Simultaneous Determination of Five Cytochrome P450 Probe Substrates and Their Metabolites and Organic Anion Transporting Polypeptide Probe Substrate in Human Plasma Using Liquid Chromatography-Tandem Mass Spectrometry


 ,
,  and
and 
Abstract
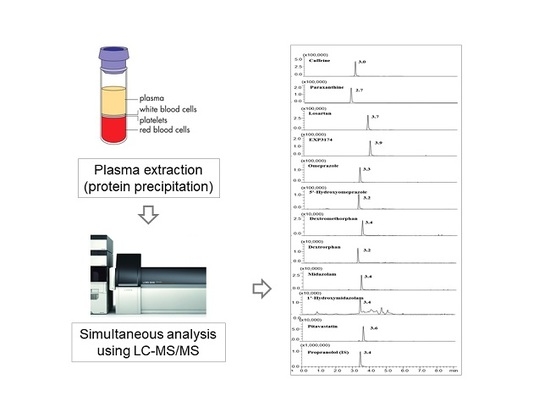
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Preparation of Calibration Standard Samples
2.3. Plasma Sample Preparation
2.4. LC-MS/MS Analysis
2.5. Method Validation
2.6. Application to Pharmacokinetic Studies
3. Results and Discussion
3.1. Optimization of Analytical Conditions and Sample Preparation
3.2. Method Validation
3.3. Clinical Applications
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Michalets, E.L. Update: Clinically significant cytochrome P450 drug interactions. Pharmacotherapy 1998, 18, 84–112. [Google Scholar] [PubMed]
- Nelson, D.R. The cytochrome P450 homepage. Hum. Genom. 2009, 4, 59–65. [Google Scholar]
- Zhang, S.; Song, N.; Li, Q.; Fan, H.; Liu, C. Liquid chromatography/tandem mass spectrometry method for simultaneous evaluation of activities of five cytochrome P450s using a five-drug cocktail and application to cytochrome p450 phenotyping studies in rats. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 871, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Paine, M.F.; Khalighi, M.; Fisher, J.M.; Shen, D.D.; Kunze, K.L.; Marsh, C.L.; Perkins, J.D.; Thummel, K.E. Characterization of interintestinal and intraintestinal variations in human CYP3A-dependent metabolism. J. Pharmacol. Exp. Ther. 1997, 283, 1552–1562. [Google Scholar] [PubMed]
- Indiaina University. P450 Drug Interaction Table. Available online: http://medicine.iupui.edu/clinpharm/ddis/main-table (accessed on 18 May 2018).
- U.S. Food and Drug Administration. Drug Interactions & Labeling. Available online: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling (accessed on 18 May 2018).
- International Transporter Consortium; Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Hartkoorn, R.C.; Kwan, W.S.; Shallcross, V.; Chaikan, A.; Liptrott, N.; Egan, D.; Sora, E.S.; James, C.E.; Gibbons, S.; Bray, P.G.; et al. Hiv protease inhibitors are substrates for OATP1A2, OATP1B1 and OATP1B3 and lopinavir plasma concentrations are influenced by SLCO1B1 polymorphisms. Pharmacogenet. Genom. 2010, 20, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Maeda, K.; Sugiyama, Y. Transporter-mediated drug–drug interactions involving oatp substrates: Predictions based on in vitro inhibition studies. Clin. Pharmacol. Ther. 2012, 91, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Ieiri, I.; Tsunemitsu, S.; Maeda, K.; Ando, Y.; Izumi, N.; Kimura, M.; Yamane, N.; Okuzono, T.; Morishita, M.; Kotani, N.; et al. Mechanisms of pharmacokinetic enhancement between ritonavir and saquinavir; micro/small dosing tests using midazolam (CYP3A4), fexofenadine (P-glycoprotein), and pravastatin (OATP1B1) as probe drugs. J. Clin. Pharmacol. 