Overview of the Manufacturing Methods of Solid Dispersion Technology for Improving the Solubility of Poorly Water-Soluble Drugs and Application to Anticancer Drugs

 and
and 
Abstract
:1. Introduction
2. Solid Dispersions
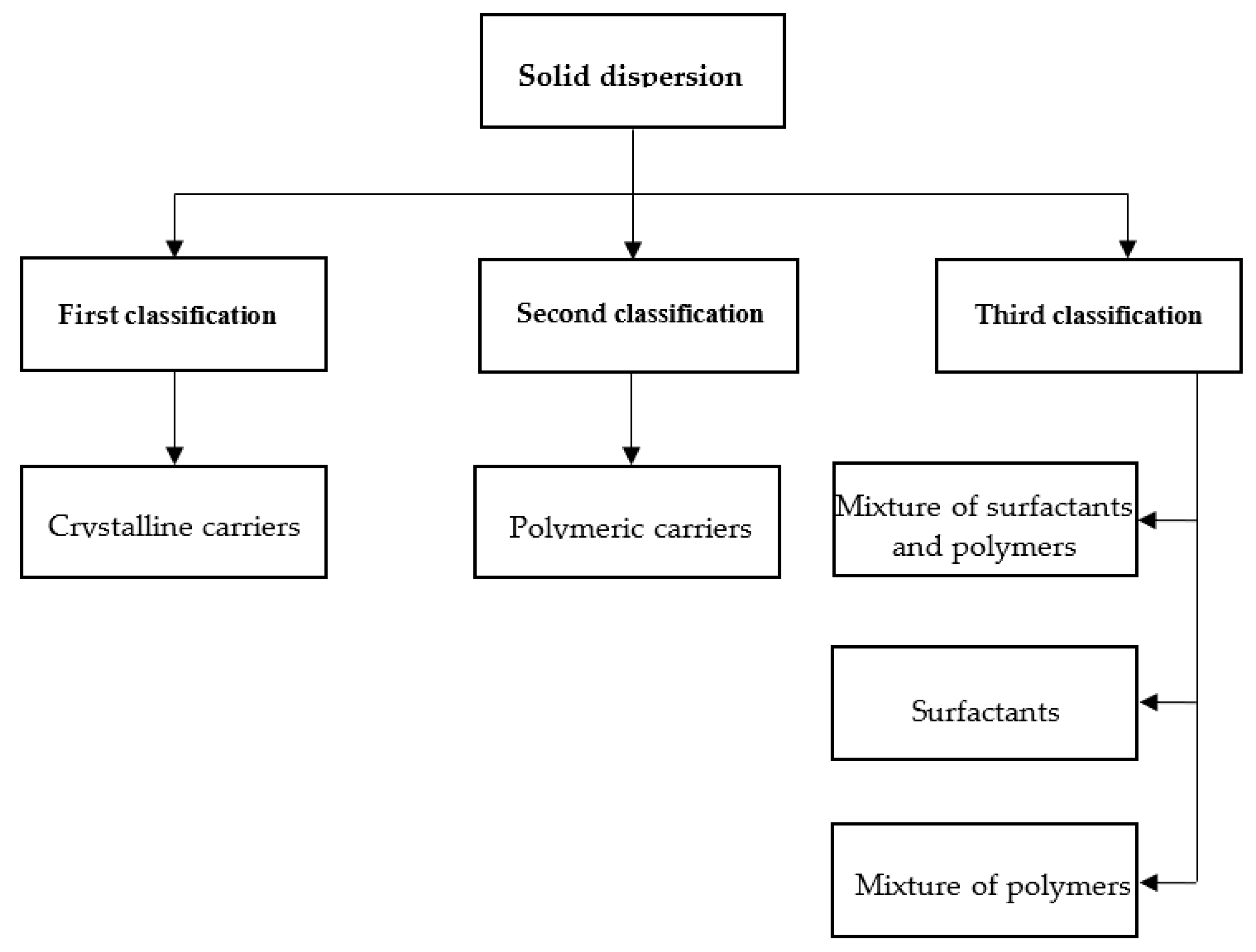
2.1. Carrier-Based Class of Solid Dispersion
2.1.1. First Class of SD
2.1.2. Second Class of SD
2.1.3. Third Class of SD
2.2. Structure-Based Class of Solid Dispersion
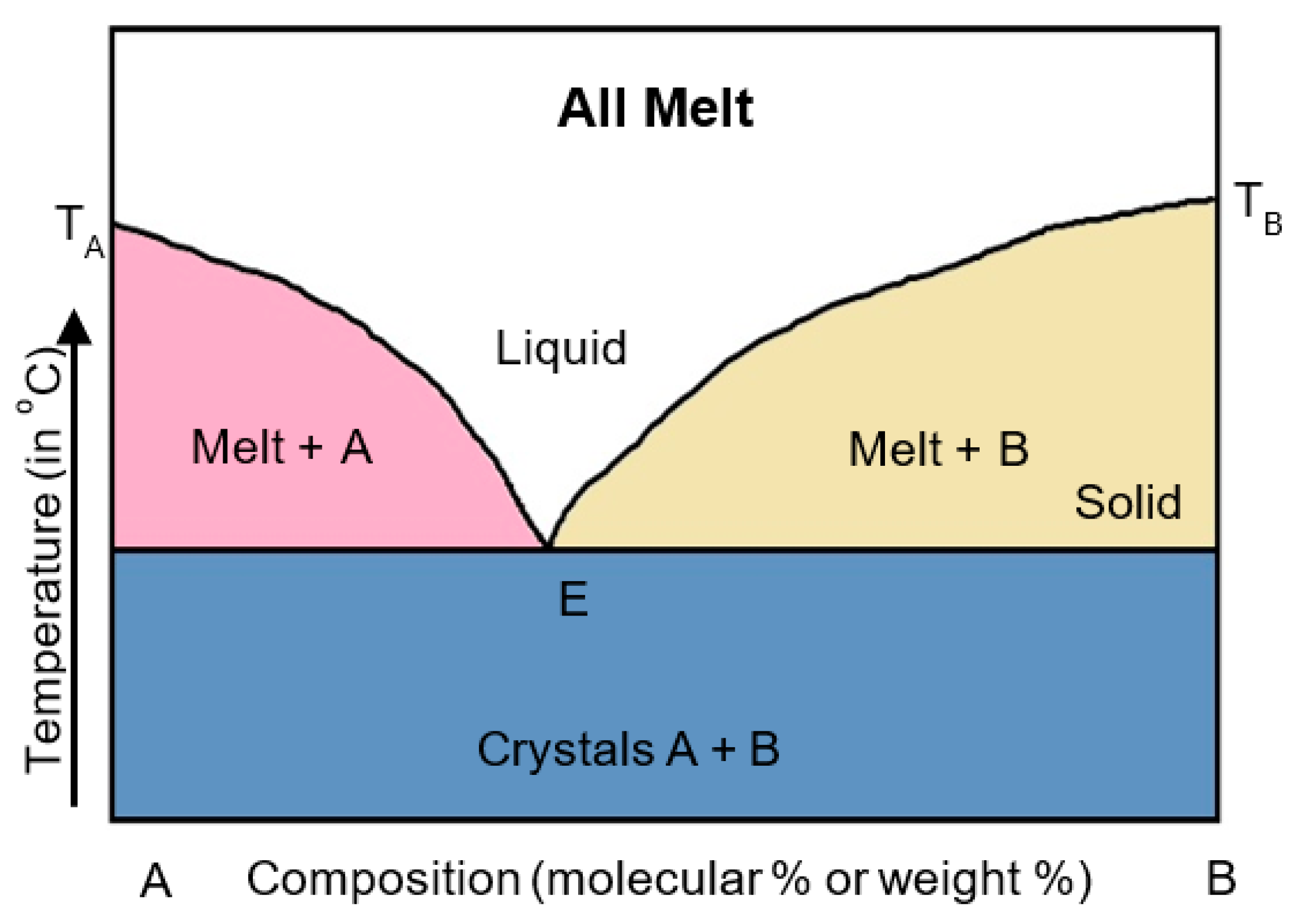
2.2.1. Eutectic Mixtures
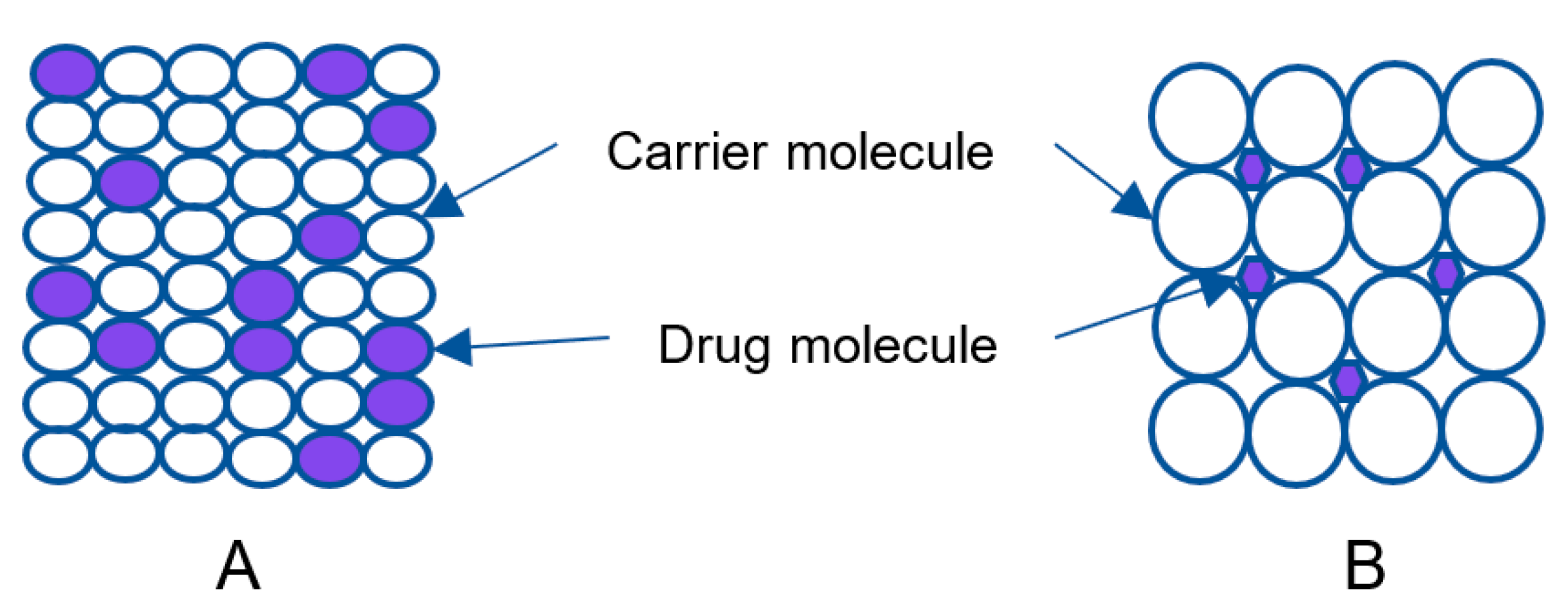
2.2.2. Solid Solution
2.2.3. Glass Solution/Glass Suspension
2.3. Advantages of Solid Dispersions
- One of the most important advantages of SD is drugs interacting with hydrophilic carriers can decrease agglomeration and release in a supersaturation state, resulting in rapid absorption and improved BA [84].
- SD can improve drug wettability and increase the surface area, resulting in enhanced aqueous solubility of drugs.
- SD can be produced as a solid oral dosage form, which is more convenient for patients than other forms like liquid products.
- In addition, SD showed an advantage compared to salt formulation, cocrystallization, and other methods. For example, salt formulations use ionized active pharmaceutical ingredients (APIs) (cationic or anionic form) and are widely used in the pharmaceutical industry due to the broad capacity of design according to desired drug properties. However, not all drugs can ionize with all cations/anions, and phase dissociation or stability issue is inherent in salt formation or cocrystallization. Salt formulation showed several disadvantages such as reduced solubility and dissolution rate, resulting in decreased relative BA (common ion effect for HCl salts); greater regulatory scrutiny for strong acid salts isolated from alkyl alcohols; and increased hygroscopicity, e.g., for Na and, K salts, spray-drying/lyophilization can dissociate strong acid salts. The disadvantages of salt formulation can be resolved when the formulation is produced using an SD.
- Practically, dissolution of drugs is a prerequisite for complete absorption to have the desired therapeutic effect of anticancer drugs after oral administration. Most of the anticancer drugs exhibit poor aqueous solubility causes of dissolution limit resulting low BA and high variability in blood concentration. The limitation of drug dissolution can improve by SD, a technique that induces supersaturated drug dissolution and with that it enhances in vivo absorption.
2.4. Disadvantages of Solid Dispersions
- Physical instability.
- SDs show changes in crystallinity and decreased dissolution rate with aging.
