Feasibility of Using Gluconolactone, Trehalose and Hydroxy-Propyl Gamma Cyclodextrin to Enhance Bendroflumethiazide Dissolution Using Lyophilisation and Physical Mixing Techniques




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
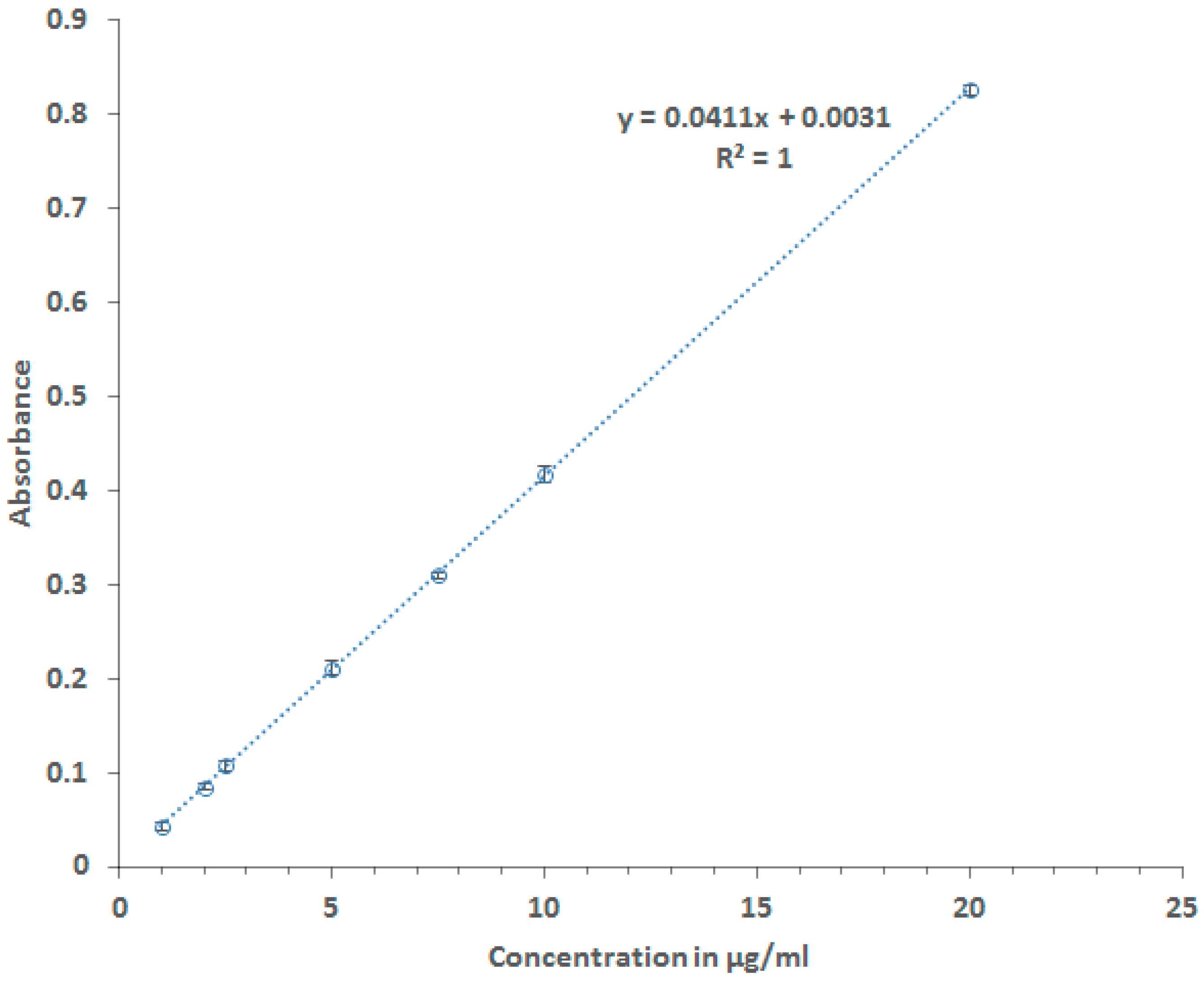
2.2.1. Calibration Curve
2.2.2. Physical Mixing
2.2.3. Lyophilisation-Freeze Drying
3. Characterisation of Physically Mixed and Lyophilised Bendroflumethiazide Samples
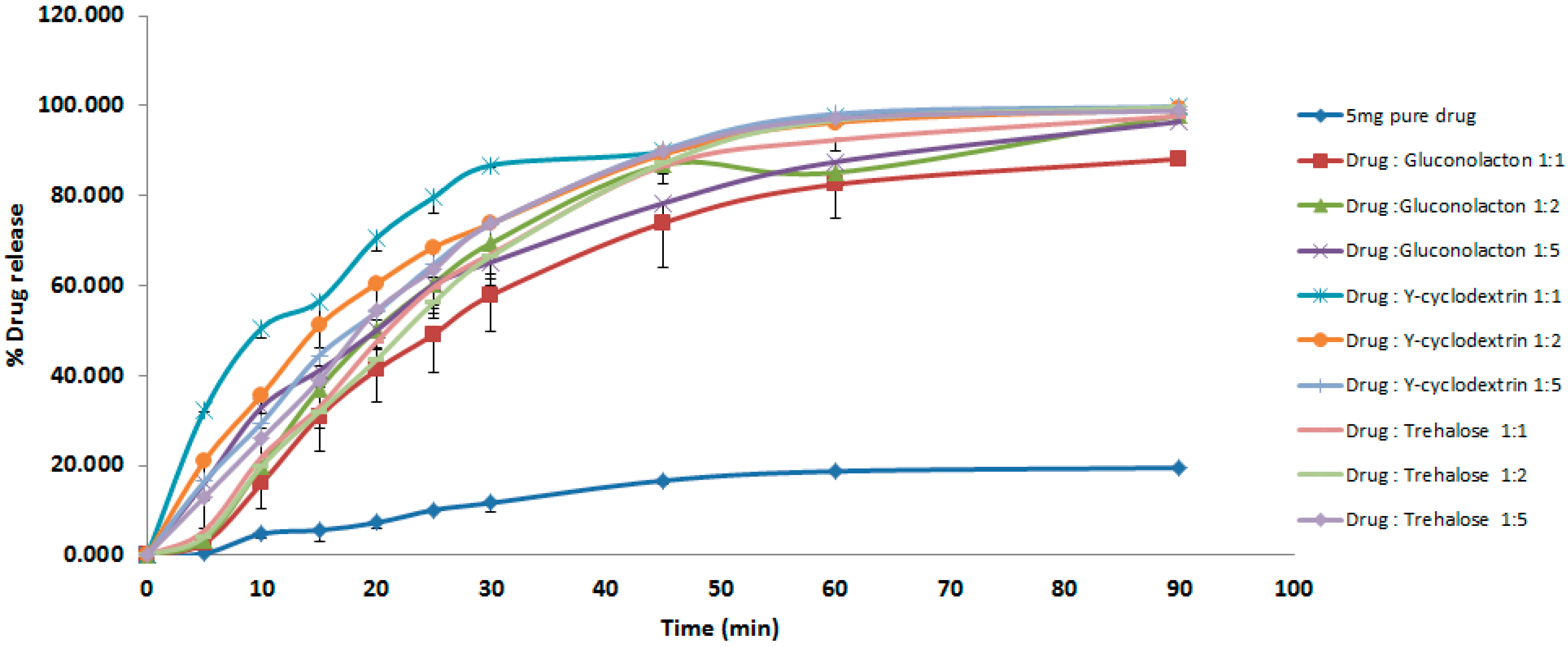
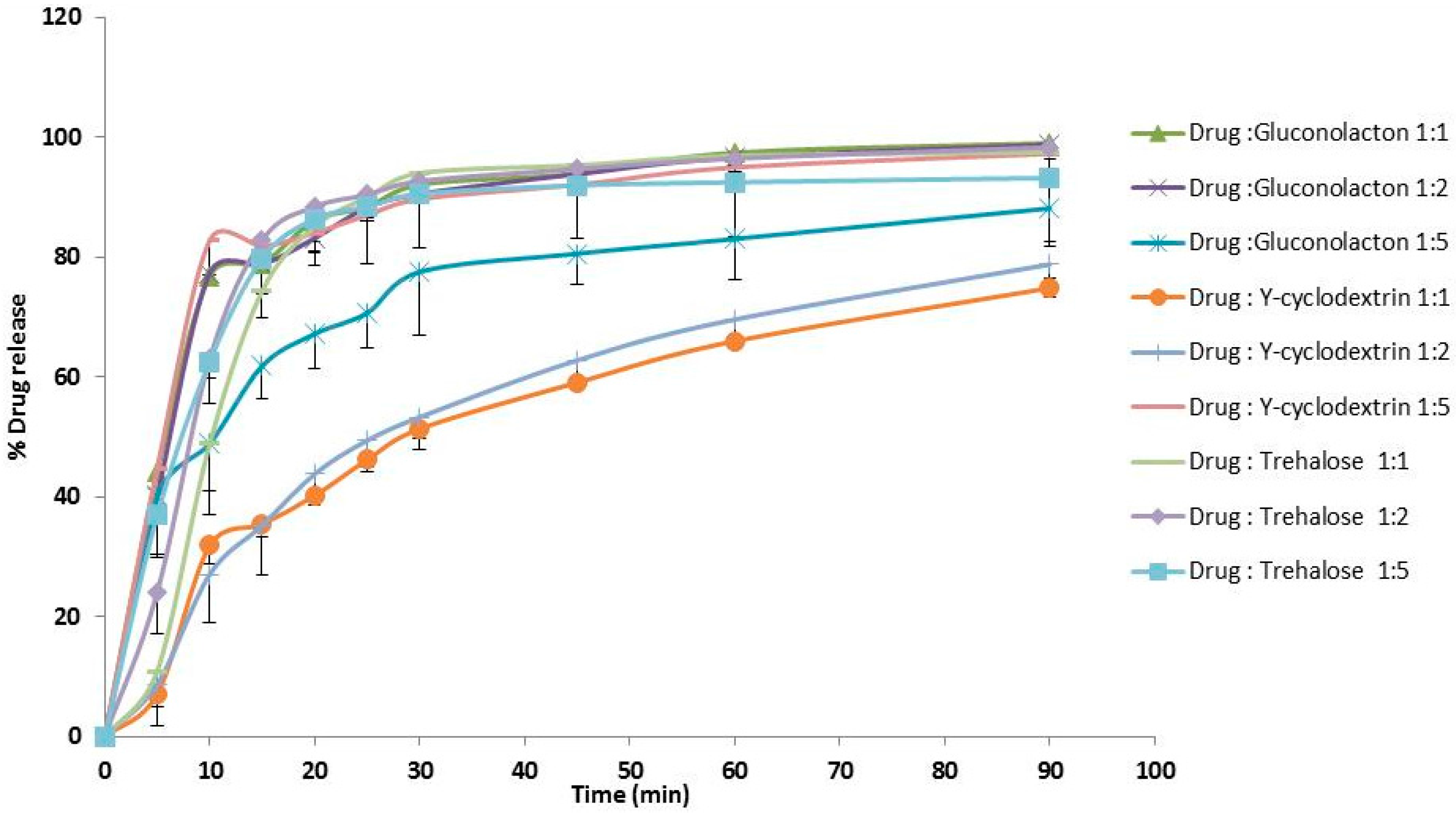
3.1. Dissolution Studies
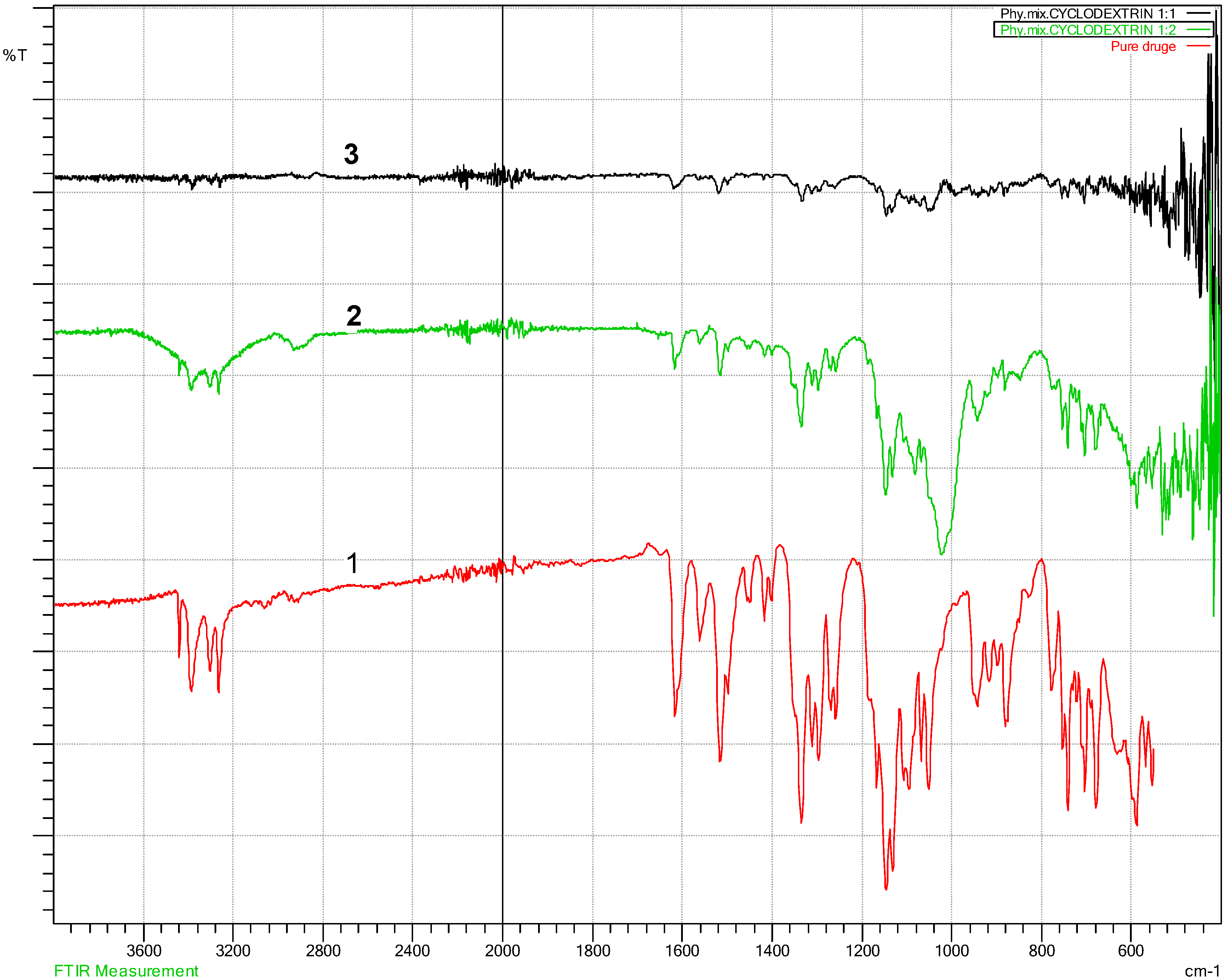
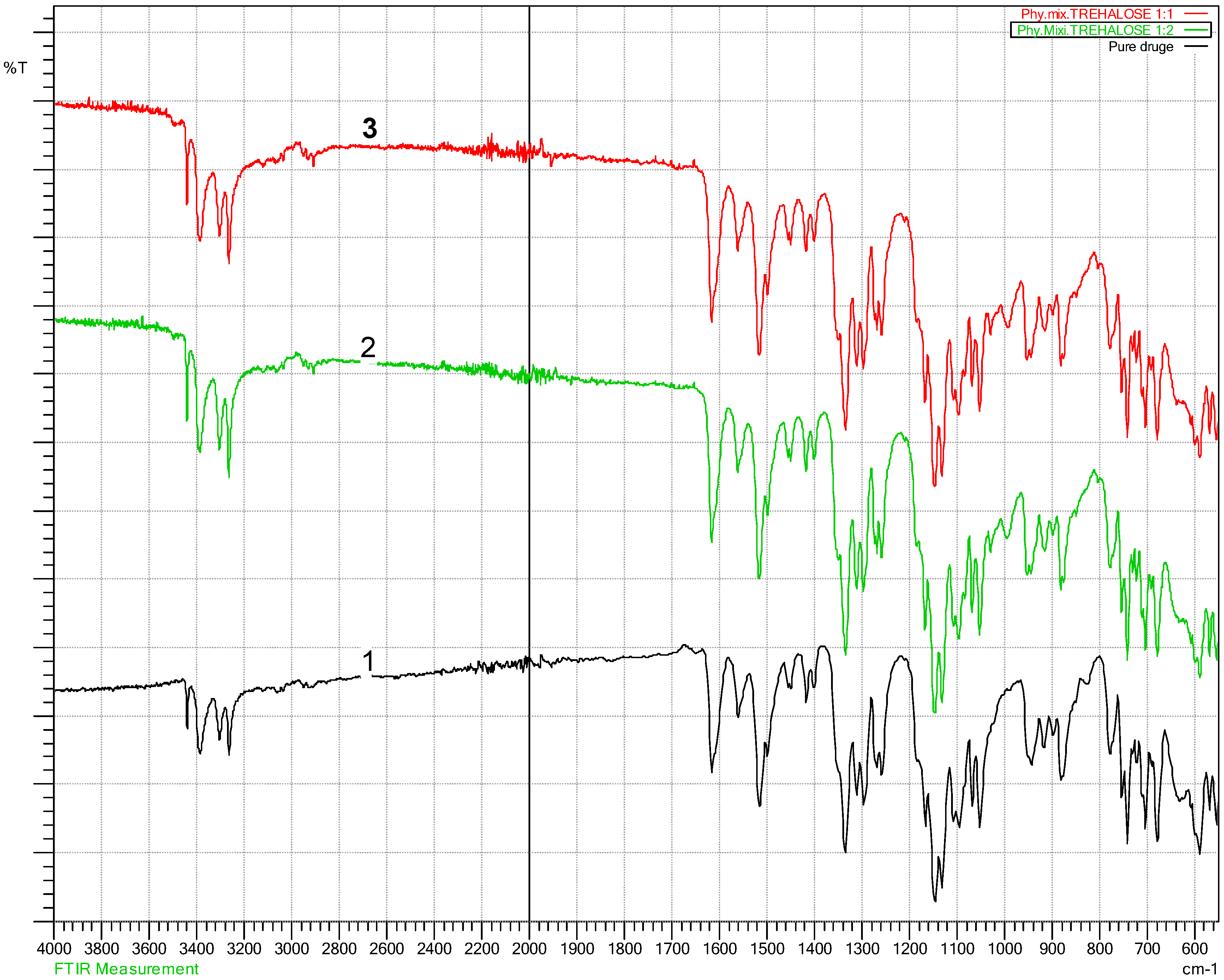
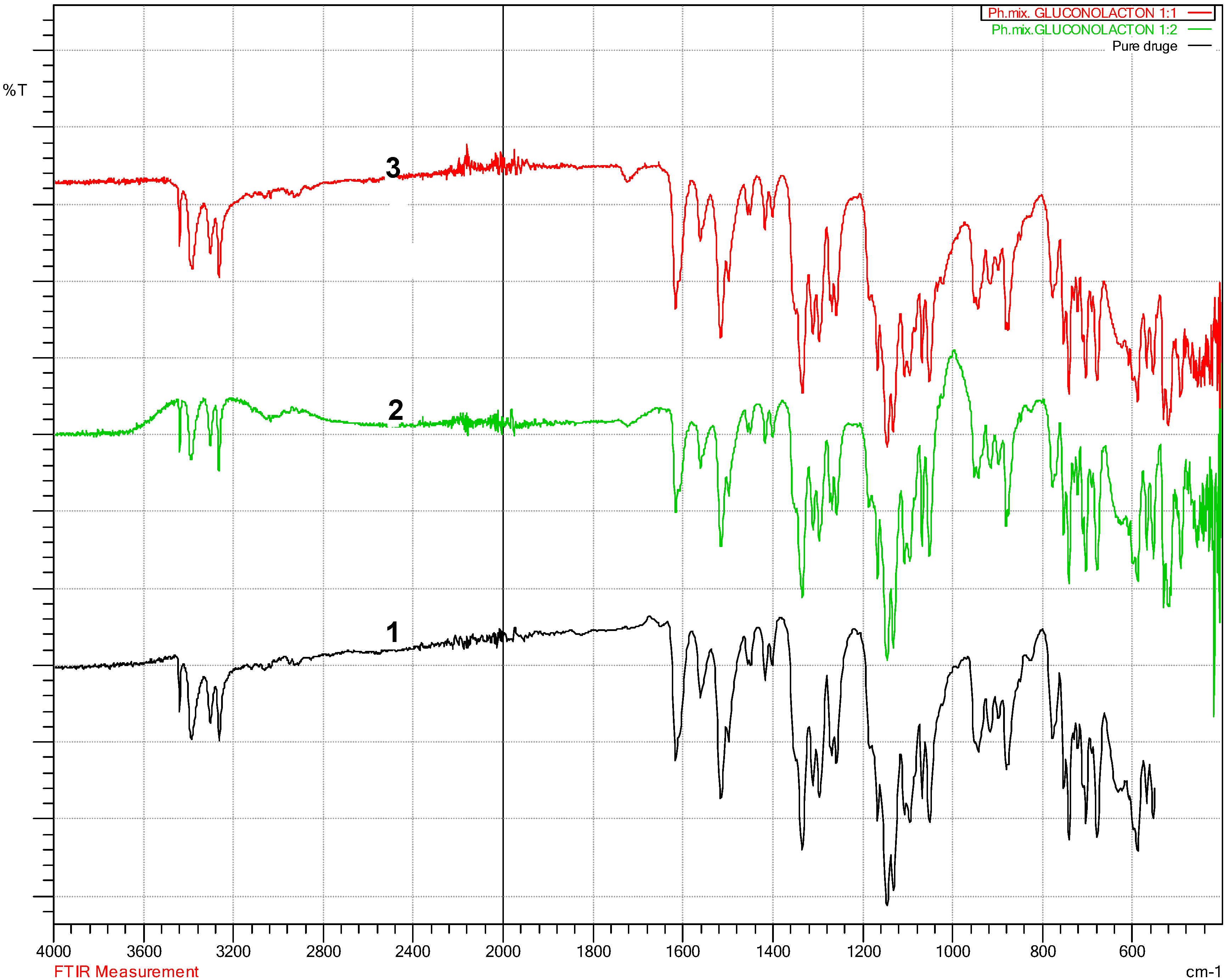
3.2. Fourier Transform Infra-Red Spectroscopy (FT-IR)
3.3. Scanning Electron Microscopy—SEM
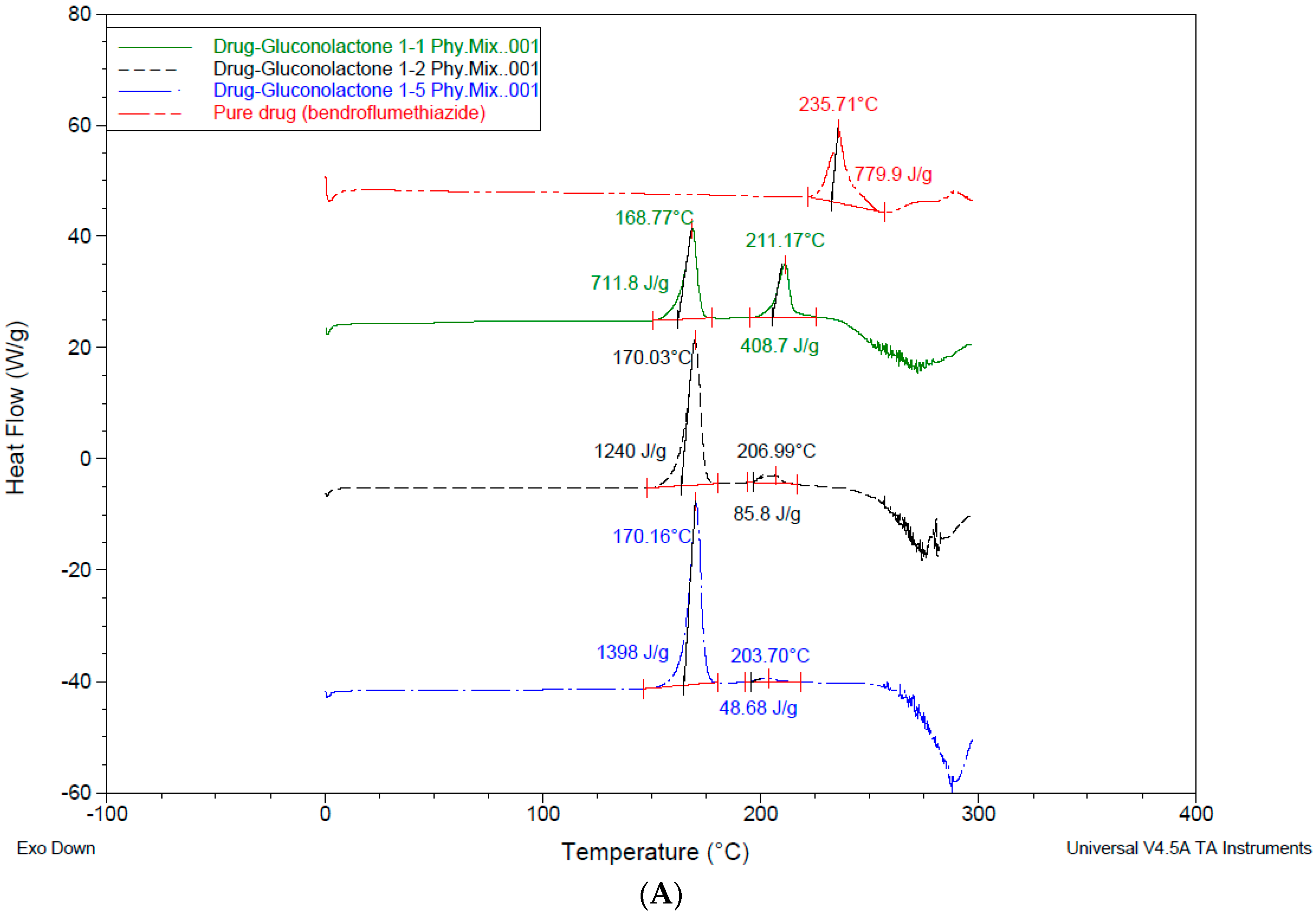
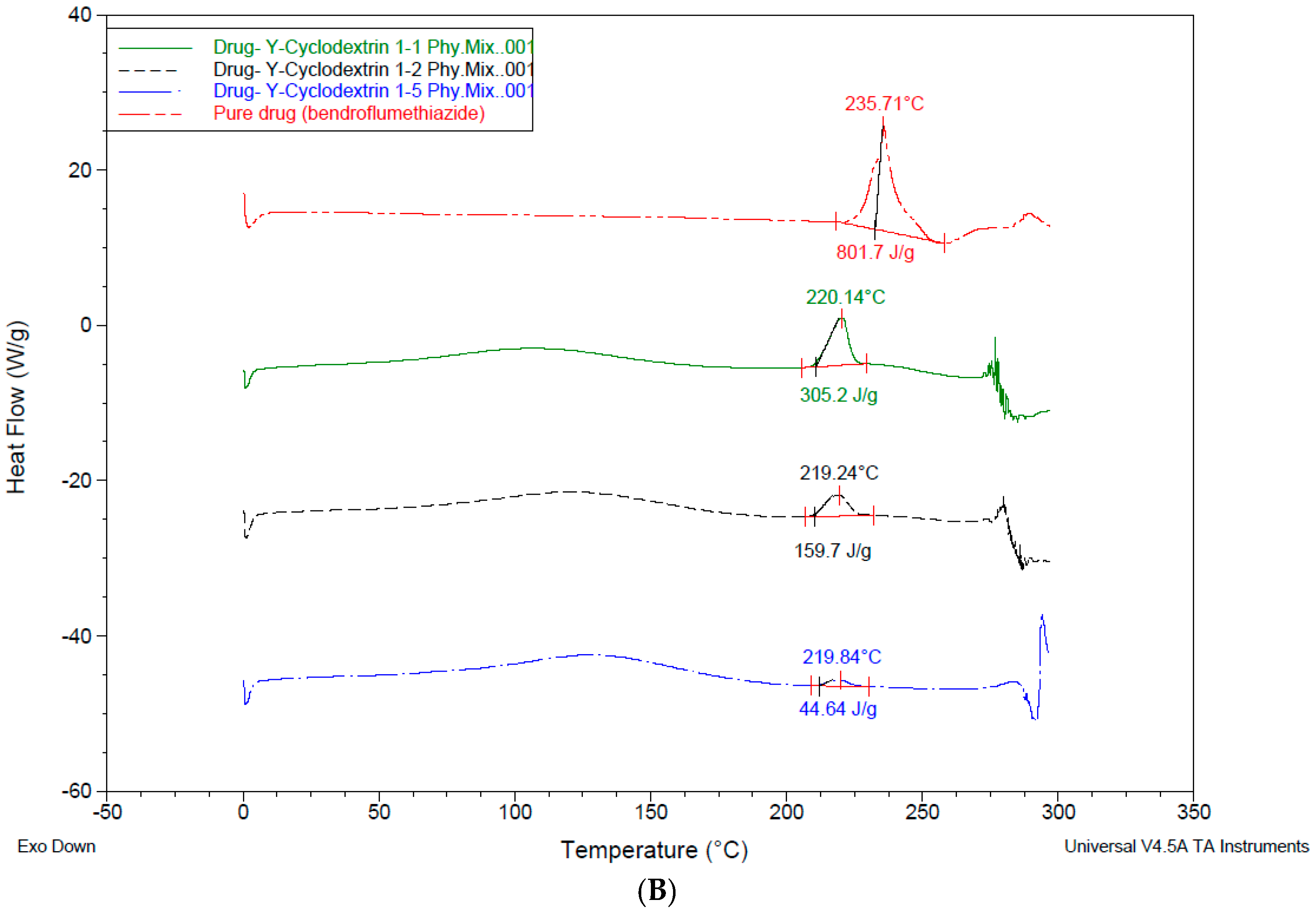