2013, 53, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.G.; Kim, Y.; Jeon, J.Y.; Kim, D.S. Effect of fermented red ginseng on cytochrome P450 and P-glycoprotein activity in healthy subjects, as evaluated using the cocktail approach. Br. J. Clin. Pharmacol. 2016, 82, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.S.; Park, S.J.; Shinde, D.D.; Shin, J.G.; Kim, D.H. High-sensitivity liquid chromatography-tandem mass spectrometry for the simultaneous determination of five drugs and their cytochrome P450-specific probe metabolites in human plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 895–896, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Bosilkovska, M.; Deglon, J.; Samer, C.; Walder, B.; Desmeules, J.; Staub, C.; Daali, Y. Simultaneous LC-MS/MS quantification of P-glycoprotein and cytochrome P450 probe substrates and their metabolites in dbs and plasma. Bioanalysis 2014, 6, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.Y.; Song, I.S.; Sunwoo, Y.E.; Shon, J.H.; Liu, K.H.; Cha, I.J.; Shin, J.G. Development of the “Inje cocktail” for high-throughput evaluation of five human cytochrome P450 isoforms in vivo. Clin. Pharmacol. Ther. 2007, 82, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.; Tao, X.; Zhu, L.; Stonier, M.; Lutz, J.D.; Masson, E.; Zhang, S.; Ganguly, B.; Tzogas, Z.; Lubin, S.; et al. Use of a cocktail probe to assess potential drug interactions with cytochrome P450 after administration of belatacept, a costimulatory immunomodulator. Br. J. Clin. Pharmacol. 2017, 83, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Streetman, D.S.; Bleakley, J.F.; Kim, J.S.; Nafziger, A.N.; Leeder, J.S.; Gaedigk, A.; Gotschall, R.; Kearns, G.L.; Bertino, J.S., Jr. Combined phenotypic assessment of CYP1A2, CYP2C19, CYP2D6, CYP3A, N-acetyltransferase-2, and xanthine oxidase with the “Cooperstown cocktail”. Clin. Pharmacol. Ther. 2000, 68, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Zgheib, N.K.; Frye, R.F.; Tracy, T.S.; Romkes, M.; Branch, R.A. Validation of incorporating flurbiprofen into the pittsburgh cocktail. Clin. Pharmacol. Ther. 2006, 80, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.; Andersson, K.; Dalen, P.; Mirghani, R.A.; Muirhead, G.J.; Nordmark, A.; Tybring, G.; Wahlberg, A.; Yasar, U.; Bertilsson, L. The Karolinska cocktail for phenotyping of five human cytochrome P450 enzymes. Clin. Pharmacol. Ther. 2003, 73, 517–528. [Google Scholar] [CrossRef]