- Due to their thermodynamic instability, SD is sensitive to temperature and humidity during storage. These factors can promote phase separation and crystallization of SD by increasing the overall molecular mobility, decreasing the glass transition temperature (Tg) or disrupting interactions between the drug and carrier, resulting in a decreased solubility and dissolution rate of the drug.
- Patients suffering from cancer should continue to use anticancer drugs during treatment. However, the instability of SD during the period of storage can affect drug quality and the effectiveness of treatment.
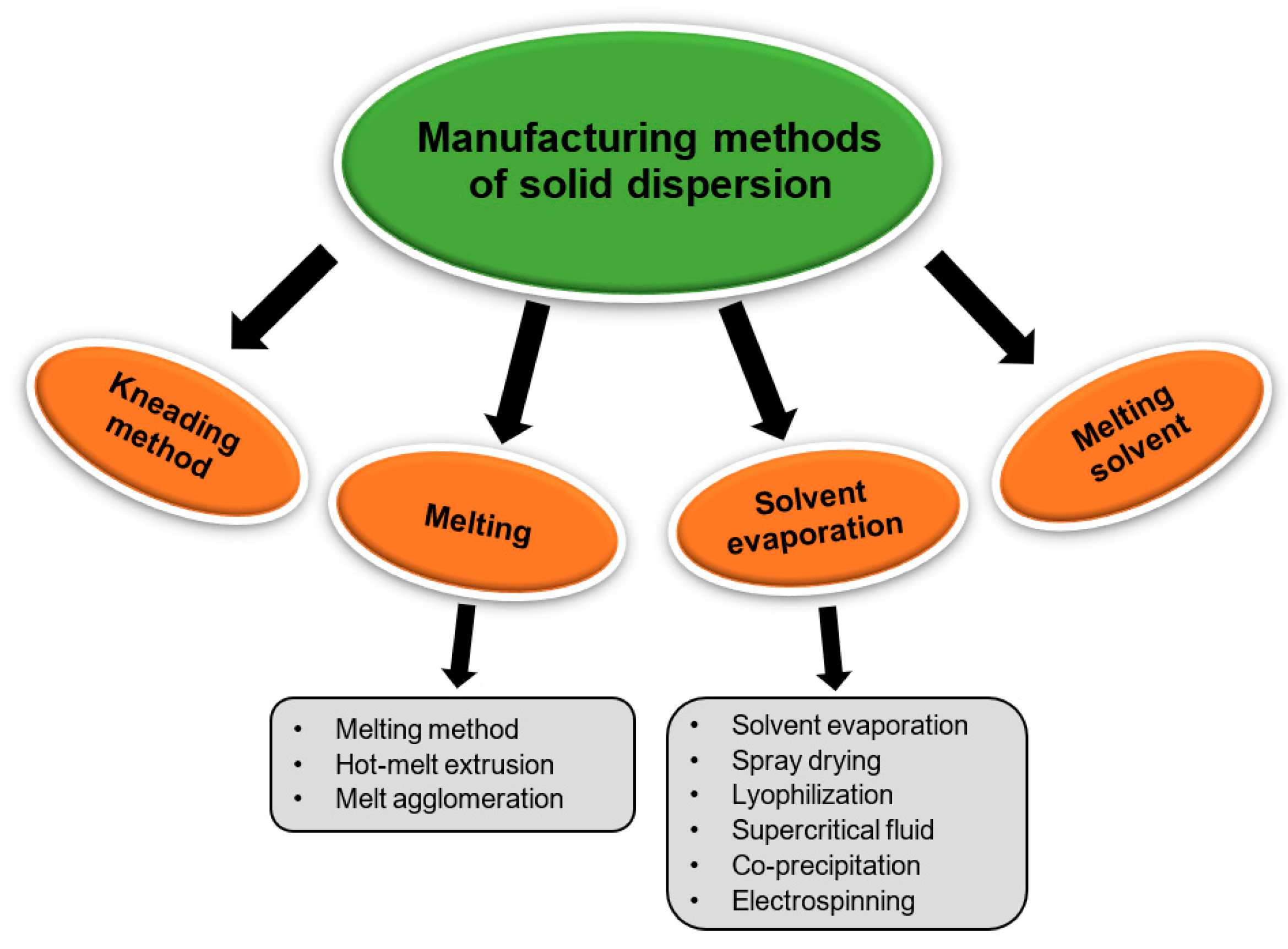
2.5. Preparation Methods for Solid Dispersions
2.5.1. Melting Method/Fusion Method
2.5.2. Solvent Evaporation Method
2.5.3. Melting Solvent Method (Melt Evaporation)
2.5.4. Melt Agglomeration Process
2.5.5. Hot-Melt Extrusion Method
2.5.6. Lyophilization Techniques/Freeze-Drying
2.5.7. Electrospinning Method
2.5.8. Co-Precipitation
2.5.9. Supercritical Fluid (SCF) Technology
2.5.10. Spray-Drying Method
2.5.11. Kneading Method
2.5.12. Suitable Methods for Production of SDs of Anticancer Drugs
2.5.13. Lab Scale and Industrial Scale Manufacturing Processes
2.6. Use of SD for Improving Poorly Soluble Anticancer Drugs
2.7. Future Prospects
3. Conclusions
Funding
Conflicts of Interest
References
- Shewach, D.S.; Kuchta, R.D. Introduction to cancer chemotherapeutics. Chem. Rev. 2009, 109, 2859–2861. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Shin, H.; Han, J.; Na, K. Combination of photodynamic therapy (PDT) and anti-tumor immunity in cancer therapy. J. Pharm. Investig. 2018, 48, 143–151. [Google Scholar] [CrossRef]
- Arias, J.L. Drug targeting strategies in cancer treatment: An overview. Mini Rev. Med. Chem. 2011, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-M.; Lin, H.-Y.; Chan, M.-H. Preparation, characterization, and in vitro evaluation of folate-modified mesoporous bioactive glass for targeted anticancer drug carriers. J. Mater. Chem. B 2013, 1, 6147. [Google Scholar] [CrossRef]
- Vinothini, K.; Rajendran, N.K.; Ramu, A.; Elumalai, N.; Rajan, M. Folate receptor targeted delivery of paclitaxel to breast cancer cells via folic acid conjugated graphene oxide grafted methyl acrylate nanocarrier. Biomed. Pharmacother. 2019, 110, 906–917. [Google Scholar] [CrossRef]
- Voeikov, R.; Abakumova, T.; Grinenko, N.; Melnikov, P.; Bespalov, V.; Stukov, A.; Chekhonin, V.; Klyachko, N.; Nukolova, N. Dioxadet-loaded nanogels as a potential formulation for glioblastoma treatment. J. Pharm. Investig. 2017, 47, 75–83. [Google Scholar] [CrossRef]
- Huang, R.; Wang, Q.; Zhang, X.; Zhu, J.; Sun, B. Trastuzumab-cisplatin conjugates for targeted delivery of cisplatin to HER2-overexpressing cancer cells. Biomed. Pharmacother. 2015, 72, 17–23. [Google Scholar] [CrossRef]
- Le, Q.-V.; Choi, J.; Oh, Y.-K. Nano delivery systems and cancer immunotherapy. J. Pharm. Investig. 2018, 48, 527–539. [Google Scholar] [CrossRef]
