Combined Effect of Anticancer Agents and Cytochrome C Decorated Hybrid Nanoparticles for Liver Cancer Therapy

 , , ,
, , , 
 and
and 
Abstract
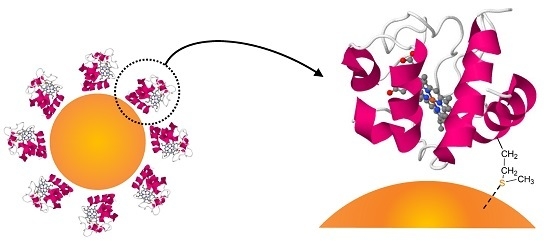
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Synthesis and Decoration of Hybrid Nanoparticles
2.2. Subculture of Cell Lines
2.3. Cellular Uptake of Cytochrome C
2.4. Cell Viability Evaluation
2.4.1. MTT Assay
2.4.2. Trypan Blue Exclusion
2.5. Apoptosis Detection
2.5.1. Caspase-3 Assay
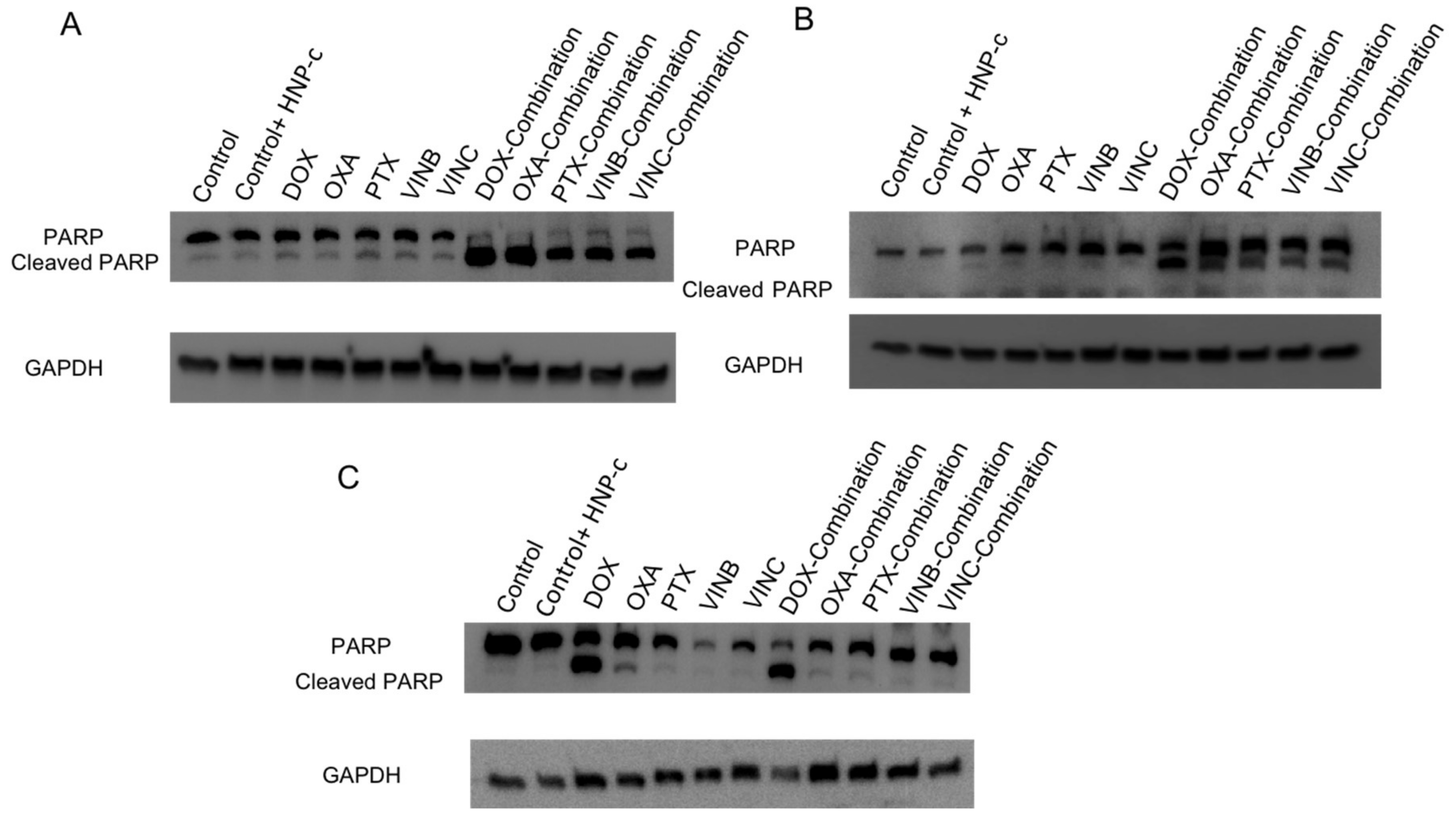
2.5.2. Western Blotting for Detection of Poly (ADP-ribose) polymerase (PARP)
2.5.3. Terminal Deoxynucleotidyl Transferase (dUTP) Nick End Labelling (TUNEL) Assay
2.6. Imaging Studies
2.6.1. Fluorescent Microscope
2.6.2. Nanoparticle Internalisation Imaging
3. Results and Discussion
3.1. Cellular Internalisation
3.2. Cytotoxicity
3.3. Apoptosis Detection
3.3.1. Caspase-3 Assay
3.3.2. Western Blotting for Detection of PARP
3.3.3. Terminal Deoxynucleotidyl Transferase (dUTP) Nick end Labelling (TUNEL) Assay
3.4. Fluorescent Microscopy
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ananthakrishnan, A.; Gogineni, V.; Saeian, K. Epidemiology of primary and secondary liver cancers. Semin. Interv. Radiol. 2006, 23, 47–63. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Marrero, J.A.; Rudolph, L.; Reddy, K.R. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology 2008, 134, 1752–1763. [Google Scholar] [CrossRef] [PubMed]
- Cancer Research UK. Liver Cancer. 2015. Available online: http://www.cancerresearchuk.org/about-cancer/liver-cancer/survival/ (accessed on 20 November 2017).