- Grangeon, A.; Gravel, S.; Gaudette, F.; Turgeon, J.; Michaud, V. Highly sensitive LC-MS/MS methods for the determination of seven human CYP450 activities using small oral doses of probe-drugs in human. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1040, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Shelepova, T.; Nafziger, A.N.; Victory, J.; Kashuba, A.D.; Rowland, E.; Zhang, Y.; Sellers, E.; Kearns, G.; Leeder, J.S.; Gaedigk, A.; et al. Effect of a triphasic oral contraceptive on drug-metabolizing enzyme activity as measured by the validated Cooperstown 5 + 1 cocktail. J. Clin. Pharmacol. 2005, 45, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Prueksaritanont, T.; Chu, X.; Evers, R.; Klopfer, S.O.; Caro, L.; Kothare, P.A.; Dempsey, C.; Rasmussen, S.; Houle, R.; Chan, G.; et al. Pitavastatin is a more sensitive and selective organic anion-transporting polypeptide 1b clinical probe than rosuvastatin. Br. J. Clin. Pharmacol. 2014, 78, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Ikeda, Y.; Fujita, T.; Yoshida, K.; Azuma, Y.; Haruyama, Y.; Yamane, N.; Kumagai, Y.; Sugiyama, Y. Identification of the rate-determining process in the hepatic clearance of atorvastatin in a clinical cassette microdosing study. Clin. Pharmacol. Ther. 2011, 90, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Stopfer, P.; Giessmann, T.; Hohl, K.; Sharma, A.; Ishiguro, N.; Taub, M.E.; Zimdahl-Gelling, H.; Gansser, D.; Wein, M.; Ebner, T.; et al. Pharmacokinetic evaluation of a drug transporter cocktail consisting of digoxin, furosemide, metformin, and rosuvastatin. Clin. Pharmacol. Ther. 2016, 100, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Maeda, K.; Shitara, Y.; Sugiyama, Y. Contribution of OATP2 (PATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J. Pharmacol. Exp. Ther. 2004, 311, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, S.; Maeda, K.; Wang, Y.; Sugiyama, Y. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab. Dispos. 2008, 36, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Kusuhara, H.; Sugiyama, D.; Ito, K.; Ueda, S.; Endou, H.; Sugiyama, Y. Functional involvement of rat organic anion transporter 3 (rOAT3; SLC22A8) in the renal uptake of organic anions. J. Pharmacol. Exp. Ther. 2002, 300, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Nishizato, Y.; Ieiri, I.; Suzuki, H.; Kimura, M.; Kawabata, K.; Hirota, T.; Takane, H.; Irie, S.; Kusuhara, H.; Urasaki, Y.; et al. Polymorphisms of OATP-C (SLC21A6) and OAT3 (SLC22A8) genes: Consequences for pravastatin pharmacokinetics. Clin. Pharmacol. Ther. 2003, 73, 554–565. [Google Scholar] [CrossRef]
- Jindal, K.; Narayanam, M.; Singh, S. A systematic strategy for the identification and determination of pharmaceuticals in environment using advanced lc-ms tools: Application to ground water samples. J. Pharm. Biomed. Anal. 2015, 108, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Ardjomand-Woelkart, K.; Kollroser, M.; Li, L.; Derendorf, H.; Butterweck, V.; Bauer, R. Development and validation of a LC-MS/MS method based on a new 