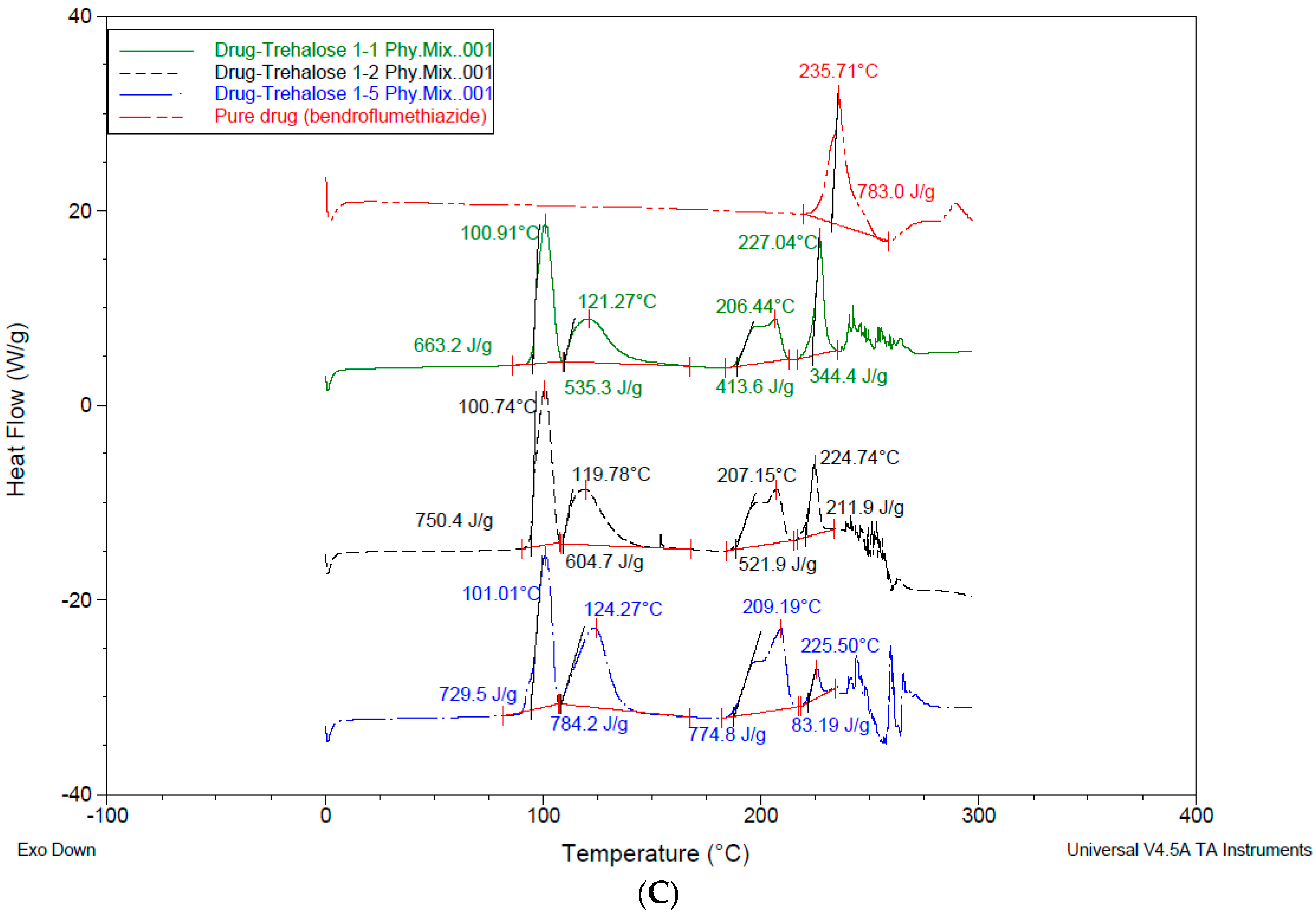
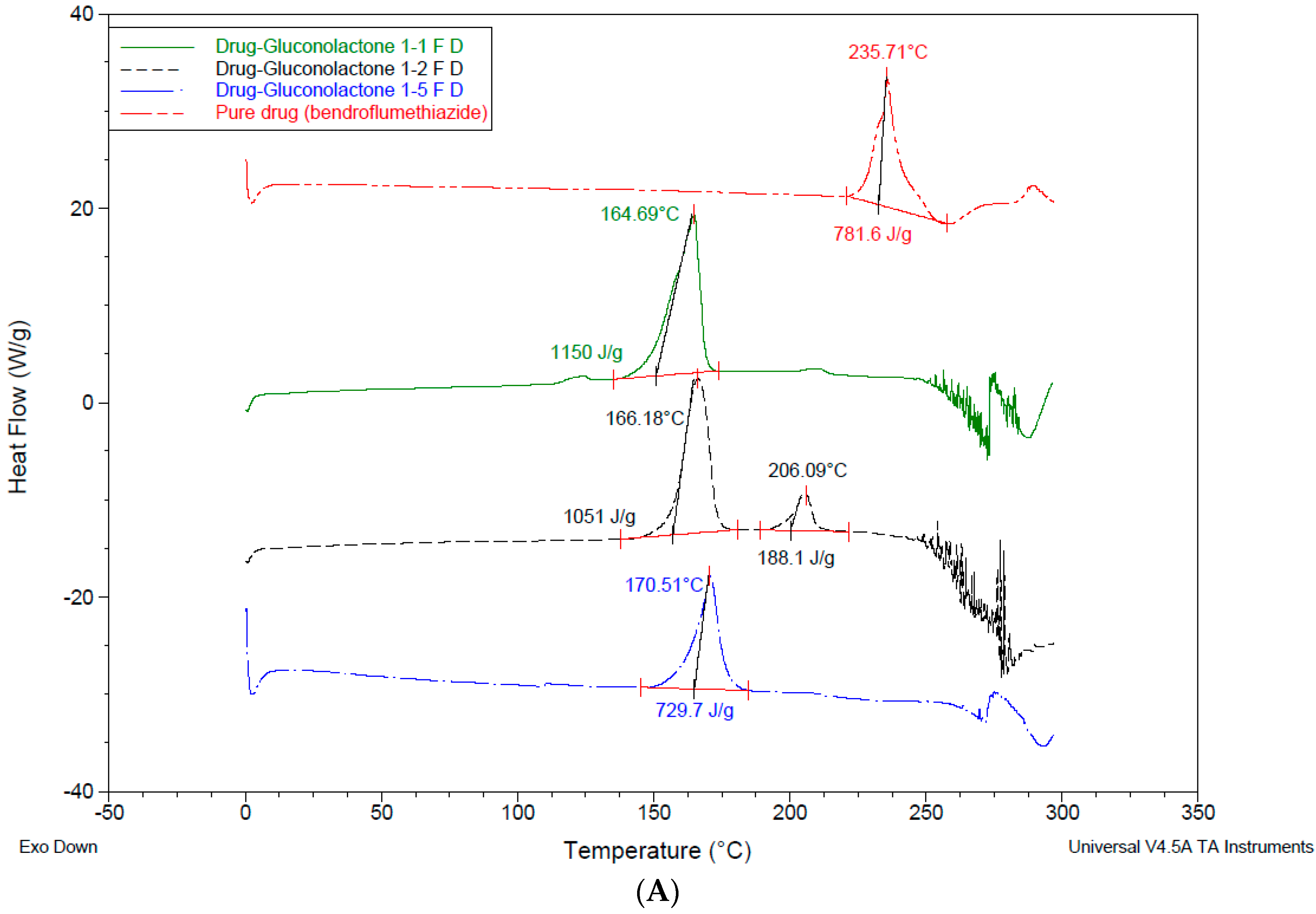
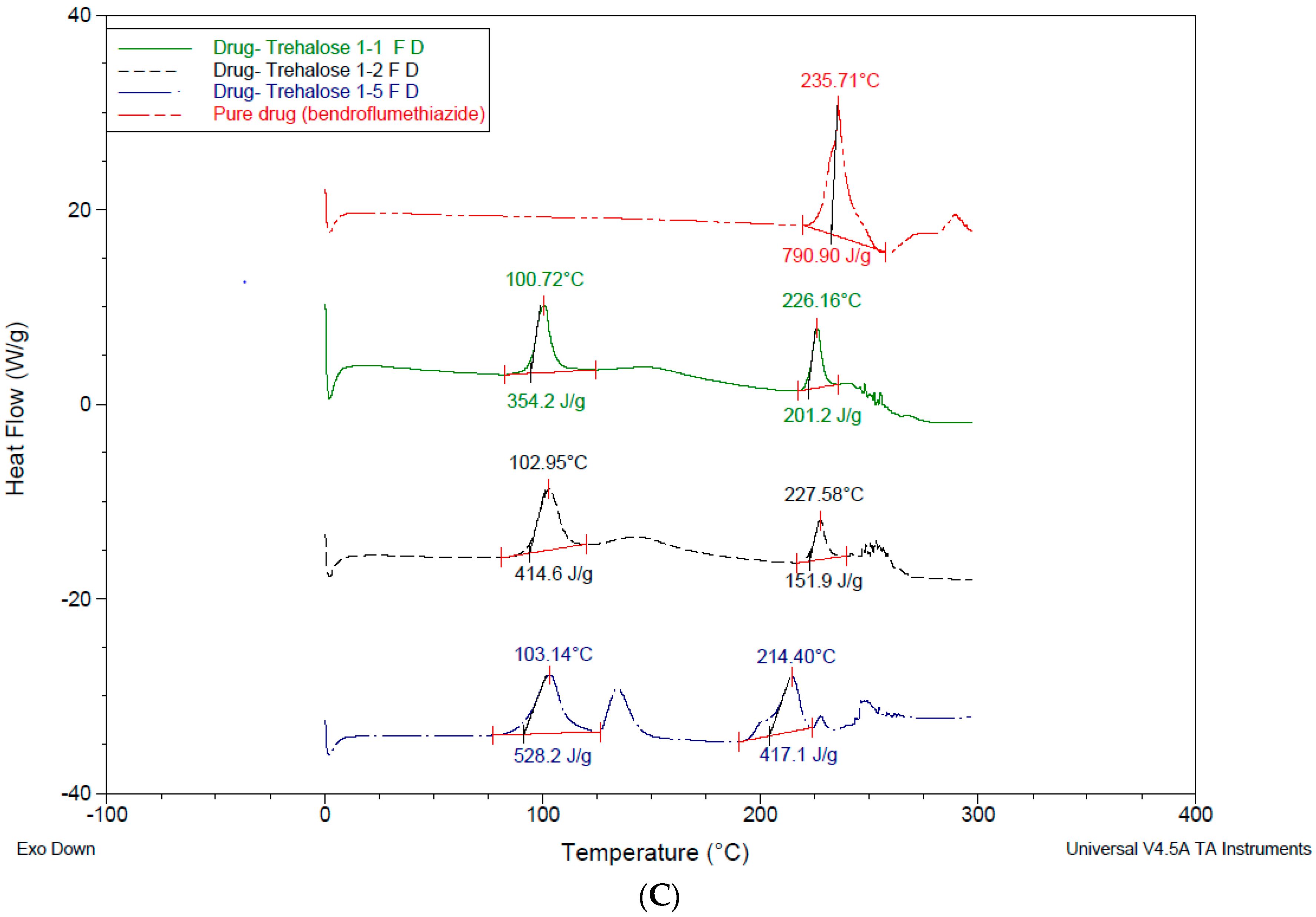
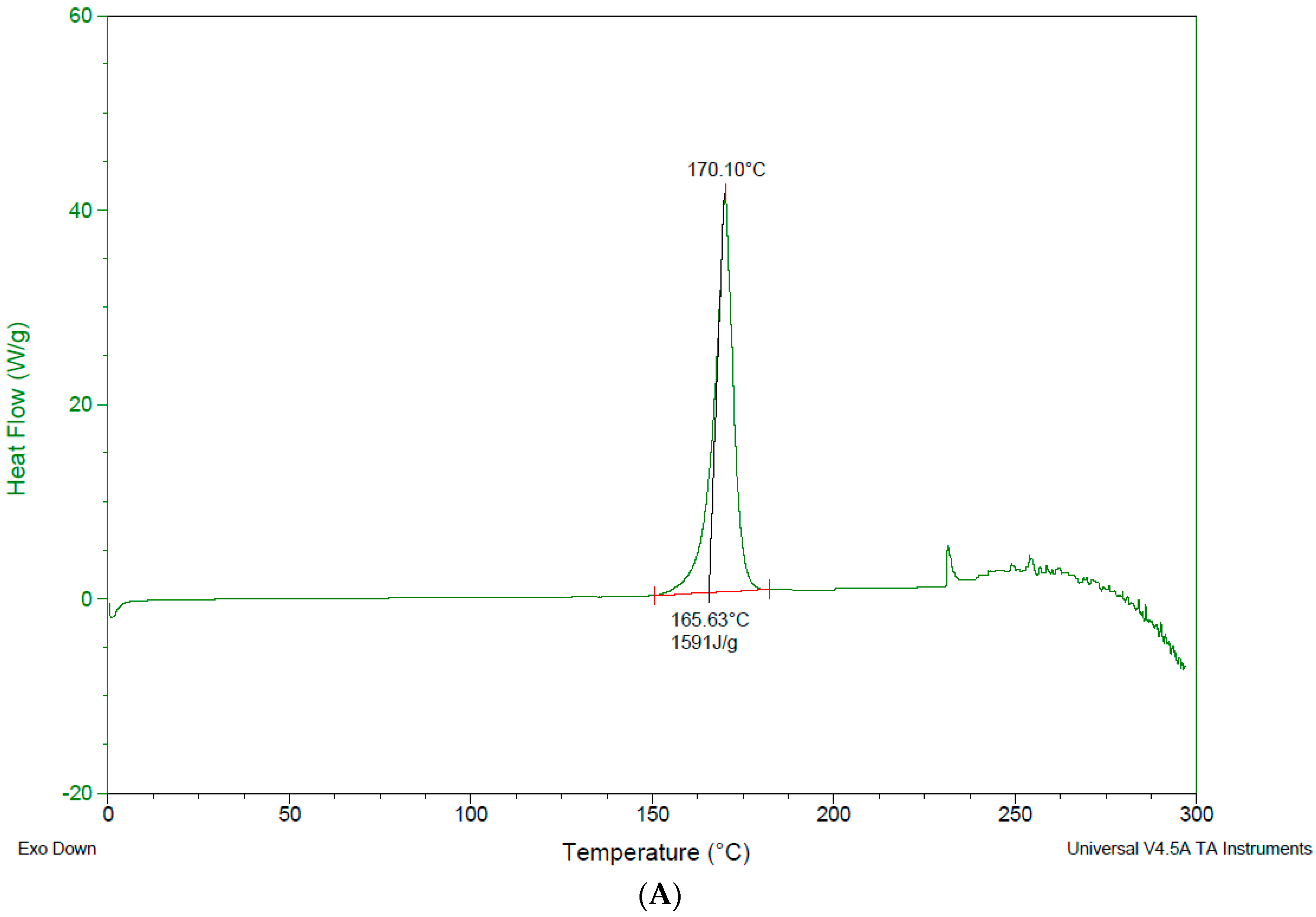
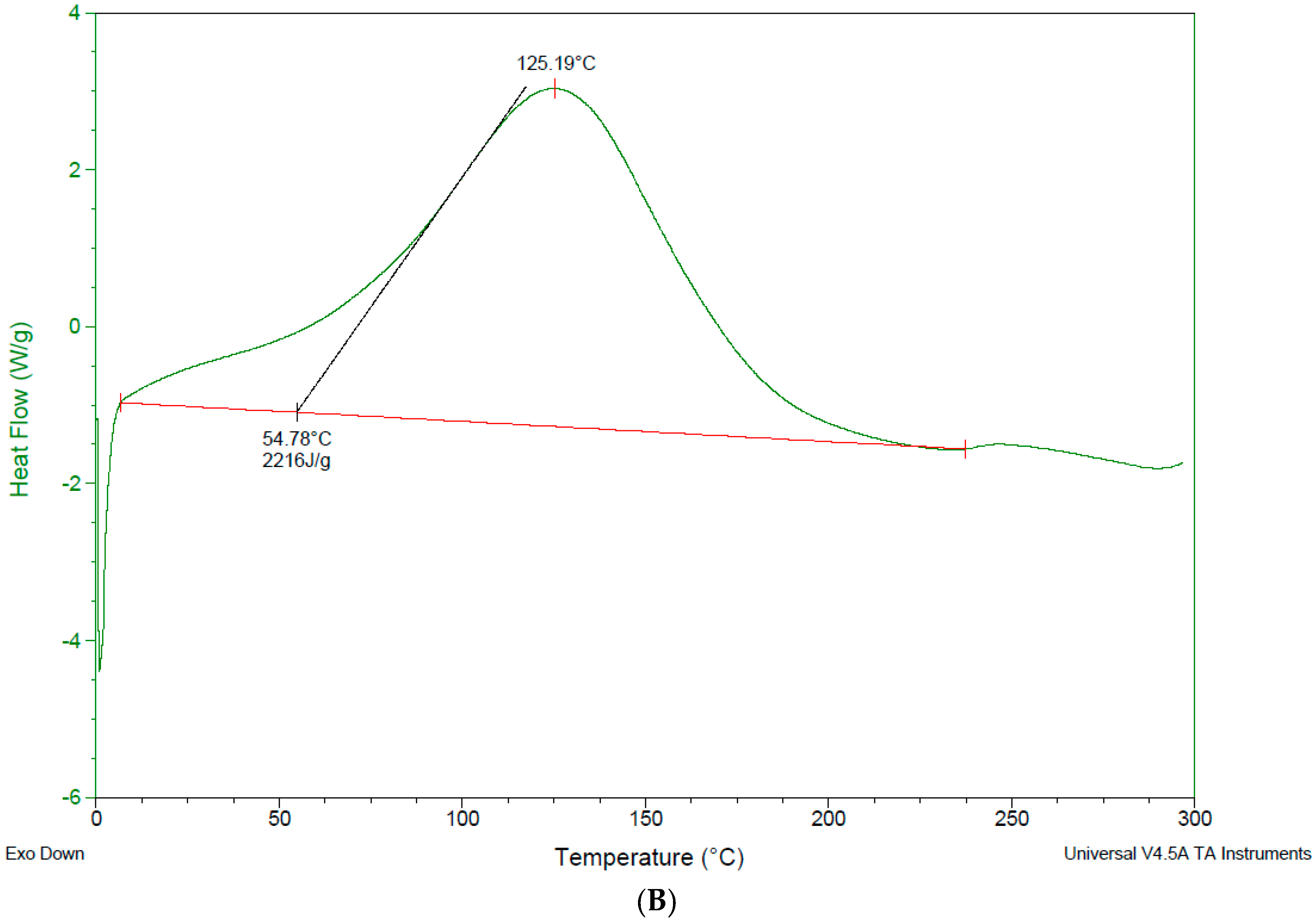
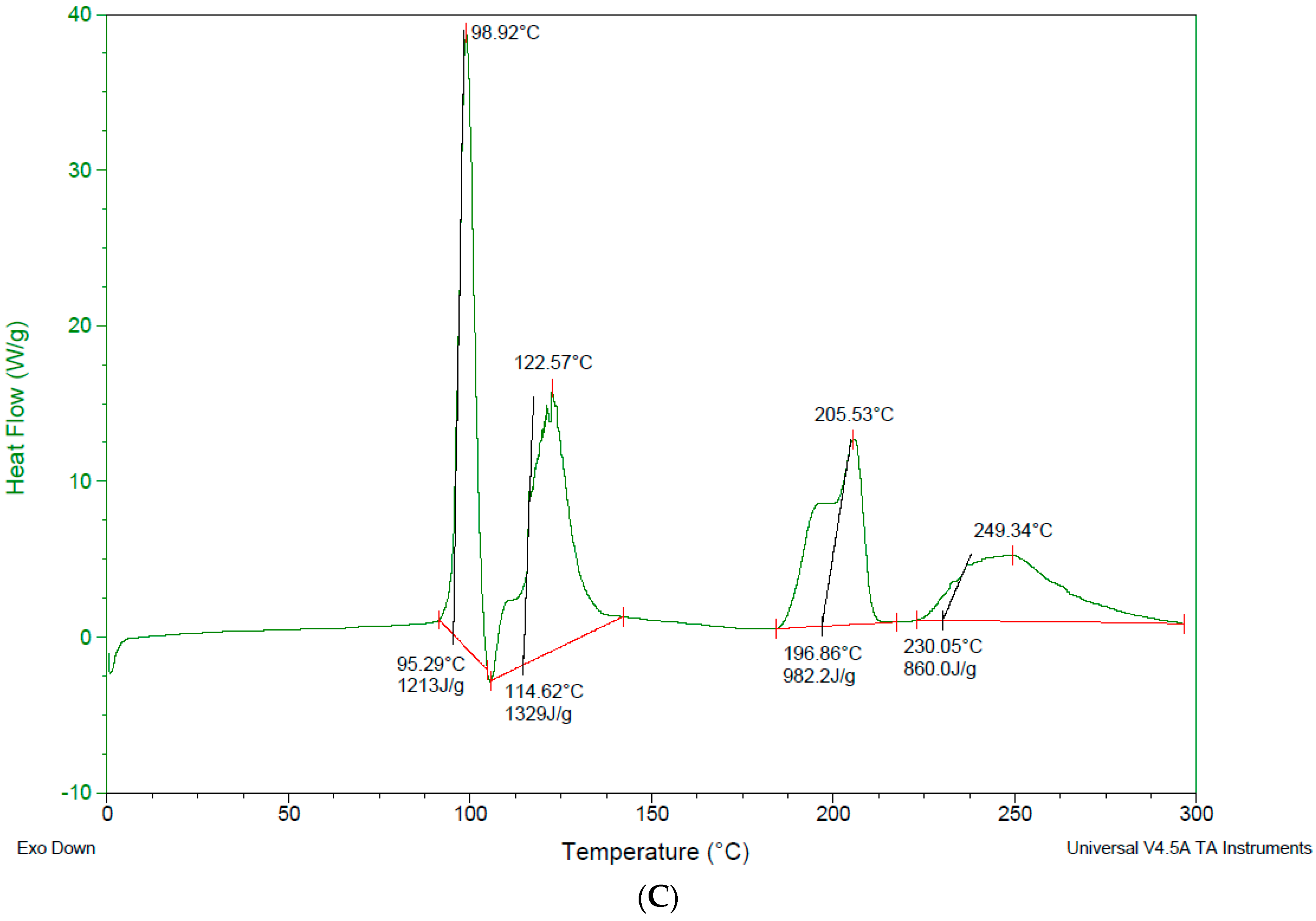
3.4. Differential Scanning Calorimetry—DSC
3.5. Statistical Analysis
4. Results and Discussion
4.1. Dissolution Studies
4.2. Fourier Transform Infra-Red Spectroscopy (FT-IR) Results
4.3. Differential Scanning Calorimetry (DSC)
4.4. Scanning Electron Microscopy—SEM
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Vemula, V.R.; Lagishetty, V.; Lingala, S. Solubility enhancement techniques. Int. J. Pharm. Sci. Rev. Res. 2010, 5, 41–51. [Google Scholar]
- Kesarwani, P.; Rastogi, S.; Bhalla, V.; Arora, V. Solubility Enhancement of Poorly Water Soluble Drugs: A Review. Int. J. Pharm. Sci. Res. 2014, 5, 3123. [Google