- Park, O.; Yu, G.; Jung, H.; Mok, H. Recent studies on micro-/nano-sized biomaterials for cancer immunotherapy. J. Pharm. Investig. 2017, 47, 11–18. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, D.Y. Photothermal therapy with gold nanoparticles as an anticancer medication. J. Pharm. Investig. 2017, 47, 19–26. [Google Scholar] [CrossRef]
- Kirtane, A.R.; Narayan, P.; Liu, G.; Panyam, J. Polymer-surfactant nanoparticles for improving oral bioavailability of doxorubicin. J. Pharm. Investig. 2017, 47, 65–73. [Google Scholar] [CrossRef]
- Valicherla, G.R.; Dave, K.M.; Syed, A.A.; Riyazuddin, M.; Gupta, A.P.; Singh, A.; Wahajuddin; Mitra, K.; Datta, D.; Gayen, J.R. Formulation optimization of docetaxel loaded self-emulsifying drug delivery system to enhance bioavailability and anti-tumor activity. Sci. Rep. 2016, 6, 26895. [Google Scholar] [CrossRef]
- Fda, Cder Taxol (paclitaxel) injection. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/020262s049lbl.pdf (accessed on 24 January 2019).
- Nolvadex (Tamoxifen citrate). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/1998/17970.pdf (accessed on 4 September 2018).
- Singla, A.K.; Garg, A.; Aggarwal, D. Paclitaxel and its formulations. Int. J. Pharm. 2002, 235, 179–192. [Google Scholar] [CrossRef]
- Gréen, H.; Khan, M.S.; Jakobsen-Falk, I.; Åvall-Lundqvist, E.; Peterson, C. Impact of CYP3A5*3 and CYP2C8-HapC on paclitaxel/carboplatin-Induced myelosuppression in patients with ovarian cancer. J. Pharm. Sci. 2011, 100, 4205–4209. [Google Scholar] [CrossRef]
- Picard, M. Management of hypersensitivity reactions to taxanes. Immunol. Allergy Clin. North Am. 2017, 37, 679–693. [Google Scholar] [CrossRef]
- Gornstein, E.L.; Schwarz, T.L. Neurotoxic mechanisms of paclitaxel are local to the distal axon and independent of transport defects. Exp. Neurol. 2017, 288, 153–166. [Google Scholar] [CrossRef]
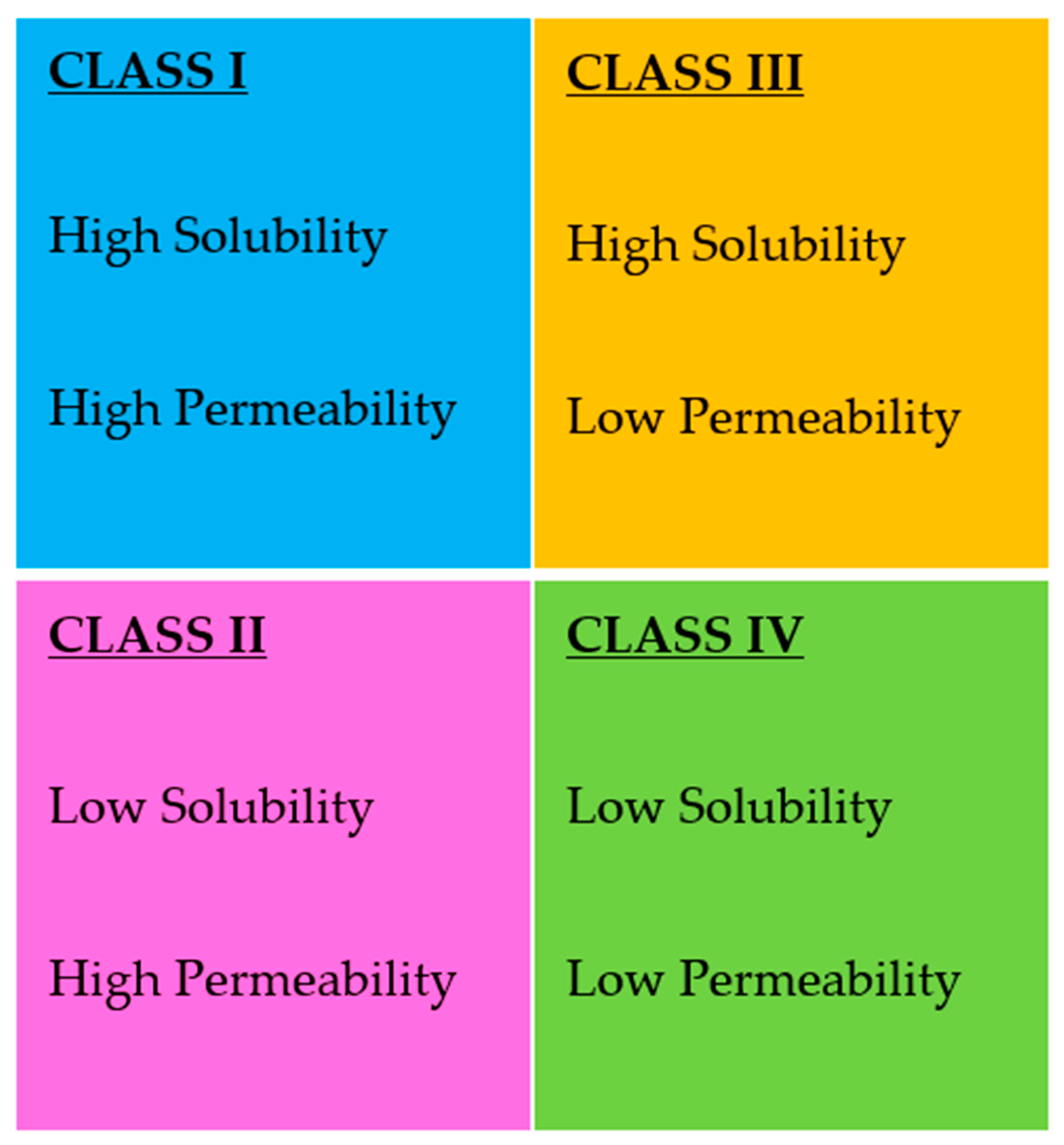
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef]
- Takagi, T.; Ramachandran, C.; Bermejo, M.; Yamashita, S.; Yu, L.X.; Amidon, G.L. A provisional biopharmaceutical classification of the top 200 oral drug products in the united states, great britain, spain, and japan. Mol. Pharm. 2006, 3, 631–643. [Google Scholar] [CrossRef]
- Rodriguez-Aller, M.; Guillarme, D.; Veuthey, J.-L.; Gurny, R. Strategies for formulating and delivering poorly water-soluble drugs. J. Drug Deliv. Sci. Technol. 2015, 30, 342–351. [Google Scholar] [CrossRef]
- Guidance for Industry, Waiver of in Vivo Bioavailability and Bioequivalence Studies for Immediate Release Solid Oral Dosage Forms Based on A Biopharmaceutics Classification System . Available online: https://www.fda.gov/downloads/Drugs/Guidances/ucm070246.pdf (accessed on 9 January 2019).