96-well hybrid-spe-precipitation technique for quantification of CYP450 substrates/metabolites in rat plasma. Anal. Bioanal. Chem. 2011, 400, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Miao, L.; Lin, L.; Ren, C.Y.; Liu, J.X.; Cui, Y.M. Repeated administration of sailuotong, a fixed combination of panax ginseng, ginkgo biloba, and crocus sativus extracts for vascular dementia, alters CYP450 activities in rats. Phytomedicine 2018, 38, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Uchida, S.; Inui, N.; Takeuchi, K.; Watanabe, H.; Namiki, N. Simultaneous LC-MS/MS analysis of the plasma concentrations of a cocktail of 5 cytochrome P450 substrate drugs and their metabolites. Biol. Pharm. Bull. 2014, 37, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Snyder, B.D.; Rowland, A.; Polasek, T.M.; Miners, J.O.; Doogue, M.P. Evaluation of felodipine as a potential perpetrator of pharmacokinetic drug-drug interactions. Eur. J. Clin. Pharmacol. 2014, 70, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Han, F.; Guo, P.; Zhao, H.; Lin, Z.J.; Huang, M.Q.; Bertelsen, K.; Weng, N. Simultaneous determination of tolbutamide, omeprazole, midazolam and dextromethorphan in human plasma by LC-MS/MS—A high throughput approach to evaluate drug-drug interactions. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Kim, H.; Cha, I.J.; Park, J.S.; Shon, J.H.; Liu, K.H.; Shin, J.G. High-throughput screening of inhibitory potential of nine cytochrome P450 enzymes in vitro using liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 2651–2658. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Ji, H.K.; Lee, T.; Liu, K.H. Simultaneous screening of activities of five cytochrome P450 and four uridine 5’-diphospho-glucuronosyltransferase enzymes in human liver microsomes using cocktail incubation and liquid chromatography-tandem mass spectrometry. Drug Metab. Dispos. 2015, 43, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.H.; Lee, Y.K.; Sunwoo, Y.E.; Yu, K.S.; Kang, W.; Lee, S.S.; Yoon, Y.R.; Shin, J.G. High-performance liquid chromatographic assay of zonisamide (1,2-benzisoxazole-3-methanesulfonamide) in human plasma using a solid-phase extraction technique. Chromatographia 2004, 59, 497–500. [Google Scholar] [CrossRef]




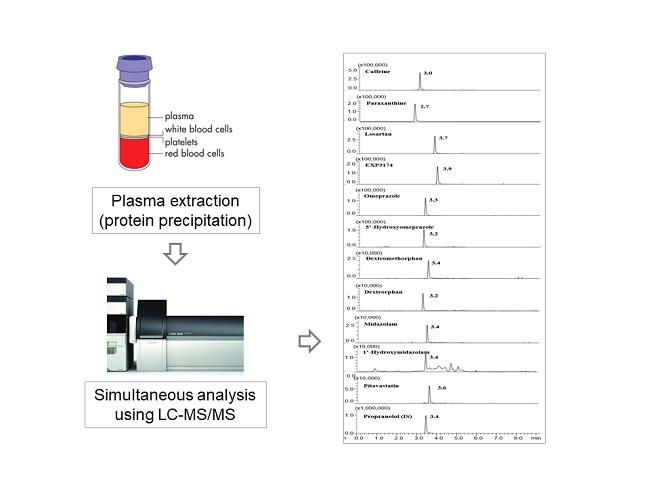{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Retention Time (min) | Calibration Range (ng/mL) | Correlation Coefficient (r2) | LOQ (ng/mL) |
|---|---|---|---|---|
| Caffeine | 2.9 | 5–4000 | 0.9982 ± 0.002 | 5.0 |
| Paraxanthine | 2.7 | 5–4000 | 0.9989 ± 0.001 | 5.0 |
| Losartan | 3.7 | 0.5–400 | 0.9989 ± 0.001 | 0.5 |
| EXP3174 | 3.8 | 0.5–400 | 0.9978 ± 0.002 | 0.5 |
| Omeprazole | 3.2 | 0.5–400 | 0.9973 ± 0.002 | 0.5 |
| 5′-Hydroxyomeprazole | 3.2 | 0.5–400 | 0.9984 ± 0.002 | 0.5 |