Scholar]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Luo, K.Q. Enhancing the solubility and bioavailability of isoflavone by particle size reduction using a supercritical carbon dioxide-based precipitation process. Chem. Eng. Res. Des. 2014, 92, 2542–2549. [Google Scholar] [CrossRef]
- Humayun, H.Y.; Shaarani, M.N.N.M.; Warrior, A.; Abdullah, B.; Salam, M.A. The Effect of Co-Solvent on the Solubility of a Sparingly Soluble Crystal of Benzoic Acid. Procedia Eng. 2016, 148, 1320–1325. [Google Scholar] [CrossRef]
- Sieger, P.; Cui, Y.; Scheuerer, S. pH-dependent solubility and permeability profiles: A useful tool for prediction of oral bioavailability. Eur. J. Pharm. Sci. 2017, 105, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Chella, N.; Shastri, N.; Tadikonda, R.R. Use of the liquisolid compact technique for improvement of the dissolution rate of valsartan. Acta Pharm. Sin. B 2012, 2, 502–508. [Google Scholar] [CrossRef]
- Elkordy, A.A.; Essa, E.A.; Dhuppad, S.; Jammigumpula, P. Liquisolid technique to enhance and to sustain griseofulvin dissolution: Effect of choice of non-volatile liquid vehicles. Int. J. Pharm. 2012, 434, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Elkordy, A.A.; Bhangale, U.; Murle, N.; Zarara, M.F. Combination of Lactose (as a carrier) with Cremophor® EL (as a liquid vehicle) to Enhance Dissolution of Griseofulvin. Powder Technol. 2013, 246, 182–186. [Google Scholar] [CrossRef]
- Javaheri, H.; Carter, P.; Elkordy, A.A. Wet granulation to overcome liquisolid technique issues of poor flowability and compactibility: A study to enhance Glibenclamide dissolution. J. Pharm. Drug Dev. 2014, 1, 501–512. [Google Scholar]
