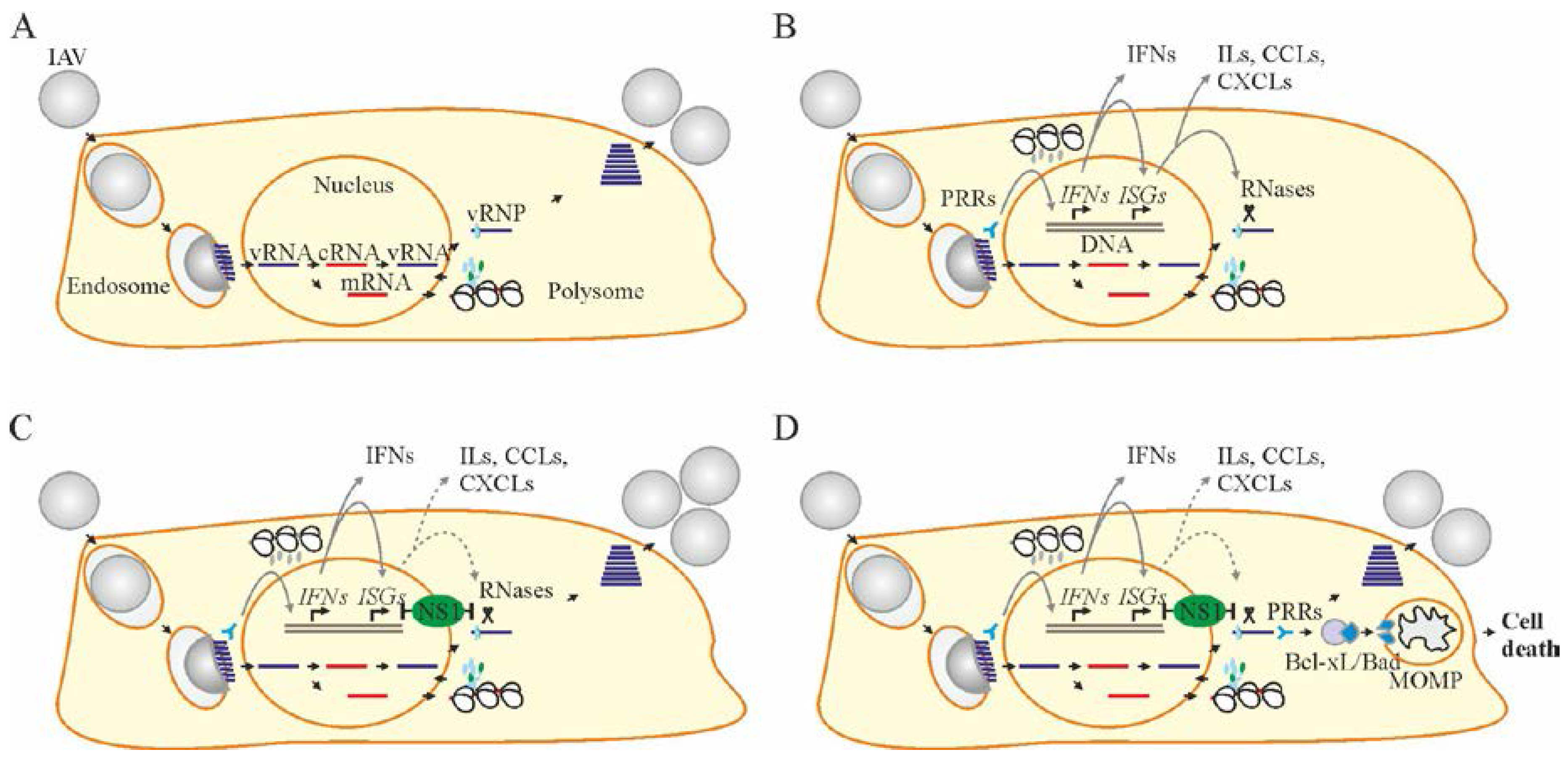
2. Influenza A Virus Structure and Replication Cycle
3. Cellular Factors Essential for Influenza A Virus Replication
4. Cellular Factors that Limit Virus Replication and Spread