- Choi, J.-S.; Lee, S.-E.; Jang, W.S.; Byeon, J.C.; Park, J.-S. Solid dispersion of dutasteride using the solvent evaporation method: Approaches to improve dissolution rate and oral bioavailability in rats. Mater. Sci. Eng. C 2018, 90, 387–396. [Google Scholar] [CrossRef]
- Xu, W.; Sun, Y.; Du, L.; Chistyachenko, Y.S.; Dushkin, A.V.; Su, W. Investigations on solid dispersions of valsartan with alkalizing agents: Preparation, characterization and physicochemical properties. J. Drug Deliv. Sci. Technol. 2018, 44, 399–405. [Google Scholar] [CrossRef]
- Choi, J.-S.; Kwon, S.-H.; Lee, S.-E.; Jang, W.S.; Byeon, J.C.; Jeong, H.M.; Park, J.-S. Use of acidifier and solubilizer in tadalafil solid dispersion to enhance the in vitro dissolution and oral bioavailability in rats. Int. J. Pharm. 2017, 526, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Mohammadian, M.; Salami, M.; Momen, S.; Alavi, F.; Emam-Djomeh, Z.; Moosavi-Movahedi, A.A. Enhancing the aqueous solubility of curcumin at acidic condition through the complexation with whey protein nanofibrils. Food Hydrocoll. 2019, 87, 902–914. [Google Scholar] [CrossRef]
- Chen, X.; McClements, D.J.; Zhu, Y.; Chen, Y.; Zou, L.; Liu, W.; Cheng, C.; Fu, D.; Liu, C. Enhancement of the solubility, stability and bioaccessibility of quercetin using protein-based excipient emulsions. Food Res. Int. 2018, 114, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Lee, S.G.; Kang, M.J.; Lee, S.; Choi, Y.W. Surface modification of lipid-based nanocarriers for cancer cell-specific drug targeting. J. Pharm. Investig. 2017, 47, 203–227. [Google Scholar] [CrossRef]
- Karashima, M.; Sano, N.; Yamamoto, S.; Arai, Y.; Yamamoto, K.; Amano, N.; Ikeda, Y. Enhanced pulmonary absorption of poorly soluble itraconazole by micronized cocrystal dry powder formulations. Eur. J. Pharm. Biopharm. 2017, 115, 65–72. [Google Scholar] [CrossRef]
- Seo, B.; Kim, T.; Park, H.J.; Kim, J.-Y.; Lee, K.D.; Lee, J.M.; Lee, Y.-W. Extension of the hansen solubility parameter concept to the micronization of cyclotrimethylenetrinitramine crystals by supercritical anti-solvent process. J. Supercrit. Fluids 2016, 111, 112–120. [Google Scholar] [CrossRef]
- Wong, J.J.L.; Yu, H.; Lim, L.M.; Hadinoto, K. A trade-off between solubility enhancement and physical stability upon simultaneous amorphization and nanonization of curcumin in comparison to amorphization alone. Eur. J. Pharm. Sci. 2018, 114, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-B.; Park, C.; Piao, Z.Z.; Amin, H.H.; Meghani, N.M.; Tran, P.H.L.; Tran, T.T.D.; Cui, J.-H.; Cao, Q.-R.; Oh, E.; et al. pH-independent controlled release tablets containing nanonizing valsartan solid dispersions for less variable bioavailability in humans. J. Drug Deliv. Sci. Technol. 2018, 46, 365–377. [Google Scholar] [CrossRef]
- Chen, B.-Q.; Kankala, R.K.; Wang, S.-B.; Chen, A.-Z. Continuous nanonization of lonidamine by modified-rapid expansion of supercritical solution process. J. Supercrit. Fluids 2018, 133, 486–493. [Google Scholar] [CrossRef]
- Reggane, M.; Wiest, J.; Saedtler, M.; Harlacher, C.; Gutmann, M.; Zottnick, S.H.; Piechon, P.; Dix, I.; Müller-Buschbaum, K.; Holzgrabe, U.; et al. Bioinspired co-crystals of imatinib providing enhanced kinetic solubility. Eur. J. Pharm. Biopharm. 2018, 128, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, B.; Gao, Y.; Zhang, J.; Shi, L. Baicalein–Nicotinamide cocrystal with enhanced solubility, dissolution, and oral bioavailability. J. Pharm. Sci. 2014, 103, 2330–2337. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Feng, X.; Williams, R.O.; Zhang, F. Characterization of amorphous solid dispersions. J. Pharm. Investig. 2018, 48, 19–41. [Google Scholar] [CrossRef]
- Zhao, Y.; Xie, X.; Zhao, Y.; Gao, Y.; Cai, C.; Zhang, Q.; Ding, Z.; Fan, Z.; Zhang, H.; Liu, M.; et al. Effect of plasticizers on manufacturing ritonavir/copovidone solid dispersions via hot-melt extrusion: Preformulation, physicochemical characterization, and pharmacokinetics in rats. Eur. J. Pharm. Sci. 2019, 127, 60–70. [Google Scholar] [CrossRef]
- Smeets, A.; Koekoekx, R.; Clasen, C.; Van den Mooter, G. Amorphous solid dispersions of darunavir: Comparison between spray drying and electrospraying. Eur. J. Pharm. Biopharm. 2018, 130, 96–107. [Google Scholar] [CrossRef]
- Sekiguchi, K.; Obi, N. Studies on absorption of eutectic mixture. I. A comparison of the behavior of eutectic mixture of sulfathiazole and that of ordinary sulfathiazole in man. Chem. Pharm. Bull. (Tokyo) 1961, 9, 866–872. [Google Scholar] [CrossRef]
- Sekiguchi, K.; Obi, N.; Ueda, Y. Studies on absorption of eutectic mixture. II. Absorption of fused conglomerates of chloramphenicol and ure in rabbits. Chem. Pharm. Bull. (Tokyo) 1964, 12, 134–144. [Google Scholar] [CrossRef]
- Levy, G. Effect of particle size on dissolution and gastrointestinal absorption rates of pharmaceuticals. Am. J. Pharm. Sci. Support. Public Health 1963, 135, 78–92. [Google Scholar]
- Kanig, J.L. Properties of fused mannitol in compressed tablets. J. Pharm. Sci. 1964, 53, 188–192. [Google Scholar] [CrossRef]
- Madgulkar, A.; Bandivadekar, M.; Shid, T.; Rao, S. Sugars as solid dispersion carrier to improve solubility and dissolution of the BCS class II drug: Clotrimazole. Drug Dev. Ind. Pharm. 2016, 42, 28–38. [Google Scholar] [CrossRef]
- Vippagunta, S.R.; Wang, Z.; Hornung, S.; Krill, S.L. Factors affecting the formation of eutectic solid dispersions and their dissolution behavior. J. Pharm. Sci. 2007, 96, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Urbanetz, N.A. Stabilization of solid dispersions of nimodipine and polyethylene glycol 2000. Eur. J. Pharm. Sci. 2006, 28, 67–76. [Google Scholar] [CrossRef]
- Haser, A.; Cao, T.; Lubach, J.; Listro, T.; Acquarulo, L.; Zhang, F. Melt extrusion vs. spray drying: The effect of processing methods on crystalline content of naproxen-povidone formulations. Eur. J. Pharm. Sci. 2017, 102, 115–125. [Google Scholar] [CrossRef]
- Simonelli, A.P.; Mehta, S.C.; Higuchi, W.I. Dissolution rates of high energy polyvinylpyrrolidone (PVP)-Sulfathiazole coprecipitates. J. Pharm. Sci. 1969, 58, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Motallae, S.; Taheri, A.; Homayouni, A. Preparation and characterization of solid dispersions of celecoxib obtained by spray-drying ethanolic suspensions containing PVP-K30 or isomalt. J. Drug Deliv. Sci. Technol. 2018, 46, 188–196. [Google Scholar] [CrossRef]