- Ma, M.-C.; Chen, Y.-Y.; Li, S.-H.; Cheng, Y.-F.; Wang, C.-C.; Chiu, T.-J.; Pei, S.-N.; Liu, C.-T.; Huang, T.-L.; Huang, C.-H.; et al. Intra-arterial chemotherapy with doxorubicin and cisplating is effective for advanced hepatocellular cell carcinoma. Sci. World J. 2014, 2014, 160138. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Ameisen, J.C. On the origin, evolution, and nature of programmed cell death: A timeline of four billion years. Cell Death Differ. 2002, 9, 367–393. [Google Scholar] [CrossRef] [PubMed]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Multiple pathways of cytochrome C release from mitochondria in apoptosis. Biochim. Biophys. Acta 2006, 1757, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Slowing, I.I.; Trewyn, B.G.; Lin, V.S.Y. Mesoporous silica nanoparticles for intracellular delivery of membrane-impermeable proteins. J. Am. Chem. Soc. 2007, 129, 8845–8849. [Google Scholar] [CrossRef] [PubMed]
- Morales-Cruz, M.; Figueroa, C.M.; González-Robles, T.; Delgado, Y.; Molina, A.; Méndez, J.; Morales, M.; Griebenow, K. Activation of caspase-dependent apoptosis by intracellular delivery of cytochrome C-based nanoparticles. J. Nanobiotechnol. 2014, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Barnett, C.M.; Gueorguieva, M.; Lees, M.R.; Mc Garvey, D.J.; Hoskins, C. Physical stability, biocompatibility and potential use of hybrid iron oxide-gold nanoparticles as drug carriers. J. Nanopart. Res. 2013, 15, 1706. [Google Scholar] [CrossRef]
- Malekigorji, M.; Hoskins, C.; Curtis, T.; Varbiro, G. Enhancement of the cytotoxic effect of anticancer agent by cytochrome C functionalised hybrid nanoparticles in hepatocellular cancer cells. J. Nanomed. Res. 2014, 1, 00010. [Google Scholar] [CrossRef]
- Malekigorji, M.; Alfahad, M.; Kong Thoo Lin, P.; Jones, S.; Curtis, A.; Hoskins, C. Thermally triggered theranostics for pancreatic cancer therapy. Nanoscale 2017, 9, 12735–12745. [Google Scholar] [CrossRef] [PubMed]
- Oluwasanmi, A.; Al-Shakarchi, W.; Manzur, A.; Aldebasi, M.H.; Elsini, R.S.; Albusair, M.K.; Haxton, K.J.; Curtis, A.D.M.; Hoskins, C. Diels alder-mediated release of gemcitabine from hybrid nanoparticles for enhanced pancreatic cancer therapy. J. Control. Release 2017, 266, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, L.; Wang, W. Current status of multimodal & combination therapy for hepatocellular carcinoma. Indian J. Med. Res. 2012, 136, 391–403. [Google Scholar] [PubMed]
- Thein, H.-H.; Isaranuwatchai, W.; Qiao, Y.; Wong, K.; Sapisochin, G.; Chan, K.K.W.; Yoshida, E.M.; Earle, C.C. Cost-effectiveness analysis of potentially curative and combination treatments for hepatocellular carcinoma with personal-level data in a Canadian setting. Cancer Med. 2017, 6, 2017–2033. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, C.; Min, Y.; Gueorguieva, M.; McDougall, C.; Volovick, A.; Prentice, P.; Wang, Z.; Melzer, A.; Cuschieri, A.; Wang, L. Hybrid gold-iron oxide nanoparticles a s a multifunctional platform for biomedical application. J. Nanobiotechnol. 2012, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Barnett, C.M.; Gueorguieva, M.; Lees, M.R.; McGarvey, D.J.; Darton, R.J.; Hoskins, C. Effect of hybrid composition on the physicochemical properties and morphology of iron oxide-gold nanoparticles. J. Nanopart. Res. 2012, 14, 1170. [Google Scholar] [CrossRef]
- Yamada, Y.; Harashima, H. Mitochondrial drug delivery systems for macromolecule and their therapeutic application to mitochondrial diseases. Adv. Drug Deliv. Rev. 2008, 60, 1439–1462. [Google Scholar] [CrossRef] [PubMed]
- Delgado, Y.; Morales-Cruz, M.; Hernádez-Román, J.; Martínez, Y.; Griebenow, K. Chemical glycosylation of cytochrome C improves physical and chemical protein stability. BMC Biochem. 2014, 15, 16. [Google Scholar] [CrossRef] [PubMed]