5. Apoptosis Is a Cellular Process That Restricts Virus Replication and Spread
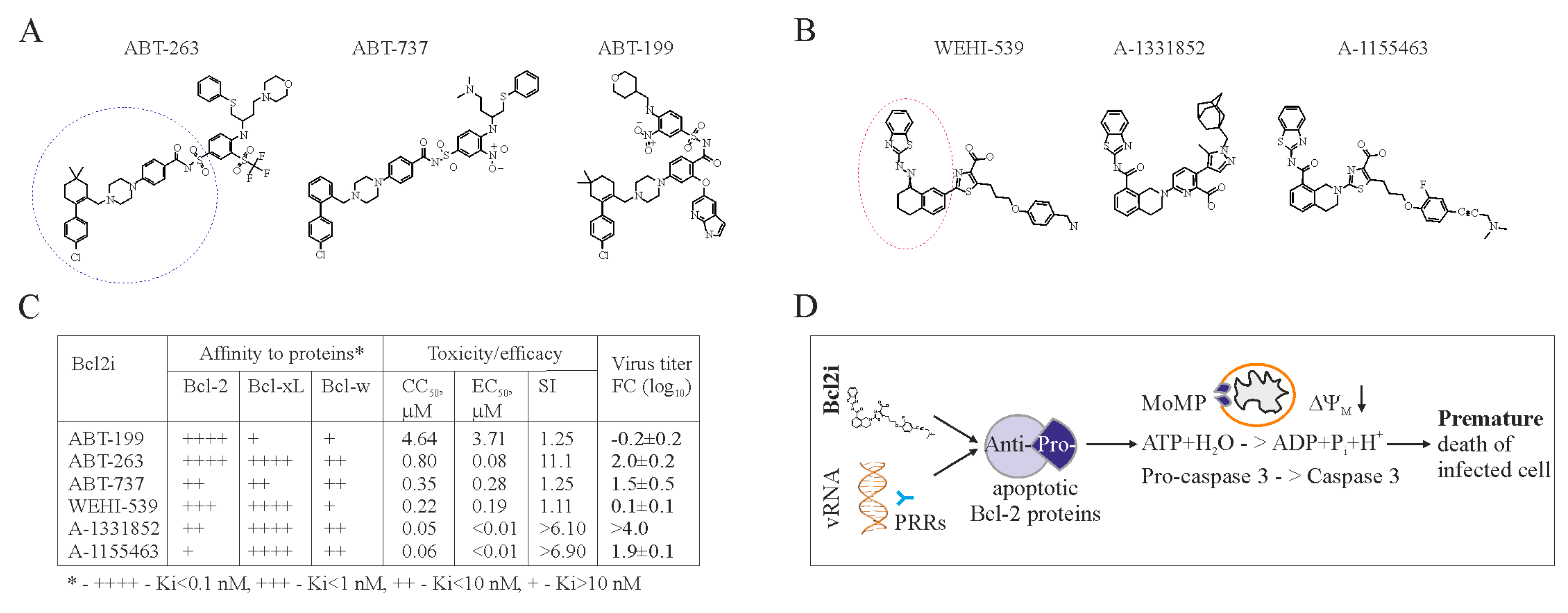
6. Apoptosis-Inducing Small Molecules
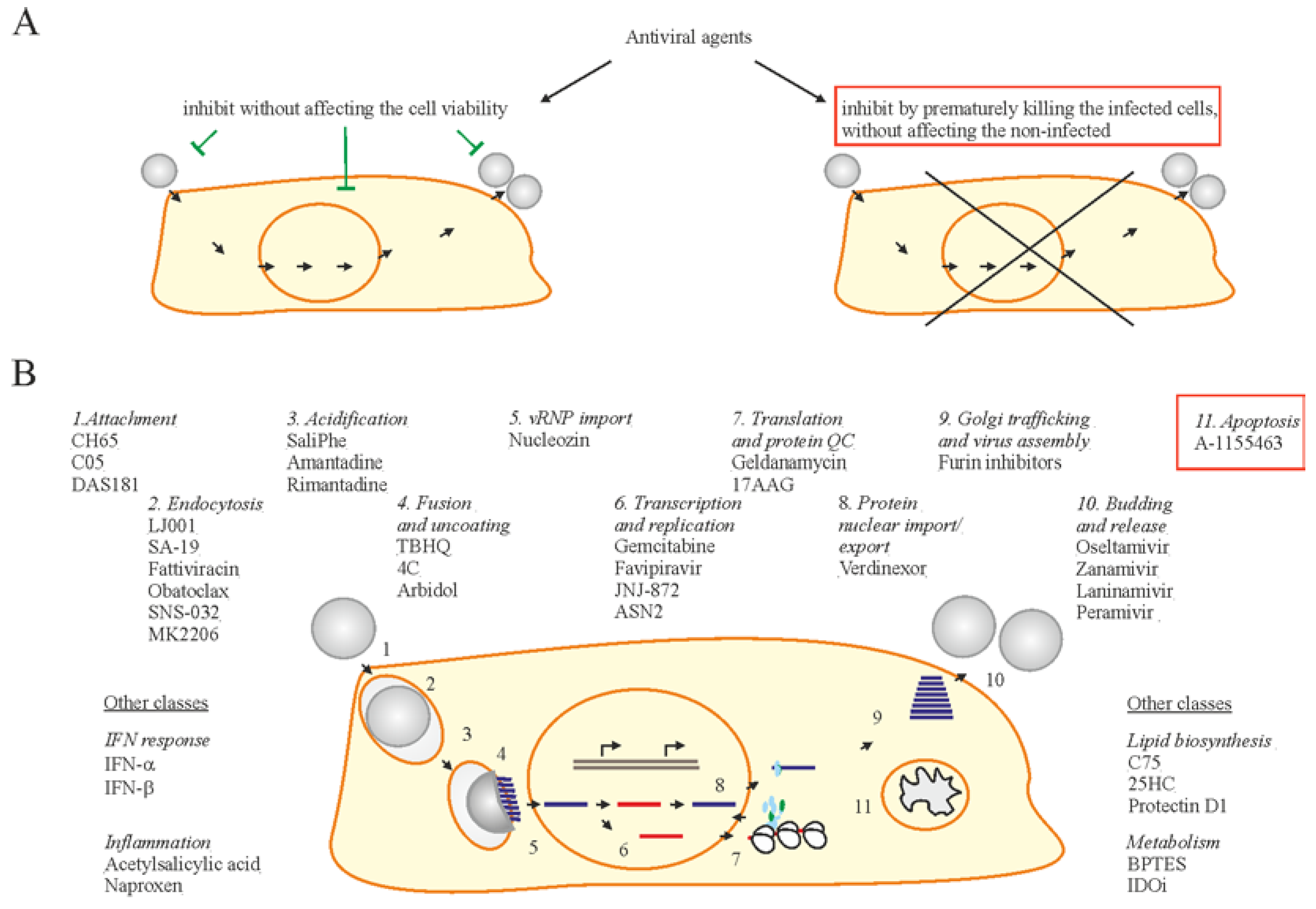
7. Accelerating Apoptosis of Infected Cells: A Novel Antiviral Strategy
8. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- WHO Influenza (Seasonal). Available online: http://wwwwhoint/mediacentre/factsheets/fs211/en/ (accessed on 8 July 2017).
- Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 386, 743–800. [Google Scholar]
- Lafond, K.E.; Nair, H.; Rasooly, M.H.; Valente, F.; Booy, R.; Rahman, M.; Kitsutani, P.; Yu, H.; Guzman, G.; Coulibaly, D.; et al. Global Role and Burden of Influenza in Pediatric Respiratory Hospitalizations, 1982–2012: A Systematic Analysis. PLoS Med. 2016, 13, e1001977. [Google Scholar] [CrossRef] [PubMed]
- Horimoto, T.; Kawaoka, Y. Influenza: Lessons from past pandemics, warnings from current incidents. Nat. Rev. Microbiol. 2005, 3, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Morens, D.M. 1918 Influenza: The mother of all pandemics. Emerg. Infect. Dis. 2006, 12, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Fineberg, H.V. Pandemic preparedness and response—Lessons from the H1N1 influenza of 2009. N. Engl. J. Med. 2014, 370, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- CDC Influenza Antiviral Medications: Summary for Clinicians. Available online: https://wwwcdcgov/flu/professionals/antivirals/summary-clinicianshtm (accessed on 8 July 2017).
- Spanakis, N.; Pitiriga, V.; Gennimata, V.; Tsakris, A. A review of neuraminidase inhibitor susceptibility in influenza strains. Expert Rev. Anti-Infect. Ther. 2014, 12, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Galvin, H.D.; Haw, T.Y.; Nutsford, A.N.; Husain, M. Drug resistance in influenza A virus: The epidemiology and management. Infect. Drug Resist. 2017, 10, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26 (Suppl. 4), D49–D53. [Google Scholar] [CrossRef] [PubMed]
- Pinsent, A.; Fraser, C.; Ferguson, N.M.; Riley, S. A systematic review of reported reassortant viral lineages of influenza A. BMC Infect. Dis. 2016, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Werner, J.L.; Steele, C. Innate receptors and cellular defense against pulmonary infections. J. Immunol. 2014, 193, 3842–3850. [Google Scholar] [CrossRef] [PubMed]
- Mansour, D.E.; El-Shazly, A.A.; Elawamry, A.I.; Ismail, A.T. Comparison of ocular findings in patients with H1N1 influenza infection versus patients receiving influenza vaccine during a pandemic. Ophthalmic Res. 2012, 48, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Geiler, J.; Klassert, D.; Doerr, H.W.; Cinatl, J., Jr. Infection of human retinal pigment epithelial cells with influenza A viruses. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5419–5425. [Google Scholar] [CrossRef] [PubMed]
- Edinger, T.O.; Pohl, M.O.; Stertz, S. Entry of influenza A virus: Host factors and antiviral targets. J. Gen. Virol. 2014, 95, 263–277. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Whittaker, G.R. Fusion of Enveloped Viruses in Endosomes. Traffic 2016, 17, 593–614. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, I.; Miyake, Y.; Nobs, S.P.; Schneider, C.; Horvath, P.; Kopf, M.; Matthias, P.; Helenius, A.; Yamauchi, Y. Influenza A virus uses the aggresome processing machinery for host cell entry. Science 2014, 346, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Pumroy, R.A.; Ke, S.; Hart, D.J.; Zachariae, U.; Cingolani, G. Molecular determinants for nuclear import of influenza A PB2 by importin alpha isoforms 3 and 7. Structure 2015, 23, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Te Velthuis, A.J.; Fodor, E. Influenza virus RNA polymerase: Insights into the mechanisms of viral RNA synthesis. Nat. Rev. Microbiol. 2016, 14, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Reguera, J.; Gerlach, P.; Cusack, S. Towards a structural understanding of RNA synthesis by negative strand RNA viral polymerases. Curr. Opin. Struct. Biol. 2016, 36, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Lakdawala, S.S.; Fodor, E.; Subbarao, K. Moving On Out: Transport and Packaging of Influenza Viral RNA into Virions. Annu. Rev. Virol. 2016, 3, 411–427. [Google Scholar] [CrossRef] [PubMed]