- Ghanavati, R.; Taheri, A.; Homayouni, A. Anomalous dissolution behavior of celecoxib in PVP/Isomalt solid dispersions prepared using spray drier. Mater. Sci. Eng. C 2017, 72, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-S.; Lee, S.-E.; Jang, W.S.; Byeon, J.C.; Park, J.-S. Tadalafil solid dispersion formulations based on PVP/VA S-630: Improving oral bioavailability in rats. Eur. J. Pharm. Sci. 2017, 106, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Eloy, J.O.; Marchetti, J.M. Solid dispersions containing ursolic acid in poloxamer 407 and PEG 6000: A comparative study of fusion and solvent methods. Powder Technol. 2014, 253, 98–106. [Google Scholar] [CrossRef]
- Otto, D.P.; Otto, A.; de Villiers, M.M. Experimental and mesoscale computational dynamics studies of the relationship between solubility and release of quercetin from PEG solid dispersions. Int. J. Pharm. 2013, 456, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Reginald-Opara, J.N.; Attama, A.; Ofokansi, K.; Umeyor, C.; Kenechukwu, F. Molecular interaction between glimepiride and soluplus ® -PEG 4000 hybrid based solid dispersions: Characterisation and anti-diabetic studies. Int. J. Pharm. 2015, 496, 741–750. [Google Scholar] [CrossRef]
- Jiménez de los Santos, C.J.; Pérez-Martínez, J.I.; Gómez-Pantoja, M.E.; Moyano, J.R. Enhancement of albendazole dissolution properties using solid dispersions with Gelucire 50/13 and PEG 15000. J. Drug Deliv. Sci. Technol. 2017, 42, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Ceballos, A.; Cirri, M.; Maestrelli, F.; Corti, G.; Mura, P. Influence of formulation and process variables on in vitro release of theophylline from directly-compressed Eudragit matrix tablets. Farm. 2005, 60, 913–918. [Google Scholar] [CrossRef]
- Huang, J.; Wigent, R.; Bentzley, C.; Schwartz, J. Nifedipine solid dispersion in microparticles of ammonio methacrylate copolymer and ethylcellulose binary blend for controlled drug delivery. Effect of drug loading on release kinetics. Int. J. Pharm. 2006, 319, 44–54. [Google Scholar] [CrossRef]
- Fan, N.; He, Z.; Ma, P.; Wang, X.; Li, C.; Sun, J.; Sun, Y.; Li, J. Impact of HPMC on inhibiting crystallization and improving permeability of curcumin amorphous solid dispersions. Carbohydr. Polym. 2018, 181, 543–550. [Google Scholar] [CrossRef]
- Fan, N.; Ma, P.; Wang, X.; Li, C.; Zhang, X.; Zhang, K.; Li, J.; He, Z. Storage stability and solubilization ability of HPMC in curcumin amorphous solid dispersions formulated by Eudragit E100. Carbohydr. Polym. 2018, 199, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhang, H.; He, H.; Zhang, X.; Wang, Q.; Li, K.; He, Z. Cooperative effect of polyvinylpyrrolidone and HPMC E5 on dissolution and bioavailability of nimodipine solid dispersions and tablets. Asian J. Pharm. Sci. 2018. [Google Scholar] [CrossRef]
- Wang, S.; Liu, C.; Chen, Y.; Zhang, Z.; Zhu, A.; Qian, F. A high-sensitivity HPLC-ELSD method for HPMC-AS quantification and its application in elucidating the release mechanism of HPMC-AS based amorphous solid dispersions. Eur. J. Pharm. Sci. 2018, 122, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Liu, L.; Li, X.; Ma, J.; Liu, R.; Wang, S. Extended tacrolimus release via the combination of lipid-based solid dispersion and HPMC hydrogel matrix tablets. Asian J. Pharm. Sci. 2018. [Google Scholar] [CrossRef]
- Verreck, G.; Decorte, A.; Heymans, K.; Adriaensen, J.; Liu, D.; Tomasko, D.; Arien, A.; Peeters, J.; Van den Mooter, G.; Brewster, M.E. Hot stage extrusion of p-amino salicylic acid with EC using CO2 as a temporary plasticizer. Int. J. Pharm. 2006, 327, 45–50. [Google Scholar] [CrossRef]
- Ohara, T.; Kitamura, S.; Kitagawa, T.; Terada, K. Dissolution mechanism of poorly water-soluble drug from extended release solid dispersion system with ethylcellulose and hydroxypropylmethylcellulose. Int. J. Pharm. 2005, 302, 95–102. [Google Scholar] [CrossRef]
- Rashid, R.; Kim, D.W.; ud Din, F.; Mustapha, O.; Yousaf, A.M.; Park, J.H.; Kim, J.O.; Yong, C.S.; Choi, H.-G. Effect of hydroxypropylcellulose and tween 80 on physicochemical properties and bioavailability of ezetimibe-loaded solid dispersion. Carbohydr. Polym. 2015, 130, 26–31. [Google Scholar] [CrossRef] [PubMed]
- García-Zubiri, Í.X.; González-Gaitano, G.; Isasi, J.R. Thermal stability of solid dispersions of naphthalene derivatives with β-cyclodextrin and β-cyclodextrin polymers. Thermochim. Acta 2006, 444, 57–64. [Google Scholar] [CrossRef]
- Franco, P.; Reverchon, E.; De Marco, I. PVP/ketoprofen coprecipitation using supercritical antisolvent process. Powder Technol. 2018, 340, 1–7. [Google Scholar] [CrossRef]
- Dhandapani, N.V.; El-gied, A.A. Solid dispersions of cefixime using β-cyclodextrin: Characterization and in vitro evaluation. Int. J. Pharmacol. Pharm. Sci. 2016, 10, 1523–1527. [Google Scholar]
- Jafari, E. Preparation, characterization and dissolution of solid dispersion of diclofenac sodium using Eudragit E-100. J. Appl. Pharm. Sci. 2013, 3, 167–170. [Google Scholar]
- van Drooge, D.J.; Hinrichs, W.L.J.; Visser, M.R.; Frijlink, H.W. Characterization of the molecular distribution of drugs in glassy solid dispersions at the nano-meter scale, using differential scanning calorimetry and gravimetric water vapour sorption techniques. Int. J. Pharm. 2006, 310, 220–229. [Google Scholar] [CrossRef]
- Srinarong, P.; Hämäläinen, S.; Visser, M.R.; Hinrichs, W.L.J.; Ketolainen, J.; Frijlink, H.W. Surface-active derivative of inulin (inutec® SP1) is a superior carrier for solid dispersions with a high drug load. J. Pharm. Sci. 2011, 100, 2333–2342. [Google Scholar] [CrossRef]
- Majerik, V.; Charbit, G.; Badens, E.; Horváth, G.; Szokonya, L.; Bosc, N.; Teillaud, E. Bioavailability enhancement of an active substance by supercritical antisolvent precipitation. J. Supercrit. Fluids 2007, 40, 101–110. [Google Scholar] [CrossRef]
- Damian, F.; Blaton, N.; Naesens, L.; Balzarini, J.; Kinget, R.; Augustijns, P.; Van den Mooter, G. Physicochemical characterization of solid dispersions of the antiviral agent UC-781 with polyethylene glycol 6000 and Gelucire 44/14. Eur. J. Pharm. Sci. 2000, 10, 311–322. [Google Scholar] [CrossRef]
- Li, F.; Hu, J.; Deng, J.; Su, H.; Xu, S.; Liu, J. In vitro controlled release of sodium ferulate from compritol 888 ATO-based matrix tablets. Int. J. Pharm. 2006, 324, 152–157. [Google Scholar] [CrossRef]
- Panda, T.; Das, D.; Panigrahi, L. Formulation development of solid dispersions of bosentan using Gelucire 50/13 and poloxamer 188. J. Appl. Pharm. Sci. 2016, 6, 027–033. [Google Scholar] [CrossRef]