| Dextromethorphan | 3.4 | 0.05–40 | 0.9982 ± 0.002 | 0.05 |
| Dextrorphan | 3.1 | 0.05–40 | 0.9979 ± 0.002 | 0.05 |
| Midazolam | 3.4 | 0.05–40 | 0.9985 ± 0.001 | 0.05 |
| 1′-Hydroxymidazolam | 3.4 | 0.05–100 | 0.9938 ± 0.006 | 0.05 |
| Pitavastatin | 3.5 | 0.5–400 | 0.9987 ± 0.001 | 0.5 |
| Analyte | SRM Transition Ions (m/z) [13,29] | CE (eV) | |
|---|---|---|---|
| Precursor Ion | Product Ion | ||
| Caffeine | 195.0 | 138.0 | 20 |
| Paraxanthine | 181.0 | 124.0 | 20 |
| Losartan | 423.0 | 207.0 | 23 |
| EXP3174 | 437.0 | 235.0 | 20 |
| Omeprazole | 346.0 | 198.0 | 15 |
| 5′-Hydroxyomeprazole | 362.0 | 214.0 | 15 |
| Dextromethorphan | 272.0 | 171.0 | 37 |
| Dextrorphan | 258.0 | 157.0 | 40 |
| Midazolam | 326.0 | 291.0 | 27 |
| 1′-Hydroxymidazolam | 342.0 | 324.0 | 23 |
| Pitavastatin | 422.0 | 290.0 | 33 |
| Propranolol (IS) | 261.0 | 184.0 | 20 |
| Analyte | Nominal Concentration (ng/mL) | Intra-Day (n = 5) | Inter-Day (n = 6) | ||||
|---|---|---|---|---|---|---|---|
| Measured (ng/mL) * | RSD ** (%) | Accuracy (%) | Measured (ng/mL) * | RSD ** (%) | Accuracy (%) | ||
| Caffeine | 20.0 | 22.2 ± 0.4 | 2.0 | 110.7 | 21.8 ± 0.8 | 3.5 | 109.2 |
| 100.0 | 110.2 ± 2.2 | 2.0 | 110.2 | 103.6 ± 4.0 | 3.9 | 103.6 | |
| 1000 | 1071.0 ± 26.5 | 2.5 | 107.1 | 975.8 ± 28.0 | 2.8 | 97.6 | |
| Paraxanthine | 20.0 | 22.2 ± 0.5 | 2.0 | 110.9 | 21.5 ± 0.8 | 3.7 | 107.3 |
| 100.0 | 113.9 ± 0.8 | 0.7 | 113.9 | 102.6 ± 4.2 | 4.1 | 102.6 | |
| 1000 | 1073.8 ± 25.8 | 2.4 | 107.4 | 997.1 ± 23.9 | 2.4 | 99.7 | |
| Losartan | 2.0 | 2.1 ± 0.0 | 1.5 | 104.8 | 2.1 ± 0.1 | 5.5 | 103.0 |
| 10.0 | 10.7 ± 0.2 | 1.8 | 107.1 | 10.0 ± 0.2 | 1.8 | 100.4 | |
| 100 | 102.2 ± 0.8 | 0.8 | 102.2 | 99.0 ± 1.9 | 1.9 | 99.0 | |
| EXP3174 | 2.0 | 2.1 ± 0.0 | 1.9 | 106.1 | 2.0 ± 0.1 | 4.0 | 101.1 |
| 10.0 | 10.4 ± 0.2 | 1.7 | 104.3 | 10.0 ± 0.3 | 3.0 | 100.0 | |
| 100 | 99.1 ± 1.2 | 1.5 | 99.1 | 98.9 ± 4.6 | 4.7 | 98.9 | |
| Omeprazole | 2.0 | 2.1 ± 0.1 | 3.3 | 104.8 | 2.0 ± 0.1 | 5.6 | 102.0 |
| 10.0 | 11.2 ± 0.2 | 1.3 | 111.8 | 10.1 ± 0.7 | 6.5 | 101.4 | |
| 100 | 106.5 ± 1.0 | 1.0 | 106.5 | 96.7 ± 6.6 | 6.8 | 96.7 | |
| 5′-Hydroxyomeprazole | 2.0 | 2.0 ± 0.1 | 4.3 | 101.6 | 2.0 ± 0.1 | 2.7 | 102.0 |
| 10.0 | 10.7 ± 0.2 | 1.8 | 106.6 | 10.1 ± 0.4 | 3.6 | 100.6 | |
| 100 | 101.2 ± 1.3 | 1.3 | 101.2 | 96.1 ± 3.1 | 3.2 | 96.1 | |
| Dextromethorphan | 0.2 | 0.2 ± 0.0 | 7.3 | 97.1 | 0.2 ± 0.0 | 5.0 | 100.0 |
| 1.0 | 1.2 ± 0.0 | 2.2 | 117.3 | 1.0 ± 0.0 | 3.1 | 101.8 | |
| 10 | 11.1 ± 0.1 | 1.0 | 111.3 | 10.2 ± 0.1 | 1.1 | 101.7 | |
| Dextrorphan | 0.2 | 0.2 ± 0.0 | 11.3 | 100.2 | 0.2 ± 0.0 | 8.8 | 103.0 |
| 1.0 | 1.1 ± 0.0 | 3.3 | 106.5 | 1.0 ± 0.0 | 2.6 | 103.4 | |
| 10 | 10.7 ± 0.3 | 2.5 | 106.5 | 10.2 ± 0.1 | 1.3 | 101.6 | |
| Midazolam | 0.2 | 0.2 ± 0.0 | 6.3 | 103.1 | 0.2 ± 0.0 | 2.7 | 102.0 |
| 1.0 | 1.1 ± 0.0 | 3.5 | 111.1 | 1.0 ± 0.0 | 1.6 | 100.8 | |
