Dual Acting Polymeric Nano-Aggregates for Liver Cancer Therapy




Abstract
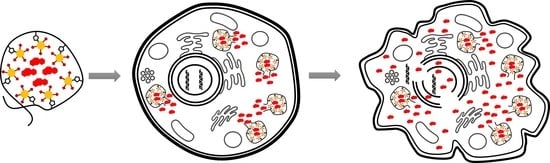
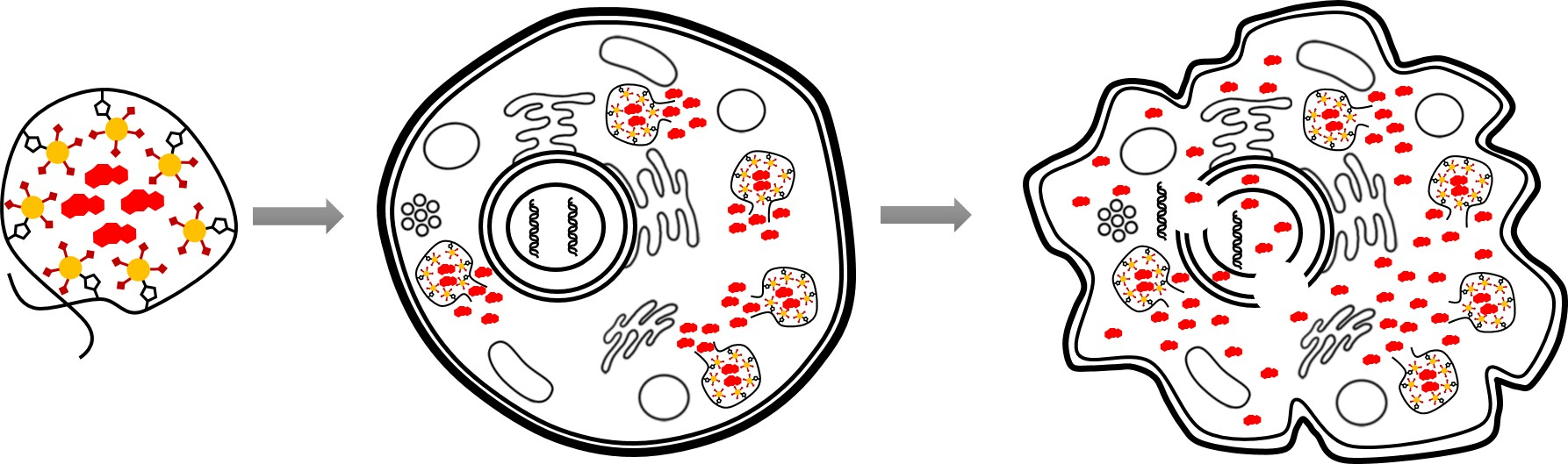
:
1. Introduction
2. Materials and Methods
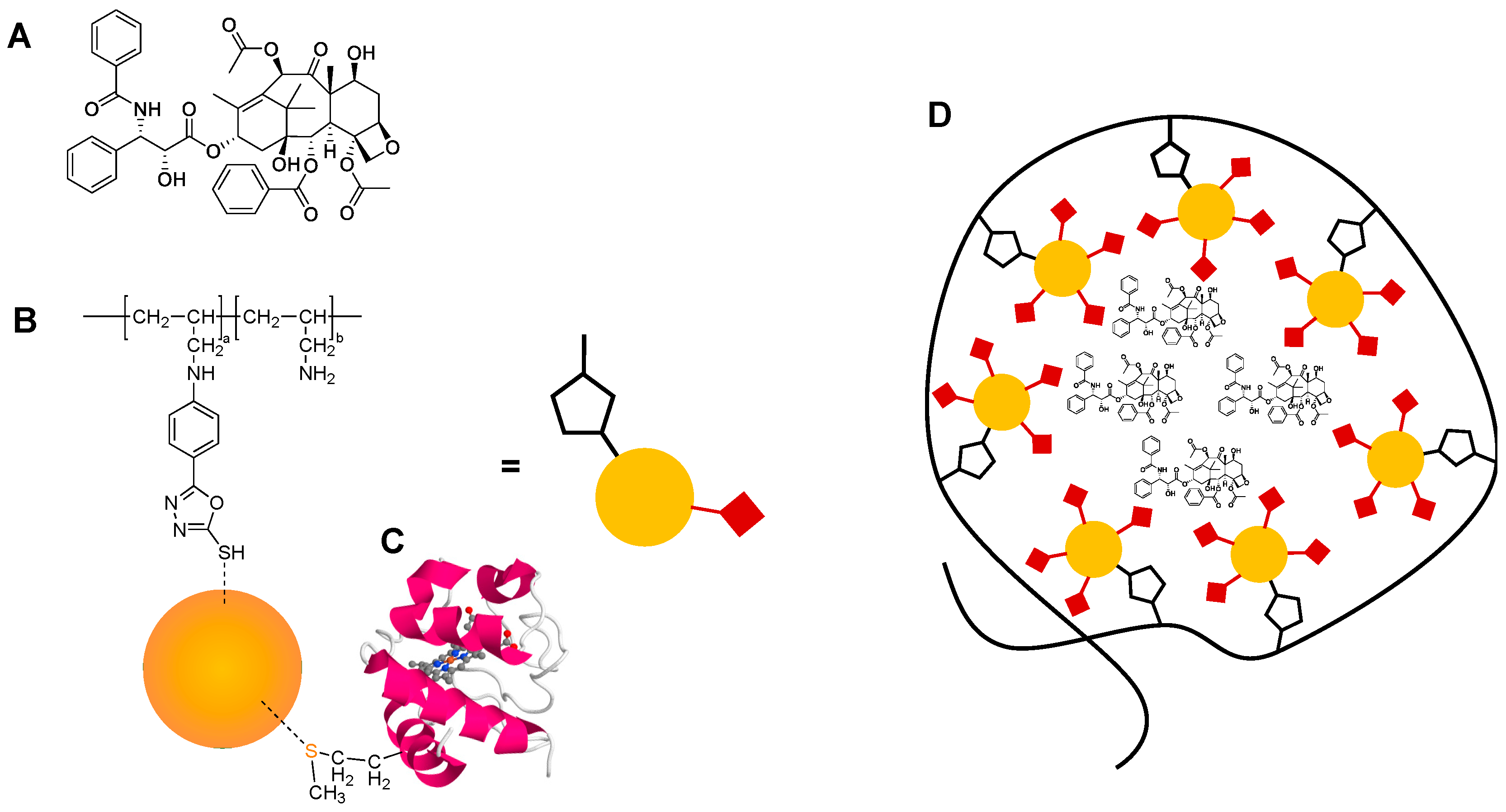
2.1. Poly(allylamine)-oxadiazole (PAA-Ox5) Synthesis and Characterisation
2.2. HNP Synthesis and Characterisation
2.3. PAA-Ox5-HNP Conjugation
2.4. Characterisation of PAA-Ox5 and PAA-Ox5-HNP Nano-Aggregates
2.5. Attachment of Cytochrome C onto the HNPs in the PAA-Ox5-HNP
2.6. Paclitaxel (PTX) Loading and Release of Nano-Aggregates
2.7. Biological Characterisation of Nano-Aggregates and Formulations
3. Results
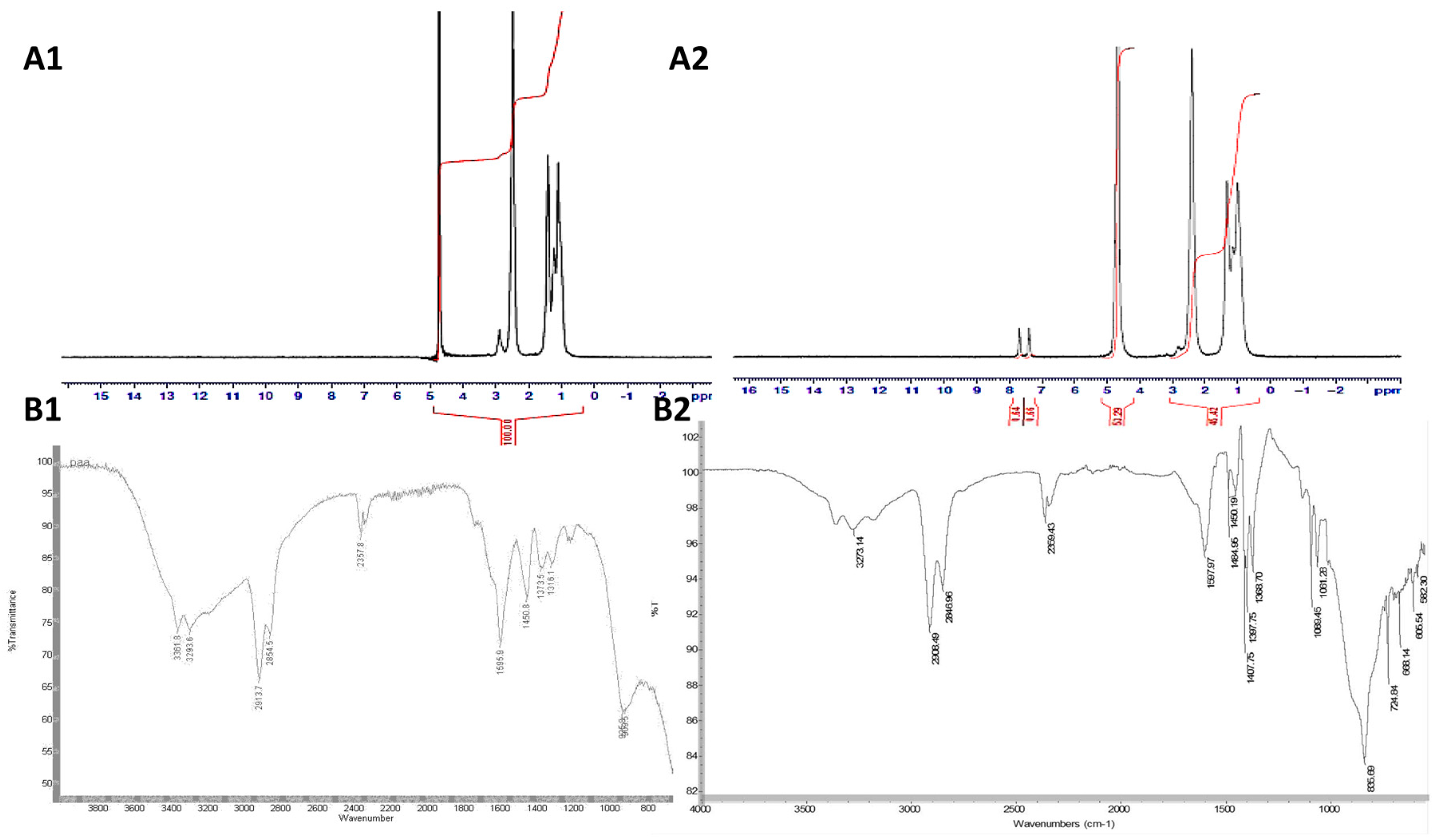
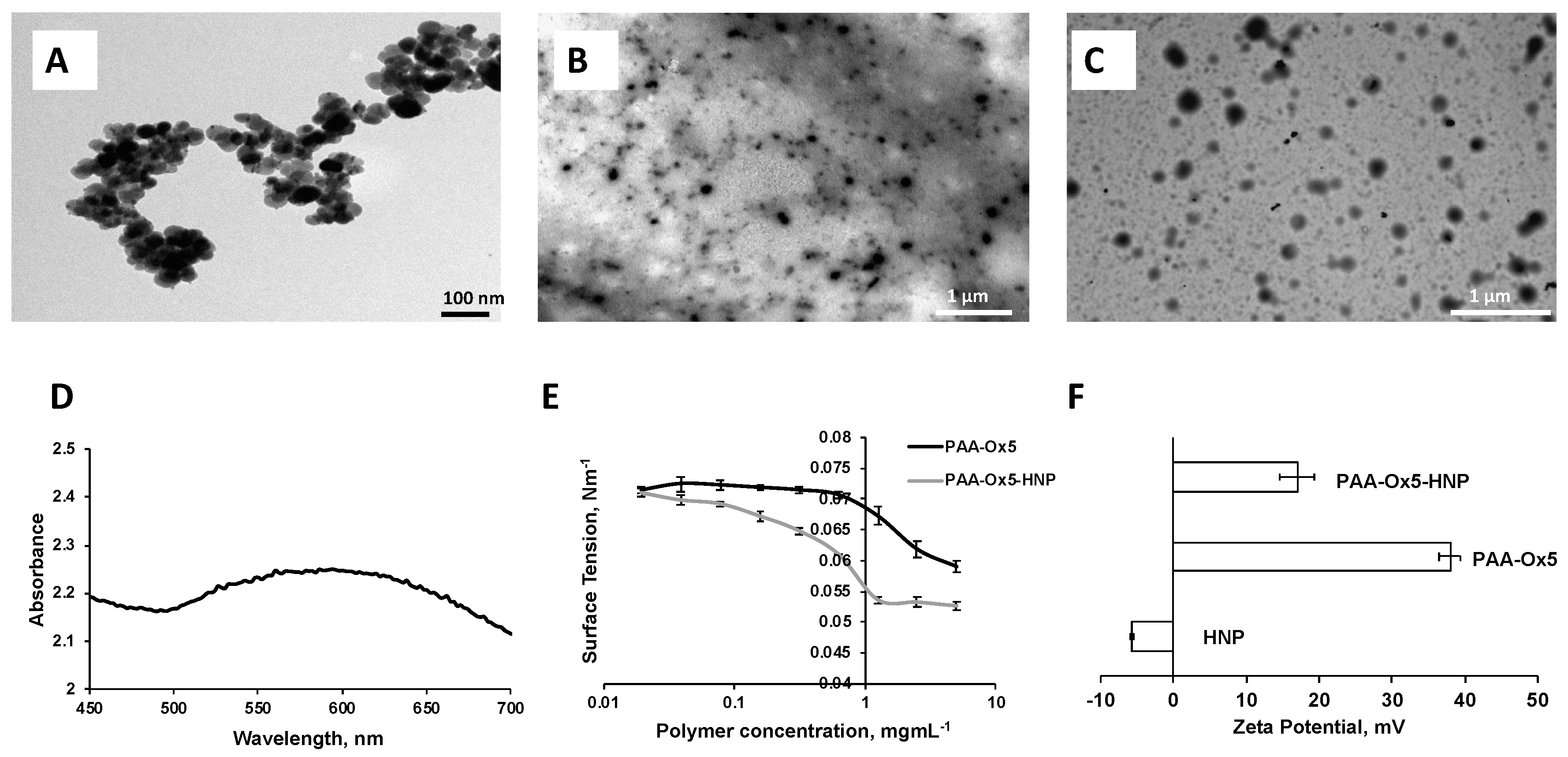
3.1. Synthesis and Characterisation of PAA-Ox5
3.2. Synthesis and Characterisation HNPs
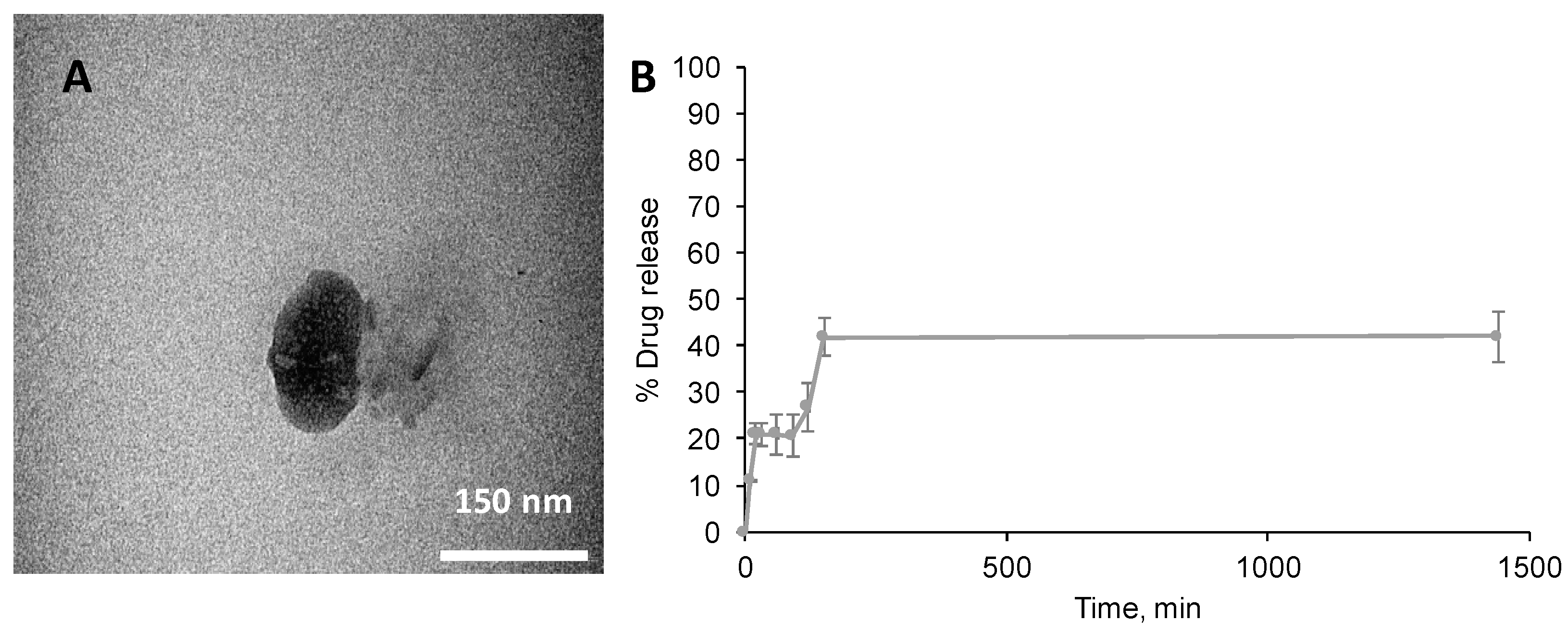
3.3. Characterisation of the Nano-Aggregates
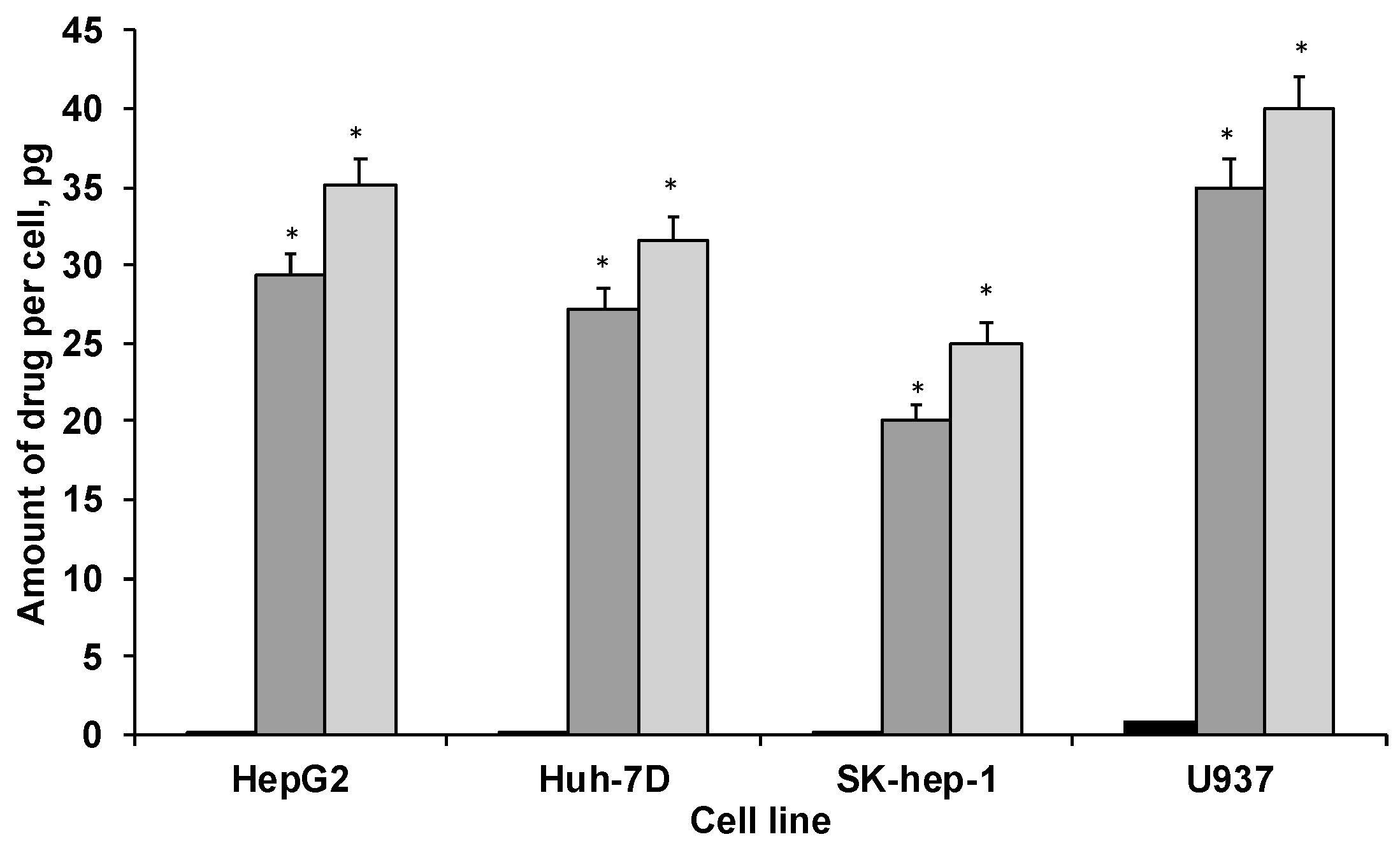
3.4. Quantification of Cytochrome C onto the HNPs in the PAA-Ox5-HNP
3.5. PTX Loading and Release of Nano-Aggregates
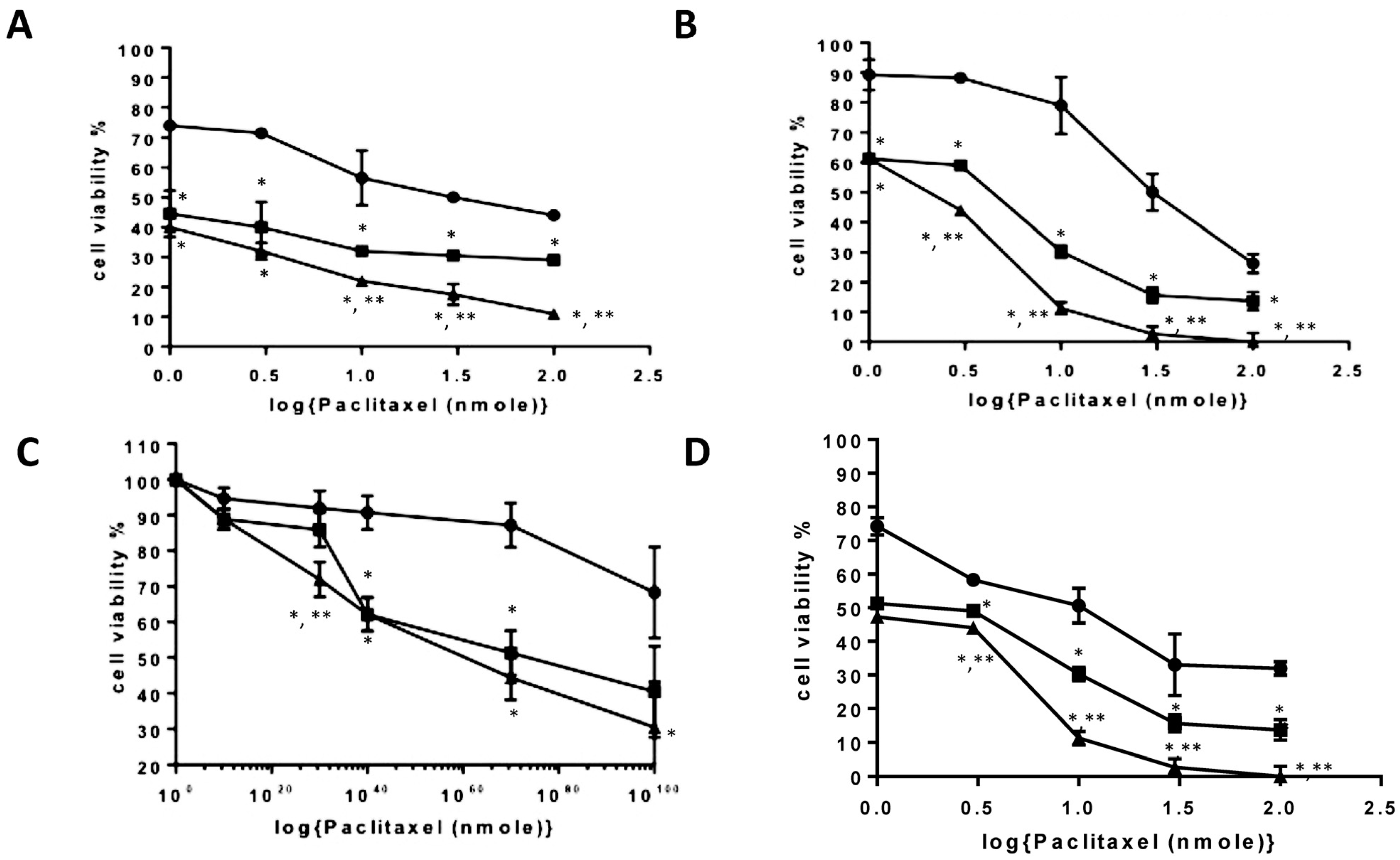
3.6. Biological Characterisation of Nano-Aggregates and Formulations