- Karolewicz, B.; Gajda, M.; Pluta, J.; Górniak, A. Dissolution study and thermal analysis of fenofibrate–Pluronic F127 solid dispersions. J. Therm. Anal. Calorim. 2016, 125, 751–757. [Google Scholar] [CrossRef]
- Chen, Y.; Shi, Q.; Chen, Z.; Zheng, J.; Xu, H.; Li, J.; Liu, H. Preparation and characterization of emulsified solid dispersions containing docetaxel. Arch. Pharm. Res. 2011, 34, 1909–1917. [Google Scholar] [CrossRef]
- Elgindy, N.; Elkhodairy, K.; Molokhia, A.; Elzoghby, A. Lyophilization monophase solution technique for preparation of amorphous flutamide dispersions. Drug Dev. Ind. Pharm. 2011, 37, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.-Y.; Lou, H.; Hageman, M.J. Preparation of lapatinib ditosylate solid dispersions using solvent rotary evaporation and hot melt extrusion for solubility and dissolution enhancement. Int. J. Pharm. 2018, 552, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.H.; Gibaldi, M.; Kanig, J.L. Increasing dissolution rates and gastrointestinal absorption of drugs via solid solutions and eutectic mixtures. I. Theoretical considerations and discussion of the literature. J. Pharm. Sci. 1965, 54, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Leuner, C. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Bhatnagar, P.; Dhote, V.; Mahajan, S.C.; Mishra, P.K.; Mishra, D.K. Solid dispersion in pharmaceutical drug development: From basics to clinical applications. Curr. Drug Deliv. 2014, 11, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Serajuddin, A.T. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef]
- Sarkari, M.; Brown, J.; Chen, X.; Swinnea, S.; Williams, R.O.; Johnston, K.P. Enhanced drug dissolution using evaporative precipitation into aqueous solution. Int. J. Pharm. 2002, 243, 17–31. [Google Scholar] [CrossRef]
- Vo, C.L.-N.; Park, C.; Lee, B.-J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Radhakrishnan, P.; Singh, S.K.; Verma, P.R.P. Furosemide - soluplus® solid dispersion: Development and characterization. Recent Pat. Drug Deliv. Formul. 2018, 11, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Lu, F.; Hou, J.; Shen, Y.; Guo, S. Incorporation of paclitaxel solid dispersions with poloxamer188 or polyethylene glycol to tune drug release from poly(ϵ-caprolactone) films. Drug Dev. Ind. Pharm. 2013, 39, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Palanisamy, M.; Khanam, J. Solid dispersion of prednisolone: Solid state characterization and improvement of dissolution profile. Drug Dev. Ind. Pharm. 2011, 37, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Chamsai, B.; Limmatvapirat, S.; Sungthongjeen, S.; Sriamornsak, P. Enhancement of solubility and oral bioavailability of manidipine by formation of ternary solid dispersion with d-α-tocopherol polyethylene glycol 1000 succinate and copovidone. Drug Dev. Ind. Pharm. 2017, 43, 2064–2075. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, V.; Suchandrasen; Prasad, V.P.R. Physicochemical characterization and in vitro dissolution behavior of olanzapine-mannitol solid dispersions. Brazilian J. Pharm. Sci. 2012, 48, 243–255. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, A.K.; Singh, S. Physicochemical characterization and dissolution study of solid dispersions of diacerein with polyethylene glycol 6000. Drug Dev. Ind. Pharm. 2011, 37, 1181–1191. [Google Scholar] [CrossRef]
- Adeli, E. Preparation and evaluation of azithromycin binary solid dispersions using various polyethylene glycols for the improvement of the drug solubility and dissolution rate. Brazilian J. Pharm. Sci. 2016, 52, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Shuai, S.; Yue, S.; Huang, Q.; Wang, W.; Yang, J.; Lan, K.; Ye, L. Preparation, characterization and in vitro/vivo evaluation of tectorigenin solid dispersion with improved dissolution and bioavailability. Eur. J. Drug Metab. Pharmacokinet. 2016, 41, 413–422. [Google Scholar] [CrossRef]
- Daravath, B.; Tadikonda, R.R.; Vemula, S.K. Formulation and pharmacokinetics of Gelucire solid dispersions of flurbiprofen. Drug Dev. Ind. Pharm. 2015, 41, 1254–1262. [Google Scholar] [CrossRef]
- Mustapha, O.; Kim, K.S.; Shafique, S.; Kim, D.S.; Jin, S.G.; Seo, Y.G.; Youn, Y.S.; Oh, K.T.; Yong, C.S.; Kim, J.O.; et al. Comparison of three different types of cilostazol-loaded solid dispersion: Physicochemical characterization and pharmacokinetics in rats. Colloids Surfaces B Biointerfaces 2017, 154, 89–95. [Google Scholar] [CrossRef]
- Kim, S.-J.; Lee, H.-K.; Na, Y.-G.; Bang, K.-H.; Lee, H.-J.; Wang, M.; Huh, H.-W.; Cho, C.-W. A novel composition of ticagrelor by solid dispersion technique for increasing solubility and intestinal permeability. Int. J. Pharm. 2019, 555, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Al-Hamidi, H.; Obeidat, W.M.; Nokhodchi, A. The dissolution enhancement of piroxicam in its physical mixtures and solid dispersion formulations using gluconolactone and glucosamine hydrochloride as potential carriers. Pharm. Dev. Technol. 2015, 20, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, C.; He, Y.; Duan, B.; Yang, G.; Ma, W.; Zhang, Y. Factors affecting the dissolution of indomethacin solid dispersions. AAPS PharmSciTech 2017, 18, 3258–3273. [Google Scholar] [CrossRef]
- Frizon, F.; de Oliveira Eloy, J.; Donaduzzi, C.M.; Mitsui, M.L.; Marchetti, J.M. Dissolution rate enhancement of loratadine in polyvinylpyrrolidone K-30 solid dispersions by solvent methods. Powder Technol. 2013, 235, 532–539. [Google Scholar] [CrossRef]
- Cuzzucoli Crucitti, V.; Migneco, L.M.; Piozzi, A.; Taresco, V.; Garnett, M.; Argent, R.H.; Francolini, I. Intermolecular interaction and solid state characterization of abietic acid/chitosan solid dispersions possessing antimicrobial and antioxidant properties. Eur. J. Pharm. Biopharm. 2018, 125, 114–123. [Google Scholar] [CrossRef]
- Alves, L.D.S.; de La Roca Soares, M.F.; de Albuquerque, C.T.; da Silva, É.R.; Vieira, A.C.C.; Fontes, D.A.F.; Figueirêdo, C.B.M.; Soares Sobrinho, J.L.; Rolim Neto, P.J. Solid dispersion of efavirenz in PVP K-30 by conventional solvent and kneading methods. Carbohydr. Polym. 2014, 104, 166–174. [Google Scholar] [CrossRef]
- Yin, L.-F.; Huang, S.-J.; Zhu, C.-L.; Zhang, S.-H.; Zhang, Q.; Chen, X.-J.; Liu, Q.-W. In vitro and in vivo studies on a novel solid dispersion of repaglinide using polyvinylpyrrolidone as the carrier. Drug Dev. Ind. Pharm. 2012, 38, 1371–1380. [Google Scholar] [CrossRef]
- Nguyen, M.N.-U.; Van Vo, T.; Tran, P.H.-L.; Tran, T.T.-D. Zein-based solid dispersion for potential application in targeted delivery. J. Pharm. Investig. 2017, 47, 357–364. [Google Scholar] [CrossRef]