| 10 | 11.0 ± 0.1 | 0.6 | 109.9 | 10.1 ± 0.1 | 0.7 | 100.7 | |
| 1′-Hydroxymidazolam | 0.2 | 0.2 ± 0.0 | 6.3 | 88.5 | 0.1 ± 0.0 | 4.1 | 101.0 |
| 1.0 | 0.9 ± 0.0 | 3.8 | 95.3 | 1.0 ± 0.1 | 6.3 | 99.0 | |
| 10 | 10.7 ± 0.1 | 1.2 | 106.9 | 10.0 ± 0.4 | 3.5 | 100.3 | |
| Pitavastatin | 2.0 | 2.1 ± 0.1 | 2.8 | 105.3 | 2.1 ± 0.1 | 4.4 | 104.9 |
| 10.0 | 11.5 ± 0.3 | 2.9 | 114.5 | 10.5 ± 0.5 | 5.1 | 105.3 | |
| 100 | 107.5 ± 1.6 | 1.5 | 107.5 | 99.2 ± 6.2 | 6.2 | 99.2 | |
| Analyte | Nominal Concentration (ng/mL) | 4 h Short-Term Stability (25 °C) | Freeze-Thaw Stability (−80 °C/Room Temperature) | 24 h Post-Treatment Stability (4 °C) |
|---|---|---|---|---|
| Caffeine | 100 | 91.8 ± 2.1 | 99.4 ± 3.3 | 96.2 ± 0.8 |
| 1000 | 96.7 ± 7.6 | 93.9 ± 5.4 | 91.7 ± 2.3 | |
| Paraxanthine | 100 | 86.1 ± 4.6 | 94.7 ± 1.4 | 96.8 ± 4.1 |
| 1000 | 91.7 ± 7.5 | 90.0 ± 4.6 | 92.8 ± 1.5 | |
| Losartan | 10 | 94.5 ± 2.1 | 101.9 ± 2.7 | 98.4 ± 3.4 |
| 100 | 93.8 ± 3.8 | 95.6 ± 3.3 | 95.0 ± 2.0 | |
| EXP3174 | 10 | 90.5 ± 2.9 | 95.2 ± 2.6 | 98.8 ± 1.4 |
| 100 | 91.9 ± 6.3 | 92.9 ± 4.0 | 93.1 ± 1.9 | |
| Omeprazole | 10 | 89.8 ± 1.2 | 93.8 ± 1.8 | 94.7 ± 2.0 |
| 100 | 88.9 ± 6.5 | 88.4 ± 3.5 | 94.6 ± 1.6 | |
| 5′-Hydroxyomeprazole | 10 | 99.0 ± 3.7 | 99.9 ± 6.6 | 96.5 ± 2.2 |
| 100 | 91.0 ± 4.1 | 91.1 ± 4.8 | 91.0 ± 1.1 | |
| Dextromethorphan | 1 | 107.2 ± 5.3 | 102.6 ± 6.4 | 102.5 ± 2.4 |
| 10 | 100.5 ± 3.1 | 114.5 ± 3.6 | 102.5 ± 1.3 | |
| Dextrorphan | 1 | 88.3 ± 5.2 | 91.0 ± 2.0 | 95.4 ± 12.4 |
| 10 | 87.4 ± 6.3 | 92.1 ± 4.9 | 94.5 ± 4.3 | |
| Midazolam | 1 | 105.9 ± 1.3 | 106.8 ± 5.3 | 100.5 ± 3.1 |
| 10 | 104.1 ± 3.4 | 112.3 ± 1.3 | 103.6 ± 1.1 | |
| 1′-Hydroxymidazolam | 1 | 102.0 ± 5.0 | 99.7 ± 5.9 | 110.5 ± 0.3 |
| 10 | 99.9 ± 7.5 | 107.3 ± 4.8 | 102.5 ± 3.0 | |
| Pitavastatin | 10 | 91.0 ± 2.4 | 93.5 ± 3.6 | 99.1 ± 1.7 |
| 100 | 93.4 ± 9.4 | 89.7 ± 2.6 | 95.4 ± 1.2 |
| Probe Drug | Pharmacokinetic Parameters | Mean ± SD |
|---|---|---|
| Caffeine | AUC0–48 (h·ng/mL) | 27,327.6 ± 18,012.4 |
| (CYP1A2) | Cmax (ng/mL) | 2350.4 ± 843.1 |
| Tmax (h) | 0.75 (0.25–1.50) | |
| t1/2 (h) | 8.84 ± 3.01 | |
| MRT (h) | 11.1 ± 4.0 | |
| Paraxanthine | AUC0–48 (h·ng/mL) | 24,063.5 ± 13,009.8 |
| Cmax (ng/mL) | 925.3 ± 305.5 | |
| Tmax (h) | 7 (5–24) | |
| t1/2 (h) | 12.46 ± 6.03 | |
| MRT (h) | 16.8 ± 4.7 | |
| Losartan | AUC0–48 (h·ng/mL) | 387.5 ± 121.2 |
| (CYP2C9) | Cmax (ng/mL) | 172.5 ± 62.6 |
| Tmax (h) | 1.25 (0.50–2.00) | |
| t1/2 (h) | 2.14 ± 0.53 | |
| MRT (h) | 2.7 ± 0.3 | |
| EXP3174 | AUC0–48 (h·ng/mL) | 2721.6 ± 1236.2 |
| Cmax (ng/mL) | 352.1 ± 205.6 | |
| Tmax (h) | 3.5 (3.0–5.0) | |
| t1/2 (h) | 6.79 ± 0.58 | |
| MRT (h) | 8.6 ± 1.2 | |
| Omeprazole | AUC0–48 (h·ng/mL) | 1796.2 ± 2076.3 |
| (CYP2C19) | Cmax (ng/mL) | 566.1 ± 367.1 |
| Tmax (h) | 2.0 (1.5–3.0) | |