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Cancer Research UK. 2015. Available online: http://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/liver-cancer (accessed on 1 May 2018).
- Li, F.; Wang, J.-Y. Targeted delivery of drugs for liver fibrosis. Expert Opin. Drug Deliv. 2009, 6, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Park, J.O.; Stephen, Z.; Sun, C.; Veiseh, O.; Kievet, F.M.; Fang, C.; Leung, M.; Mok, H.; Zhang, M. Glypican-3 Targeting of Liver Cancer Cells Using Multifunctional Nanoparticles. Mol. Imaging 2011, 10, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, Z.; Chen, Y.; Chen, L.; Lin, L.; Li, Y. The characteristics and performance of a multifunctional nanoassembly system for the co-delivery of docetaxel and iSur-pDNA in a mouse hepatocellular carcinoma model. Biomaterials 2010, 31, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wan, G.; Guo, H.; Liu, Y.; Zhou, P.; Wang, H.; Wang, D.; Zhang, S.; Wang, Y.; Zhang, N. A multifunctional nanoparticle system combines sonodynamic therapy and chemotherapy to treat hepatocellular carcinoma. Nano Res. 2017, 10, 834–855. [Google Scholar] [CrossRef]
- Lachenmayer, A.; Toffanin, S.; Cabellos, L.; Alsinet, C.; Hoshida, Y.; Villanueva, A.; Minguez, B.; Tsai, H.-W.; Ward, S.C.; Thung, S.; et al. Combination therapy for hepatocellular carcinoma: Additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J. Hepatol. 2012, 56, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Alsaied, O.A.; Sangwan, V.; Banerjee, S.; Krosch, T.C.; Chugh, R.; Saluja, A.; Vickers, S.M.; Jensen, E.H. Sorafenib and triptolide as combination therapy for hepatocellular carcinoma. Surgery 2014, 156, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Ryoo, B.-Y.; Lim, H.Y.; Kim, D.Y.; Okusaka, T.; Ikeda, M.; Hidaka, H.; Yeon, J.-E.; Mizukoshi, E.; Morimoto, M.; et al. Resminostat and sorafenib combination therapy for advanced hepatocellular carcinoma in patients previously untreated with systemic chemotherapy. J. Clin. Oncol. 2017, 35, 252–252. [Google Scholar] [CrossRef]
- Thein, H.-H.; Qiao, Y.; Zaheen, A.; Jembere, N.; Sapisochin, G.; Chan, K.K.W.; Yoshida, E.M.; Earle, C.C. Costeffectiveness analysis of treatment with noncurative or palliative intent for hepatocellular carcinoma in the real-world setting. PLoS ONE 2017, 12, e0185198. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Almeida, J.P.M.; Chen, A.L.; Foster, A.; Drezek, R. In vivo biodistribution of nanoparticles. Nanomedicine 2011, 6, 815–835. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-N.; Poon, W.; Tavares, A.J.; McGilvray, I.D.; Chan, W.C.W. Nanoparticle–liver interactions: Cellular uptake and hepatobiliary elimination. J. Control. Release 2016, 240, 332–348. [Google Scholar] [CrossRef] [PubMed]
- Malekigorji, M.; Hoskins, C.; Curtis, T.; Varbiro, G. Enhancement of the Cytotoxic Effect of Anticancer Agent by Cytochrome c Functionalised Hybrid Nanoparticles in Hepatocellular Cancer Cells. J. Nanomed. Res. 2014, 1, 00010. [Google Scholar] [CrossRef]
- Al-Shakarchi, W.; Alsuraifi, A.; Abed, M.; Abdullah, M.; Richardson, A.; Curtis, A.; Hoskins, C. Combined effect of anticancer agent cytochrome C decorated hybrid nanoparticles for liver cancer therapy. Pharmaceutics 2018, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Kato, K.; Yasui, H.; Morizane, C.; Ikeda, M.; Ueno, H.; Muro, K.; Yamada, Y.; Okusaka, T.; Shirao, K.; et al. A phase I and pharmacokinetic study of NK105, a paclitaxelincorporating micellar nanoparticle