- Gao, N.; Guo, M.; Fu, Q.; He, Z. Application of hot melt extrusion to enhance the dissolution and oral bioavailability of oleanolic acid. Asian J. Pharm. Sci. 2017, 12, 66–72. [Google Scholar] [CrossRef] [Green Version]
- Sathigari, S.K.; Radhakrishnan, V.K.; Davis, V.A.; Parsons, D.L.; Babu, R.J. Amorphous-State characterization of efavirenz—Polymer hot-melt extrusion systems for dissolution enhancement. J. Pharm. Sci. 2012, 101, 3456–3464. [Google Scholar] [CrossRef]
- Chowdhury, N.; Vhora, I.; Patel, K.; Bagde, A.; Kutlehria, S.; Singh, M. Development of hot melt extruded solid dispersion of tamoxifen citrate and resveratrol for synergistic effects on breast cancer cells. AAPS PharmSciTech 2018, 19, 3287–3297. [Google Scholar] [CrossRef]
- Fule, R.; Amin, P. Development and evaluation of lafutidine solid dispersion via hot melt extrusion: Investigating drug-polymer miscibility with advanced characterisation. Asian J. Pharm. Sci. 2014, 9, 92–106. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Xu, T.; Zhang, D.; He, W.; Wang, S.; Jiang, T. Disulfiram thermosensitive in-situ gel based on solid dispersion for cataract. Asian J. Pharm. Sci. 2018, 13, 527–535. [Google Scholar] [CrossRef]
- Abu-Diak, O.A.; Jones, D.S.; Andrews, G.P. Understanding the performance of melt-extruded poly(ethylene oxide)–Bicalutamide solid dispersions: Characterisation of microstructural properties using thermal, spectroscopic and drug release methods. J. Pharm. Sci. 2012, 101, 200–213. [Google Scholar] [CrossRef]
- Solanki, N.G.; Lam, K.; Tahsin, M.; Gumaste, S.G.; Shah, A.V.; Serajuddin, A.T.M. Effects of surfactants on itraconazole-HPMCAS solid dispersion prepared by hot-melt extrusion I: Miscibility and drug release. J. Pharm. Sci. 2018. [Google Scholar] [CrossRef]
- Guns, S.; Dereymaker, A.; Kayaert, P.; Mathot, V.; Martens, J.A.; Van den Mooter, G. Comparison between hot-melt extrusion and spray-drying for manufacturing solid dispersions of the graft copolymer of ethylene glycol and vinylalcohol. Pharm. Res. 2011, 28, 673–682. [Google Scholar] [CrossRef]
- Alshafiee, M.; Aljammal, M.K.; Markl, D.; Ward, A.; Walton, K.; Blunt, L.; Korde, S.; Pagire, S.K.; Kelly, A.L.; Paradkar, A.; et al. Hot-melt extrusion process impact on polymer choice of glyburide solid dispersions: The effect of wettability and dissolution. Int. J. Pharm. 2019, 559, 245–254. [Google Scholar] [CrossRef]
- Altamimi, M.A.; Neau, S.H. Investigation of the in vitro performance difference of drug-soluplus® and drug-PEG 6000 dispersions when prepared using spray drying or lyophilization. Saudi Pharm. J. 2017, 25, 419–439. [Google Scholar] [CrossRef]
- Jacobsen, A.-C.; Elvang, P.A.; Bauer-Brandl, A.; Brandl, M. A dynamic in vitro permeation study on solid mono- and diacyl-phospholipid dispersions of celecoxib. Eur. J. Pharm. Sci. 2019, 127, 199–207. [Google Scholar] [CrossRef]
- Suzuki, H.; Yakushiji, K.; Matsunaga, S.; Yamauchi, Y.; Seto, Y.; Sato, H.; Onoue, S. Amorphous solid dispersion of meloxicam enhanced oral absorption in rats with impaired gastric motility. J. Pharm. Sci. 2018, 107, 446–452. [Google Scholar] [CrossRef]
- Ngo, A.N.; Thomas, D.; Murowchick, J.; Ayon, N.J.; Jaiswal, A.; Youan, B.-B.C. Engineering fast dissolving sodium acetate mediated crystalline solid dispersion of docetaxel. Int. J. Pharm. 2018, 545, 329–341. [Google Scholar] [CrossRef]
- Sonali, D.; Tejal, S.; Vaishali, T.; Tejal, G. Silymarin-solid dispersions: Characterization and influence of preparation methods on dissolution. Acta Pharm. 2010, 60, 427–443. [Google Scholar] [CrossRef] [Green Version]
- Dhumal, R.; Shimpi, S.; Paradkar, A. Development of spray-dried co-precipitate of amorphous celecoxib containing storage and compression stabilizers. Acta Pharm. 2007, 57, 287–300. [Google Scholar] [CrossRef]
- Hou, H.H.; Rajesh, A.; Pandya, K.M.; Lubach, J.W.; Muliadi, A.; Yost, E.; Jia, W.; Nagapudi, K. Impact of method of preparation of amorphous solid dispersions on mechanical properties: Comparison of coprecipitation and spray drying. J. Pharm. Sci. 2019, 108, 870–879. [Google Scholar] [CrossRef]
- Adeli, E. The use of supercritical anti-solvent (SAS) technique for preparation of irbesartan-Pluronic® F-127 nanoparticles to improve the drug dissolution. Powder Technol. 2016, 298, 65–72. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, Y.; Liu, D.; Gao, Y.; Qian, S. Preparation of apigenin nanocrystals using supercritical antisolvent process for dissolution and bioavailability enhancement. Eur. J. Pharm. Sci. 2013, 48, 740–747. [Google Scholar] [CrossRef]
- Moneghini, M.; Kikic, I.; Voinovich, D.; Perissutti, B.; Filipović-Grčić, J. Processing of carbamazepine–PEG 4000 solid dispersions with supercritical carbon dioxide: Preparation, characterisation, and in vitro dissolution. Int. J. Pharm. 2001, 222, 129–138. [Google Scholar] [CrossRef]
- Tabbakhian, M.; Hasanzadeh, F.; Tavakoli, N.; Jamshidian, Z. Dissolution enhancement of glibenclamide by solid dispersion: Solvent evaporation versus a supercritical fluid-based solvent -antisolvent technique. Res. Pharm. Sci. 2014, 9, 337–350. [Google Scholar]
- Djuris, J.; Milovanovic, S.; Medarevic, D.; Dobricic, V.; Dapčević, A.; Ibric, S. Selection of the suitable polymer for supercritical fluid assisted preparation of carvedilol solid dispersions. Int. J. Pharm. 2019, 554, 190–200. [Google Scholar] [CrossRef]
- Herbrink, M.; Schellens, J.H.M.; Beijnen, J.H.; Nuijen, B. Improving the solubility of nilotinib through novel spray-dried solid dispersions. Int. J. Pharm. 2017, 529, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Al-Zoubi, N.; Odah, F.; Obeidat, W.; Al-Jaberi, A.; Partheniadis, I.; Nikolakakis, I. Evaluation of spironolactone solid dispersions prepared by co-spray drying with soluplus ® and polyvinylpyrrolidone and influence of tableting on drug release. J. Pharm. Sci. 2018, 107, 2385–2398. [Google Scholar] [CrossRef]
- Pradhan, R.; Kim, S.Y.; Yong, C.S.; Kim, J.O. Preparation and characterization of spray-dried valsartan-loaded Eudragit® E PO solid dispersion microparticles. Asian J. Pharm. Sci. 2016, 11, 744–750. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, R.; Tran, T.H.; Choi, J.Y.; Choi, I.S.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Development of a rebamipide solid dispersion system with improved dissolution and oral bioavailability. Arch. Pharm. Res. 2015, 38, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Pawar, J.N.; Shete, R.T.; Gangurde, A.B.; Moravkar, K.K.; Javeer, S.D.; Jaiswar, D.R.; Amin, P.D. Development of amorphous dispersions of artemether with hydrophilic polymers via spray drying: Physicochemical and in silico studies. Asian J. Pharm. Sci. 2016, 11, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Paudel, A.; Van den Mooter, G. Influence of solvent composition on the miscibility and physical stability of naproxen/PVP K 25 solid dispersions prepared by cosolvent spray-drying. Pharm. Res. 2012, 29, 251–270. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Alim, M.; Agrawal, V.K. Solubility and dissolution enhancement of domperidone using 2-hydroxypropyl- β - cyclodextrin by kneading method. Asian J. Pharm. 2017, 11, 168–175. [Google Scholar]