- Belanov, S.S.; Bychkov, D.; Benner, C.; Ripatti, S.; Ojala, T.; Kankainen, M.; Kai Lee, H.; Wei-Tze Tang, J.; Kainov, D.E. Genome-Wide Analysis of Evolutionary Markers of Human Influenza A(H1N1)pdm09 and A(H3N2) Viruses May Guide Selection of Vaccine Strain Candidates. Genome Biol. Evol. 2015, 7, 3472–3483. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.H.; Kakkola, L.; Nagaraj, A.S.; Cheltsov, A.V.; Anastasina, M.; Kainov, D.E. Emerging cellular targets for influenza antiviral agents. Trends Pharmacol. Sci. 2012, 33, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Soderholm, S.; Fu, Y.; Gaelings, L.; Belanov, S.; Yetukuri, L.; Berlinkov, M.; Cheltsov, A.V.; Anders, S.; Aittokallio, T.; Nyman, T.A.; et al. Multi-Omics Studies towards Novel Modulators of Influenza A Virus-Host Interaction. Viruses 2016, 8, E269. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.L.; Stertz, S. Role of Host Genes in Influenza Virus Replication. Curr. Top. Microbiol. Immunol. 2017, 1–99. [Google Scholar] [CrossRef]
- Powell, J.D.; Waters, K.M. Influenza-Omics and the Host Response: Recent Advances and Future Prospects. Pathogens 2017, 6, E25. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.L. Current and novel antiviral strategies for influenza infection. Curr. Opin. Virol. 2016, 18, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kawaoka, Y. Influenza virus-host interactomes as a basis for antiviral drug development. Curr. Opin. Virol. 2015, 14, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Batra, J.; Lal, S.K. Interplay between influenza A virus and host factors: Targets for antiviral intervention. Arch. Virol. 2015, 160, 1877–1891. [Google Scholar] [CrossRef] [PubMed]
- Keener, A.B. Host with the most: Targeting host cells instead of pathogens to fight infectious disease. Nat. Med. 2017, 23, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Zheng, D.; Lee, Y.H.; Chan, T.K.; Kumar, Y.; Ho, W.E.; Chen, J.Z.; Tannenbaum, S.R.; Ong, C.N. Metabolomics Investigation Reveals Metabolite Mediators Associated with Acute Lung Injury and Repair in a Murine Model of Influenza Pneumonia. Sci. Rep. 2016, 6, 26076. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Munz, C. Autophagy and Mammalian Viruses: Roles in Immune Response, Viral Replication, and Beyond. Adv. Virus Res. 2016, 95, 149–195. [Google Scholar] [PubMed]
- Chlanda, P.; Zimmerberg, J. Protein-lipid interactions critical to replication of the influenza A virus. FEBS Lett. 2016, 590, 1940–1954. [Google Scholar] [CrossRef] [PubMed]
- Tisoncik-Go, J.; Gasper, D.J.; Kyle, J.E.; Eisfeld, A.J.; Selinger, C.; Hatta, M.; Morrison, J.; Korth, M.J.; Zink, E.M.; Kim, Y.M.; et al. Integrated Omics Analysis of Pathogenic Host Responses during Pandemic H1N1 Influenza Virus Infection: The Crucial Role of Lipid Metabolism. Cell Host Microbe 2016, 19, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Proia, R.L.; Hla, T. Emerging biology of sphingosine-1-phosphate: Its role in pathogenesis and therapy. J. Clin. Investig. 2015, 125, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Gaelings, L.; Soderholm, S.; Belanov, S.; Nandania, J.; Nyman, T.A.; Matikainen, S.; Anders, S.; Velagapudi, V.; Kainov, D.E. JNJ872 inhibits influenza A virus replication without altering cellular antiviral responses. Antivir. Res. 2016, 133, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Killip, M.J.; Fodor, E.; Randall, R.E. Influenza virus activation of the interferon system. Virus Res. 2015, 209, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowie, A.G.; Unterholzner, L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008, 8, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Sanchez, D.J.; Aliyari, R.; Lu, S.; Cheng, G. Systematic identification of type I and type II interferon-induced antiviral factors. Proc. Natl. Acad. Sci. USA 2012, 109, 4239–4244. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Soderholm, S.; Anastasina, M.; Islam, M.M.; Tynell, J.; Poranen, M.M.; Bamford, D.H.; Stenman, J.; Julkunen, I.; Sauliene, I.; De Brabander, J.K.; et al. Immuno-modulating properties of saliphenylhalamide, SNS-032, obatoclax, and gemcitabine. Antivir. Res. 2016, 126, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchjorsen, J.; Kristiansen, H.; Christiansen, R.; Rintahaka, J.; Matikainen, S.; Paludan, S.R.; Hartmann, R. Differential regulation of the OASL and OAS1 genes in response to viral infections. J. Interferon Cytokine Res. 2009, 29, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, S.; Wolff, T. Influenza A virus TRIMs the type I interferon response. Cell Host Microbe 2009, 5, 420–421. [Google Scholar] [CrossRef] [PubMed]
- Gaelings, L.; Soderholm, S.; Bugai, A.; Fu, Y.; Nandania, J.; Schepens, B.; Lorey, M.B.; Tynell, J.; Vande Ginste, L.; Le Goffic, R.; et al. Regulation of kynurenine biosynthesis during influenza virus infection. FEBS J. 2017, 284, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Dudek, S.E.; Nitzsche, K.; Ludwig, S.; Ehrhardt, C. Influenza A viruses suppress cyclooxygenase-2 expression by affecting its mRNA stability. Sci. Rep. 2016, 6, 27275. [Google Scholar] [CrossRef] [PubMed]
- Meunier, E.; Broz, P. Interferon-inducible GTPases in cell autonomous and innate immunity. Cell. Microbiol. 2016, 18, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Gold, E.S.; Diercks, A.H.; Podolsky, I.; Podyminogin, R.L.; Askovich, P.S.; Treuting, P.M.; Aderem, A. 25-Hydroxycholesterol acts as an amplifier of inflammatory signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 10666–10671. [Google Scholar] [CrossRef] [PubMed]
- Ayllon, J.; Garcia-Sastre, A. The NS1 protein: a multitasking virulence factor. Curr. Top. Microbiol. Immunol. 2015, 386, 73–107. [Google Scholar] [PubMed]
- Anastasina, M.; Schepens, B.; Soderholm, S.; Nyman, T.A.; Matikainen, S.; Saksela, K.; Saelens, X.; Kainov, D.E. The C terminus of NS1 protein of influenza A/WSN/1933(H1N1) virus modulates antiviral responses in infected human macrophages and mice. J. Gen. Virol. 2015, 96, 2086–2091. [Google Scholar] [CrossRef] [PubMed]
- Anastasina, M.; Le May, N.; Bugai, A.; Fu, Y.; Soderholm, S.; Gaelings, L.; Ohman, T.; Tynell, J.; Kyttanen, S.; Barboric, M.; et al. Influenza virus NS1 protein binds cellular DNA to block transcription of antiviral genes. Biochim. Biophys. Acta 2016, 1859, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Bornholdt, Z.A.; Prasad, B.V. X-ray structure of NS1 from a highly pathogenic H5N1 influenza virus. Nature 2008, 456, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Min, J.Y.; Li, S.; Sen, G.C.; Krug, R.M. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology 2007, 363, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Min, J.Y.; Krug, R.M. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2′-5′ oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 7100–7105. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Min, J.Y.; Krug, R.M.; Sen, G.C. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 2006, 349, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Baskin, C.R.; Bielefeldt-Ohmann, H.; Tumpey, T.M.; Sabourin, P.J.; Long, J.P.; Garcia-Sastre, A.; Tolnay, A.E.; Albrecht, R.; Pyles, J.A.; Olson, P.H.; et al. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc. Natl. Acad. Sci. USA 2009, 106, 3455–3460. [Google Scholar] [CrossRef] [PubMed]