formulation. Br. J. Cancer 2007, 97, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.S.; Balasubramanian, S.V.; Straubinger, R.M. Pharmaceutical and Physical Properties of Paclitaxel (Taxol †) Complexes with Cyclodextrins. J. Pharm. Sci. 1995, 84, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Guan, H.; Chen, X.; Lu, C.; Chen, L.; Hu, X.; Shi, Q.; Jing, X. A novel polymer–paclitaxel conjugate based on amphiphilic triblock copolymer. J. Control. Release 2007, 117, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, J.Z.; Hu, J.; Guo, Q.; Yang, D. Preparation of amphiphilic copolymers for covalent loading of paclitaxel for drug delivery system. J. Polym. Sci. Part A Polym. Chem. 2014, 52, 366–374. [Google Scholar] [CrossRef]
- Mu, M.; Konno, T.; Inoue, Y.; Ishihara, K. Solubilization of poorly water-soluble compounds using amphiphilic phospholipid polymers with different molecular architectures. Colloids Surf. B 2017, 158, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.S.; Almeida, A.; Prezotti, F.; Cury, B.; Campana-Filho, S.P.; Sarmento, B. Synthesis and characterization of 3,6-O,O’-dimyristoyl chitosan micelles for oral delivery of paclitaxel. Colloids Surf. B 2017, 152, 220–228. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. 2013. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/fda-nanoparticle-paclitaxel (accessed on 2 May 2018).
- Barnett, C.M.; Lees, M.R.; Curtis, A.D.M.; Kong Thoo Lin, P.; Cheng, W.P.; Hoskins, C. Poly(allylamine) Magnetomicelles for Image Guided Drug Delivery. Pharm. Nanotechnol. 2013, 1, 224–238. [Google Scholar] [CrossRef]
- Barnett, C.M.; Gueorguieva, M.; Lees, M.R.; McGarvey, D.J.; Darton, R.J.; Hoskins, C. Effect of hybrid composition on the physicochemical properties and morphology of iron oxide-gold nanoparticles. J. Nanopart. Res. 2012, 14, 1170. [Google Scholar] [CrossRef]
- Malekigorji, M.; Alfahad, M.; Kong Thoo Lin, P.; Jones, S.; Curtis, A.; Hoskins, C. Thermally triggered theranostics for pancreatic cancer therapy. Nanoscale 2017, 9, 12735–12745. [Google Scholar] [CrossRef] [PubMed]
- Oluwasanmi, A.; Al-Shakarchi, W.; Manzur, A.; Aldebasi, M.H.; Elsini, R.S.; Albusair, M.K.; Haxton, K.J.; Curtis, A.D.M.; Hoskins, C. Diels alder-mediated release of gemcitabine from hybrid nanoparticles for enhanced pancreatic cancer therapy. J. Control. Release 