- Volkova, T.V.; Perlovich, G.L.; Terekhova, I.V. Enhancement of dissolution behavior of antiarthritic drug leflunomide using solid dispersion methods. Thermochim. Acta 2017, 656, 123–128. [Google Scholar] [CrossRef]
- Kaur, S.; Jena, S.K.; Samal, S.K.; Saini, V.; Sangamwar, A.T. Freeze dried solid dispersion of exemestane: A way to negate an aqueous solubility and oral bioavailability problems. Eur. J. Pharm. Sci. 2017, 107, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, D.O.; Healy, A.M.; Corrigan, O.I. The effect of spray drying solutions of bendroflumethiazide/polyethylene glycol on the physicochemical properties of the resultant materials. Int. J. Pharm. 2003, 262, 125–137. [Google Scholar] [CrossRef]
- Agrawal, A.G.; Kumar, A.; Gide, P.S. Self emulsifying drug delivery system for enhanced solubility and dissolution of glipizide. Colloids Surf. B Biointerfaces 2015, 126, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Cerdeira, A.M.; Mazzotti, M.; Gander, B. Miconazole nanosuspensions: Influence of formulation variables on particle size reduction and physical stability. Int. J. Pharm. 2010, 396, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Uhl, P.; Pantze, S.; Storck, P.; Parmentier, J.; Witzigmann, D.; Hofhaus, G.; Huwyler, J.; Mier, W.; Fricker, G. Oral delivery of vancomycin by tetraether lipid liposomes. Eur. J. Pharm. Sci. 2017, 108, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, K.; Vinarov, Z.; Tcholakova, S. Improving Ibuprofen solubility by surfactant-facilitated self-assembly into mixed micelles. J. Drug Deliv. Sci. Technol. 2016, 36, 208–215. [Google Scholar] [CrossRef]
- Loh, G.O.K.; Tan, Y.T.F.; Peh, K.-K. Enhancement of norfloxacin solubility via inclusion complexation with β-cyclodextrin and its derivative hydroxypropyl-β-cyclodextrin. Asian J. Pharm. Sci. 2016, 11, 536–546. [Google Scholar] [CrossRef]
- Kate, L.; Gokarna, V.; Borhade, V.; Prabhu, P.; Deshpande, V.; Pathak, S.; Sharma, S.; Patravale, V. Bioavailability enhancement of atovaquone using hot melt extrusion technology. Eur. J. Pharm. Sci. 2016, 86, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Crum, M.; Elkordy, A.A.; Zarara, M.; Elkordy, E.A. In situ lyophilisation of nifedipine directly in hard gelatine capsules. Pharm. Dev. Technol. 2013, 18, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Kadam, S.V.; Shinkar, D.M.; Saudagar, R.B. Review on Solubility Enhancement Techniques. IJPBS 2013, 3, 462–475. [Google Scholar]
- British National Formulary (BNF) 70. Available online: https://pharm.reviews/images/statyi/british-national-formulary-2015.pdf (accessed on 1 November 2017).