- Yallapu, M.M.; Othman, S.F.; Curtis, E.T.; Gupta, B.K.; Jaggi, M.; Chauhan, S.C. Multi-functional magnetic nanoparticles for magnetic resonance imaging and cancer therapy. Biomaterials 2011, 32, 1890–1905. [Google Scholar] [CrossRef] [PubMed]
- Pissuwan, D.; Valenzueka, S.M.; Cortie, M.B. Therapeutic possibilities of plasmonically heated gold nanoparticles. Trends Biotechnol. 2006, 24, 62–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eun, J.R.; Jung, Y.J.; Zhang, Y.; Zhang, Y.; Tschudy-Seney, B.; Ramsamooj, R.; Wan, Y.-J.Y.; Theise, N.D.; Zern, M.A.; Duan, Y. Hepatoma SK Hep-1 Cells Exhibit Characteristics of Oncogenic Mesenchymal Stem Cells with Highly Metastatic Capacity. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- El-Kareh, A.W.; Secomb, T.W. Two-Mechanism Peak Concentration Model for Cellular Pharmacodynamics of Doxorubicin. Neoplasia 2005, 7, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.; Jänicke, R.U.; Schulze-Osthoff, K. Many cuts to ruin: A comprehensive update of caspase substrates. Cell Death Differ. 2003, 10, 76–100. [Google Scholar] [CrossRef] [PubMed]
- Gavrieli, Y.; Sherman, Y.; Ben-Sasson, S.A. Identification of programmed cell death in situ via specific labelling of nuclear DNA fragmentation. J. Cell Biol. 1992, 119, 493–501. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef] [PubMed]
- Imesch, P.; Scheiner, D.; Szabo, E.; Fink, D.; Fedier, A. Conjugates of cytochrome C and antennapedia peptide activate apoptosis and inhibit proliferation of HeLa cancer cells. Exp. Ther. Med. 2013, 6, 786–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, W.S.; Lee, M.G.; Verwilst, P.L.; Lee, J.H.; Chi, S.-G.; Kim, J.S. Mitochondria-targeted aggregation induced emission theranostics: Crucial importance of in situ activation. Chem. Sci. 2016, 7, 6050–6059. [Google Scholar] [CrossRef]
- Figueroa, C.M.; Suárez, B.N.; Molina, A.M.; Fernández, J.C.; Torres, Z.; Griebenow, K. Smart Release Nano-formulation of cytochrome C and Hyaluronic Acid Induces Apoptosis in Cancer Cells. J. Nanomed. Nanotechnol. 2017, 8, 427. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhang, Q.; Dai, L.; Shen, X.; Chen, W.; Cai, K. Phenylboronic acid-modified hollow silica nanoparticles for dual-responsive delivery of doxorubicin for targeted tumor therapy. Regen. Biomater. 2017, 4, 111–124. [Google Scholar] [CrossRef]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Shakarchi, W.; Alsuraifi, A.; Abed, M.; Abdullah, M.; Richardson, A.; Curtis, A.; Hoskins, C. Combined Effect of Anticancer Agents and Cytochrome C Decorated Hybrid Nanoparticles for Liver Cancer Therapy. Pharmaceutics 2018, 10, 48. https://doi.org/10.3390/pharmaceutics10020048
Al-Shakarchi W, Alsuraifi A, Abed M, Abdullah M, Richardson A, Curtis A, Hoskins C. Combined Effect of Anticancer Agents and Cytochrome C Decorated Hybrid Nanoparticles for Liver Cancer Therapy. Pharmaceutics. 2018; 10(2):48. https://doi.org/10.3390/pharmaceutics10020048
Chicago/Turabian StyleAl-Shakarchi, Wejdan, Ali Alsuraifi, Mohammed Abed, Marwan Abdullah, Alan Richardson, Anthony Curtis, and Clare Hoskins. 2018. "Combined Effect of Anticancer Agents and Cytochrome C Decorated Hybrid Nanoparticles for Liver Cancer Therapy" Pharmaceutics 10, no. 2: 48. https://doi.org/10.3390/pharmaceutics10020048
APA StyleAl-Shakarchi, W., Alsuraifi, A., Abed, M., Abdullah, M., Richardson, A., Curtis, A., & Hoskins, C. (2018). Combined Effect of Anticancer Agents and Cytochrome C Decorated Hybrid Nanoparticles for Liver Cancer Therapy. Pharmaceutics, 10(2), 48. https://doi.org/10.3390/pharmaceutics10020048