2017, 266, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Barnett, C.; Gueorguieva, M.; Lees, M.; McGarvey, D.; Hoskins, C. Physical stability, biocompatibility and potential use of hybrid iron oxide-gold nanoparticles as drug carriers. J. Nanopart. Res. 2013, 15, 1706. [Google Scholar] [CrossRef]
- Hoskins, C.; Ouaissi, M.; Lima, S.C.; Cheng, W.P.; Loureirio, I.; Mas, E.; Lombardo, D.; Cordeiro-da-Silva, A.; Ouaissi, A.; Kong Thoo Lin, P. In vitro and in vivo anticancer activity of a novel nano-sized formulation based on self-assembling polymers against pancreatic cancer. Pharm. Res. 2010, 12, 2694–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoskins, C.; Papachristou, A.; Ho, T.M.H.; Hine, J.; Curtis, A.D.M. Investigation into drug solubilisation potential of sulfonated calix[4]resorcinarenes. J. Nanomed. Nanotechnol. 2016, 7, 2. [Google Scholar] [CrossRef]
- Zaman, J. Addressing Solubility through Nano Based Drug Delivery Systems. Nanomed. Nanotechnol. 2016, 7, 3. [Google Scholar] [CrossRef]
- National Cancer Institute. 2017. Available online: https://www.cancer.gov/sites/nano/cancer-nanotechnology/current-treatments (accessed on 2 May 2018).