- Kash, J.C.; Tumpey, T.M.; Proll, S.C.; Carter, V.; Perwitasari, O.; Thomas, M.J.; Basler, C.F.; Palese, P.; Taubenberger, J.K.; Garcia-Sastre, A.; et al. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 2006, 443, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective Bcl-2 family inhibitors. Nat. Rev. Drug Discov. 2007, 16, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Tran, A.T.; Cortens, J.P.; Du, Q.; Wilkins, J.A.; Coombs, K.M. Influenza virus induces apoptosis via BAD-mediated mitochondrial dysregulation. J. Virol. 2013, 87, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.E.; Datan, E.; Matassov, D.; Zakeri, Z.F. Lack of Bax prevents influenza A virus-induced apoptosis and causes diminished viral replication. J. Virol. 2009, 83, 8233–8246. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, V.S.; Olsen, C.W.; Dybdahl-Sissoko, N.; Evans, D. Apoptosis: A mechanism of cell killing by influenza A and B viruses. J. Virol. 1994, 68, 3667–3673. [Google Scholar] [PubMed]
- Kakkola, L.; Denisova, O.V.; Tynell, J.; Viiliainen, J.; Ysenbaert, T.; Matos, R.C.; Nagaraj, A.; Ohman, T.; Kuivanen, S.; Paavilainen, H.; et al. Anticancer compound ABT-263 accelerates apoptosis in virus-infected cells and imbalances cytokine production and lowers survival rates of infected mice. Cell Death Dis. 2013, 4, e742. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.D.; Mansell, A.; Tate, M.D. Hero turned villain: NLRP3 inflammasome-induced inflammation during influenza A virus infection. J. Leukoc. Biol. 2017, 101, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Herold, S.; Ludwig, S.; Pleschka, S.; Wolff, T. Apoptosis signaling in influenza virus propagation, innate host defense, and lung injury. J. Leukoc. Biol. 2012, 92, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, T.; Vijayalingam, S.; Kuppuswamy, M.; Chinnadurai, G. Interaction of cellular proteins with Bcl-xL targeted to cytoplasmic inclusion bodies in adenovirus infected cells. Virology 2015, 483, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.F.; Czabotar, P.E.; Smith, B.J.; Deshayes, K.; Zobel, K.; Colman, P.M.; Fairlie, W.D. Crystal structure of ABT-737 complexed with Bcl-xL: Implications for selectivity of antagonists of the Bcl-2 family. Cell Death Differ. 2007, 14, 1711–1713. [Google Scholar] [CrossRef] [PubMed]
- Kvansakul, M.; Hinds, M.G. The Bcl-2 family: Structures, interactions and targets for drug discovery. Apoptosis 2015, 20, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective Bcl-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, C.J.; Cory, S. ABT-199, a new Bcl-2-specific BH3 mimetic, has in vivo efficacy against aggressive Myc-driven mouse lymphomas without provoking thrombocytopenia. Blood 2013, 121, 2285–2288. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Huang, D. Targeting BCL2 with BH3 Mimetics: Basic Science and Clinical Application of Venetoclax in Chronic Lymphocytic Leukemia and Related B Cell Malignancies. Clin. Pharmacol. Ther. 2017, 101, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Lessene, G.; Czabotar, P.E.; Sleebs, B.E.; Zobel, K.; Lowes, K.N.; Adams, J.M.; Baell, J.B.; Colman, P.M.; Deshayes, K.; Fairbrother, W.J.; et al. Structure-guided design of a selective Bcl-xL inhibitor. Nat. Chem. Biol. 2013, 9, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Bulanova, D.; Ianevski1, A.; Bugai, A.; Akimov, E.; Kuivanen, S.; Paavilainen, H.; Kakkola, L.; Nandania, J.; Turunen, L.; Ohman, T.; et al. Antiviral properties of anticancer Bcl-2 inhibitors. Mol. Microbiol. 2017. submitted. [Google Scholar]