- Healy, A.; Mcdonald, B.; Tajber, L.; Corrigan, O. Characterisation of excipient-free nanoporous microparticles (NPMPs) of bendroflumethiazide. Eur. J. Pharm. Biopharm. 2008, 69, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Frontini, R.; Mielck, J.B. Interactions between bendroflumethiazide and water soluble polymers. I. Solubility of bendroflumethiazide in water from solid dispersions and formation of associates under climatic stress. Eur. J. Pharm. Biopharm. 1997, 43, 185–191. [Google Scholar] [CrossRef]
- Ginés, J.M.; Arias, M.J.; Moyano, J.R.; Sánchez-Soto, P.J. Thermal investigation of crystallization of polyethylene glycols in solid dispersions containing oxazepam. Int. J. Pharm. 1996, 143, 247–253. [Google Scholar] [CrossRef]
- Madgulkar, A.; Bandivadekar, M.; Shid, T.; Rao, S. Sugars as solid dispersion carrier to improve solubility and dissolution of the BCS class II drug: Clotrimazole. Drug Dev. Ind. Pharm. 2016, 42, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Voncina, B.; Vivod, V. Cyclodextrins in textile finishing. In Eco-Friendly Textile Dyeing and Finishing; Günay, M., Ed.; InTech: London, UK, 2013. [Google Scholar]
- Brewster, M.E.; Loftsson, T. Cyclodextrins as pharmaceutical solubilizers. Adv. Drug Deliv. Rev. 2007, 59, 645–666. [Google Scholar] [CrossRef] [PubMed]
- Nokhodchi, A.; Al-Hamidi, H.; Antonijevic, M.D.; Owusu-Ware, S.; Kaialy, W. Dissolution and solid state behaviours of carbamazepine-gluconolactone solid dispersion powders: The potential use of gluconolactone as dissolution enhancer. Chem. Eng. Res. Des. 2015, 100, 452–466. [Google Scholar] [CrossRef]
- Johnson, D.H.; Wilson, W.W.; DeLucas, L.J. Protein solubilization: A novel approach. J. Chromatogr. B 2014, 971, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Aulton, M.E.; Taylor, K.M.G. Aulton’s Pharmaceutics: The Design and Manufacture of Medicines; Churchill Livingstone: London, UK, 2013. [Google Scholar]
- Al-Hamidi, H.; Obeidat, W.M.; Nokhodchi, A. The dissolution enhancement of piroxicam in its physical mixtures and solid dispersion formulations using gluconolactone and glucosamine hydrochloride as potential carriers. Pharm. Dev. Technol. 2015, 20, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Lucio, D.; Irache, J.M.; Font, M.; Martínez-Ohárriz, M.C. Supramolecular structure of glibenclamide and β-cyclodextrins complexes. Int. J. Pharm. 2017, 530, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.C. Infrared Spectral Interpretation: A Systematic Approach; CRC Press: Boca Raton, FL, USA, 1999. [Google Scholar]
- Elkordy, A.A.; Tan, X.N.; Essa, E.A. Spironolactone release from liquisolid formulations prepared with CapryolTM 90, Solutol® HS-15 and Kollicoat® SR 30 D as non-volatile liquid vehicles. Eur. J. Pharm. Biopharm. 2013, 83, 203–223. [Google Scholar] [CrossRef] [PubMed]
- Law, D.; Wang, W.; Schmitt, E.A.; Qiu, Y.; Krill, S.L.; Fort, J.J. Properties of Rapidly Dissolving Eutectic Mixtures of Poly(ethylene glycol) and Fenofibrate: The Eutectic Microstructure. J. Pharm. Sci. 2003, 92, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.B.; Das, S.S.; Singh, N.P.; Agrawal, T. Computer simulation, thermodynamic and microstructural studies of benzamide–benzoic acid eutectic system. J. Cryst. Growth 2008, 310, 2878–2884. [Google Scholar] [CrossRef]
- British Pharmacopoeia Commission. British Pharmacopoeia; TSO (The Stationery Office): Norwich, UK, 2013. [Google Scholar]
















© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saleh, A.; McGarry, K.; Chaw, C.S.; Elkordy, A.A. Feasibility of Using Gluconolactone, Trehalose and Hydroxy-Propyl Gamma Cyclodextrin to Enhance Bendroflumethiazide Dissolution Using Lyophilisation and Physical Mixing Techniques. Pharmaceutics 2018, 10, 22. https://doi.org/10.3390/pharmaceutics10010022
Saleh A, McGarry K, Chaw CS, Elkordy AA. Feasibility of Using Gluconolactone, Trehalose and Hydroxy-Propyl Gamma Cyclodextrin to Enhance Bendroflumethiazide Dissolution Using Lyophilisation and Physical Mixing Techniques. Pharmaceutics. 2018; 10(1):22. https://doi.org/10.3390/pharmaceutics10010022
Chicago/Turabian StyleSaleh, Ashraf, Kenneth McGarry, Cheng Shu Chaw, and Amal Ali Elkordy. 2018. "Feasibility of Using Gluconolactone, Trehalose and Hydroxy-Propyl Gamma Cyclodextrin to Enhance Bendroflumethiazide Dissolution Using Lyophilisation and Physical Mixing Techniques" Pharmaceutics 10, no. 1: 22. https://doi.org/10.3390/pharmaceutics10010022
APA StyleSaleh, A., McGarry, K., Chaw, C. S., & Elkordy, A. A. (2018). Feasibility of Using Gluconolactone, Trehalose and Hydroxy-Propyl Gamma Cyclodextrin to Enhance Bendroflumethiazide Dissolution Using Lyophilisation and Physical Mixing Techniques. Pharmaceutics, 10(1), 22. https://doi.org/10.3390/pharmaceutics10010022