 PAA-Ox5+PTX,
PAA-Ox5+PTX,  PAA-Ox5-HNPc+PTX. * denotes significance compared with free drug, p < 0.001.
PAA-Ox5+PTX, PAA-Ox5-HNPc+PTX. * denotes significance compared with free drug, p < 0.001.
PAA-Ox5-HNPc+PTX. * denotes significance compared with free drug, p < 0.001.
PAA-Ox5+PTX, PAA-Ox5-HNPc+PTX. * denotes significance compared with free drug, p < 0.001.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Particle Size Using Photon Correlation Spectroscopy, nm | Poly Dispersity Index | Metal Content Fe/Au | Cytochrome C Content mg/mL | PTX Content mg/mL |
|---|---|---|---|---|---|
| HNP | 948 | 1 | 2:1 | - | - |
| PAA-Ox5 | 82 | 0.55 | - | - | - |
| PAA-Ox5-HNP-c | 186 | 0.137 | 2:1 | 0.012 | - |
| PAA-Ox5+PTX | 112 | 0.232 | - | - | 0.598 |
| PAA-Ox5-HNP-c+PTX | 256 | 0.124 | 2:1 | 0.012 | 0.698 |
| HepG2 Cell Line | PTX | PAA-Ox5+PTX | PAA-Ox5-HNP-c+PTX |
|---|---|---|---|
| 24 h | - | 50 ± 3.05 nM | 40 ± 1.5 nM |
| 48 h | - | 1.99 ± 0.7 nM | 0.98 ± 1.3 nM |
| 72 h | 32 ± 4.84 nM | 1 nM | 0.7 nM |
| Huh-7D12 cell line | |||
| 24 h | - | 53 ± 1.52 nM | 11 ± 2.5 nM |
| 48 h | 37 ± 1.05 µM | 4 ± 2.61 nM | 2.21 ± 1.82 nM |
| 72 h | 31 ± 1.12 µM | 3 ± 2.5 nM | 0.9 ± 2.1 nM |
| SK-hep-1 cell line | |||
| 24 h | - | - | - |
| 48 h | - | - | 84.27 ± 3.1 nM |
| 72 h | - | 73.32 ± 1.54 nM | 57.39 ± 4.02 nM |
| U937 cell line | |||
| 24 h | - | 14 ± 1.5 nM | 3.6 ± 0.45 nM |
| 48 h | 39 ± 1.72 nM | 11 ± 1.29 nM | 2.6 ± 0.63 nM |
| 72 h | 9 ± 1 nM | 1.6 ± 1.08 nM | 1.2 ± 2.24 nM |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Shakarchi, W.; Alsuraifi, A.; Curtis, A.; Hoskins, C. Dual Acting Polymeric Nano-Aggregates for Liver Cancer Therapy. Pharmaceutics 2018, 10, 63. https://doi.org/10.3390/pharmaceutics10020063
Al-Shakarchi W, Alsuraifi A, Curtis A, Hoskins C. Dual Acting Polymeric Nano-Aggregates for Liver Cancer Therapy. Pharmaceutics. 2018; 10(2):63. https://doi.org/10.3390/pharmaceutics10020063
Chicago/Turabian StyleAl-Shakarchi, Wejdan, Ali Alsuraifi, Anthony Curtis, and Clare Hoskins. 2018. "Dual Acting Polymeric Nano-Aggregates for Liver Cancer Therapy" Pharmaceutics 10, no. 2: 63. https://doi.org/10.3390/pharmaceutics10020063
APA StyleAl-Shakarchi, W., Alsuraifi, A., Curtis, A., & Hoskins, C. (2018). Dual Acting Polymeric Nano-Aggregates for Liver Cancer Therapy. Pharmaceutics, 10(2), 63. https://doi.org/10.3390/pharmaceutics10020063