- Leverson, J.D.; Phillips, D.C.; Mitten, M.J.; Boghaert, E.R.; Diaz, D.; Tahir, S.K.; Belmont, L.D.; Nimmer, P.; Xiao, Y.; Ma, X.M.; et al. Exploiting selective Bcl-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med. 2015, 7, 279ra40. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.F.; Hasvold, L.; Wang, L.; Wang, X.; Petros, A.M.; Park, C.H.; Boghaert, E.R.; Catron, N.D.; Chen, J.; Colman, P.M.; et al. Discovery of a Potent and Selective Bcl-xL Inhibitor with in Vivo Activity. ACS Med. Chem. Lett. 2014, 5, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.P.; Sasse, F.; Bronstrup, M.; Diez, J.; Meyerhans, A. Antiviral drug discovery: broad-spectrum drugs from nature. Nat. Prod. Rep. 2015, 32, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Vigant, F.; Santos, N.C.; Lee, B. Broad-spectrum antivirals against viral fusion. Nat. Rev. Microbiol. 2015, 13, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Sun, Y.; Rao, Z. Current progress in antiviral strategies. Trends Pharmacol. Sci. 2014, 35, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Loregian, A.; Mercorelli, B.; Nannetti, G.; Compagnin, C.; Palu, G. Antiviral strategies against influenza virus: Towards new therapeutic approaches. Cell. Mol. Life Sci. 2014, 71, 3659–3683. [Google Scholar] [CrossRef] [PubMed]
- Vanderlinden, E.; Naesens, L. Emerging antiviral strategies to interfere with influenza virus entry. Med. Res. Rev. 2014, 34, 301–339. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Rao, M.; Wallis, R.S.; Kaufmann, S.H.; Rustomjee, R.; Mwaba, P.; Vilaplana, C.; Yeboah-Manu, D.; Chakaya, J.; Ippolito, G.; et al. Host-directed therapies for infectious diseases: current status, recent progress, and future prospects. Lancet Infect. Dis. 2016, 16, e47–e63. [Google Scholar] [CrossRef]
- McKimm-Breschkin, J.L.; Fry, A.M. Meeting report: 4th ISIRV antiviral group conference: Novel antiviral therapies for influenza and other respiratory viruses. Antivir. Res. 2016, 129, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Soderholm, S.; Kainov, D.E.; Ohman, T.; Denisova, O.V.; Schepens, B.; Kulesskiy, E.; Imanishi, S.Y.; Corthals, G.; Hintsanen, P.; Aittokallio, T.; et al. Phosphoproteomics to Characterize Host Response During Influenza A Virus Infection of Human Macrophages. Mol. Cell. Proteom. 2016, 15, 3203–3219. [Google Scholar] [CrossRef] [PubMed]
- Holthausen, D.J.; Lee, S.H.; Kumar, V.T.; Bouvier, N.M.; Krammer, F.; Ellebedy, A.H.; Wrammert, J.; Lowen, A.C.; George, S.; Pillai, M.R.; et al. An Amphibian Host Defense Peptide Is Virucidal for Human H1 Hemagglutinin-Bearing Influenza Viruses. Immunity 2017, 46, 587–595. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shim, J.M.; Kim, J.; Tenson, T.; Min, J.-Y.; Kainov, D.E. Influenza Virus Infection, Interferon Response, Viral Counter-Response, and Apoptosis. Viruses 2017, 9, 223. https://doi.org/10.3390/v9080223
Shim JM, Kim J, Tenson T, Min J-Y, Kainov DE. Influenza Virus Infection, Interferon Response, Viral Counter-Response, and Apoptosis. Viruses. 2017; 9(8):223. https://doi.org/10.3390/v9080223
Chicago/Turabian StyleShim, Jung Min, Jinhee Kim, Tanel Tenson, Ji-Young Min, and Denis E. Kainov. 2017. "Influenza Virus Infection, Interferon Response, Viral Counter-Response, and Apoptosis" Viruses 9, no. 8: 223. https://doi.org/10.3390/v9080223
APA StyleShim, J. M., Kim, J., Tenson, T., Min, J. -Y., & Kainov, D. E. (2017). Influenza Virus Infection, Interferon Response, Viral Counter-Response, and Apoptosis. Viruses, 9(8), 223. https://doi.org/10.3390/v9080223