| t1/2 (h) | 1.34 ± 1.14 | |
| MRT (h) | 3.5 ± 1.5 | |
| 5′-Hydroxyomeprazole | AUC0–48 (h·ng/mL) | 315.6 ± 195.3 |
| Cmax (ng/mL) | 114.1 ± 80.9 | |
| Tmax (h) | 2.0 (1.5–4.0) | |
| t1/2 (h) | 1.59 ± 1.12 | |
| MRT (h) | 3.8 ± 1.0 | |
| Dextromethorphan | AUC0–48 (h·ng/mL) | 41.36 ± 40.33 |
| (CYP2D6) | Cmax (ng/mL) | 3.70 ± 3.23 |
| Tmax (h) | 2.5 (1.0–3.0) | |
| t1/2 (h) | 8.50 ± 2.49 | |
| MRT (h) | 8.7 ± 4.3 | |
| Dextrorphan | AUC0–48 (h·ng/mL) | 63.91 ± 44.51 |
| Cmax (ng/mL) | 10.48 ± 5.32 | |
| Tmax (h) | 1.5 (1.0–3.0) | |
| t1/2 (h) | 6.44 ± 2.74 | |
| MRT (h) | 7.4 ± 1.9 | |
| Midazolam | AUC0–48 (h·ng/mL) | 18.60 ± 9.65 |
| (CYP3A) | Cmax (ng/mL) | 7.61 ± 2.26 |
| Tmax (h) | 0.5 (0.5–1.0) | |
| t1/2 (h) | 3.08 ± 1.43 | |
| MRT (h) | 2.8 ± 0.9 | |
| 1′-Hydroxymidazolam | AUC0–48 (h·ng/mL) | 17.65 ± 9.87 |
| Cmax (ng/mL) | 7.77 ± 2.77 | |
| Tmax (h) | 0.75 (0.50–1.00) | |
| t1/2 (h) | 3.32 ± 3.30 | |
| MRT (h) | 2.5 ± 0.9 | |
| Pitavastatin | AUC0–48 (h·ng/mL) | 198.1 ± 68.4 |
| (OATP1B1) | Cmax (ng/mL) | 81.31 ± 26.04 |
| Tmax (h) | 0.75 (0.50–1.50) | |
| t1/2 (h) | 13.48 ± 5.24 | |
| MRT (h) | 9.8 ± 3.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, J.-K.; Kim, H.-J.; Lee, G.-H.; Ohk, B.; Lee, S.; Song, K.-S.; Song, I.S.; Liu, K.-H.; Yoon, Y.-R. Simultaneous Determination of Five Cytochrome P450 Probe Substrates and Their Metabolites and Organic Anion Transporting Polypeptide Probe Substrate in Human Plasma Using Liquid Chromatography-Tandem Mass Spectrometry. Pharmaceutics 2018, 10, 79. https://doi.org/10.3390/pharmaceutics10030079
Heo J-K, Kim H-J, Lee G-H, Ohk B, Lee S, Song K-S, Song IS, Liu K-H, Yoon Y-R. Simultaneous Determination of Five Cytochrome P450 Probe Substrates and Their Metabolites and Organic Anion Transporting Polypeptide Probe Substrate in Human Plasma Using Liquid Chromatography-Tandem Mass Spectrometry. Pharmaceutics. 2018; 10(3):79. https://doi.org/10.3390/pharmaceutics10030079
Chicago/Turabian StyleHeo, Jae-Kyung, Hyun-Ji Kim, Ga-Hyun Lee, Boram Ohk, Sangkyu Lee, Kyung-Sik Song, Im Sook Song, Kwang-Hyeon Liu, and Young-Ran Yoon. 2018. "Simultaneous Determination of Five Cytochrome P450 Probe Substrates and Their Metabolites and Organic Anion Transporting Polypeptide Probe Substrate in Human Plasma Using Liquid Chromatography-Tandem Mass Spectrometry" Pharmaceutics 10, no. 3: 79. https://doi.org/10.3390/pharmaceutics10030079
APA StyleHeo, J. -K., Kim, H. -J., Lee, G. -H., Ohk, B., Lee, S., Song, K. -S., Song, I. S., Liu, K. -H., & Yoon, Y. -R. (2018). Simultaneous Determination of Five Cytochrome P450 Probe Substrates and Their Metabolites and Organic Anion Transporting Polypeptide Probe Substrate in Human Plasma Using Liquid Chromatography-Tandem Mass Spectrometry. Pharmaceutics, 10(3), 79. https://doi.org/10.3390/pharmaceutics10030079