- Tachibana, T.; Nakamura, A. A method for preparing an aqueous colloidal dispersion of organic materials by using water-soluble polymers: Dispersion of Β-carotene by polyvinylpyrrolidone. Kolloid-Zeitschrift Zeitschrift für Polym. 1965, 203, 130–133. [Google Scholar] [CrossRef]
- Mayersohn, M.; Gibaldi, M. New method of solid-state dispersion for increasing dissolution rates. J. Pharm. Sci. 1966, 55, 1323–1324. [Google Scholar] [CrossRef]
- Miao, L.; Liang, Y.; Pan, W.; Gou, J.; Yin, T.; Zhang, Y.; He, H.; Tang, X. Effect of supersaturation on the oral bioavailability of paclitaxel/polymer amorphous solid dispersion. Drug Deliv. Transl. Res. 2018. [Google Scholar] [CrossRef]
- Ren, F.; Jing, Q.; Tang, Y.; Shen, Y.; Chen, J.; Gao, F.; Cui, J. Characteristics of bicalutamide solid dispersions and improvement of the dissolution. Drug Dev. Ind. Pharm. 2006, 32, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Song, C.K.; Yoon, I.-S.; Kim, D.-D. Poloxamer-based solid dispersions for oral delivery of docetaxel: Differential effects of F68 and P85 on oral docetaxel bioavailability. Int. J. Pharm. 2016, 507, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Vasavada, R.C. Solubility and dissolution of etoposide from solid dispersions of PEG 8000. Drug Dev. Ind. Pharm. 1993, 19, 903–914. [Google Scholar] [CrossRef]
- Jang, S.W.; Kang, M.J. Improved oral absorption and chemical stability of everolimus via preparation of solid dispersion using solvent wetting technique. Int. J. Pharm. 2014, 473, 187–193. [Google Scholar] [CrossRef]
- Kaur, S.; Jena, S.K.; Samal, S.K.; Saini, V.; Sangamwar, A.T. Freeze dried solid dispersion of exemestane: A way to negate an aqueous solubility and oral bioavailability problems. Eur. J. Pharm. Sci. 2017, 107, 54–61. [Google Scholar] [CrossRef]
- Sodeifian, G.; Sajadian, S.A. Solubility measurement and preparation of nanoparticles of an anticancer drug (letrozole) using rapid expansion of supercritical solutions with solid cosolvent (RESS-SC). J. Supercrit. Fluids 2018, 133, 239–252. [Google Scholar] [CrossRef]
- Kim, M.-S.; Ha, E.-S.; Kim, J.-S.; Baek, I.; Yoo, J.-W.; Jung, Y.; Moon, H.R. Development of megestrol acetate solid dispersion nanoparticles for enhanced oral delivery by using a supercritical antisolvent process. Drug Des. Devel. Ther. 2015, 4269. [Google Scholar] [CrossRef]
- Li, S.; Liu, Y.; Liu, T.; Zhao, L.; Zhao, J.; Feng, N. Development and in-vivo assessment of the bioavailability of oridonin solid dispersions by the gas anti-solvent technique. Int. J. Pharm. 2011, 411, 172–177. [Google Scholar] [CrossRef]
- Tran, T.H.; Poudel, B.K.; Marasini, N.; Woo, J.S.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Development of raloxifene-solid dispersion with improved oral bioavailability via spray-drying technique. Arch. Pharm. Res. 2013, 36, 86–93. [Google Scholar] [CrossRef]
- Truong, D.H.; Tran, T.H.; Ramasamy, T.; Choi, J.Y.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Preparation and characterization of solid dispersion using a novel amphiphilic copolymer to enhance dissolution and oral bioavailability of sorafenib. Powder Technol. 2015, 283, 260–265. [Google Scholar] [CrossRef]
- Shah, N.; Iyer, R.M.; Mair, H.-J.; Choi, D.; Tian, H.; Diodone, R.; Fahnrich, K.; Pabst-Ravot, A.; Tang, K.; Scheubel, E.; et al. Improved human bioavailability of vemurafenib, a practically insoluble drug, using an amorphous polymer-stabilized solid dispersion prepared by a solvent-controlled coprecipitation process. J. Pharm. Sci. 2013, 102, 967–981. [Google Scholar] [CrossRef]
- Goldberg, A.H.; Gibaldi, M.; Kanig, J.L. Increasing dissolution rates and gastrointestinal absorption of drugs via solid solutions and eutectic mixtures III. J. Pharm. Sci. 1966, 55, 487–492. [Google Scholar] [CrossRef]
- Chen, H.; Jiang, G.; Ding, F. Monolithic osmotic tablet containing solid dispersion of 10-hydroxycamptothecin. Drug Dev. Ind. Pharm. 2009, 35, 131–137. [Google Scholar] [CrossRef]
- van Drooge, D.-J.; Hinrichs, W.L.J.; Dickhoff, B.H.J.; Elli, M.N.A.; Visser, M.R.; Zijlstra, G.S.; Frijlink, H.W. Spray freeze drying to produce a stable delta(9)-tetrahydrocannabinol containing inulin-based solid dispersion powder suitable for inhalation. Eur. J. Pharm. Sci. 2005, 26, 231–240. [Google Scholar] [CrossRef]
- Kaur, J.; Aggarwal, G.; Singh, G.; Rana, A.C. Improvement of drug solubility using solid dispersion. Int. J. Pharm. Pharm. Sci. 2012, 4, 47–53. [Google Scholar]
- Vilhelmsen, T.; Eliasen, H.; Schæfer, T. Effect of a melt agglomeration process on agglomerates containing solid dispersions. Int. J. Pharm. 2005, 303, 132–142. [Google Scholar] [CrossRef]
- Seo, A.; Holm, P.; Kristensen, H.G.; Schaefer, T. The preparation of agglomerates containing solid dispersions of diazepam by melt agglomeration in a high shear mixer. Int. J. Pharm. 2003, 259, 161–171. [Google Scholar] [CrossRef]
- Genina, N.; Hadi, B.; Löbmann, K. Hot melt extrusion as solvent-Free technique for a continuous manufacturing of drug-Loaded mesoporous silica. J. Pharm. Sci. 2018, 107, 149–155. [Google Scholar] [CrossRef]
- Betageri, G. Enhancement of dissolution of glyburide by solid dispersion and lyophilization techniques. Int. J. Pharm. 1995, 126, 155–160. [Google Scholar] [CrossRef]
- Yu, D.-G.; Li, J.-J.; Williams, G.R.; Zhao, M. Electrospun amorphous solid dispersions of poorly water-soluble drugs: A review. J. Control. Release 2018, 292, 91–110. [Google Scholar] [CrossRef]
- Pamudji, J.S.; Khairurrijal; Mauludin, R.; Sudiati, T.; Evita, M. PVA-ketoprofen nanofibers manufacturing using electrospinning method for dissolution improvement of ketoprofen. Nanotechnol. Appl. Energy Environ. 2013, 737, 166–175. [Google Scholar] [CrossRef]
- Lopez, F.L.; Shearman, G.C.; Gaisford, S.; Williams, G.R. Amorphous formulations of indomethacin and griseofulvin prepared by electrospinning. Mol. Pharm. 2014, 11, 4327–4338. [Google Scholar] [CrossRef]
- Hannay, J.B.; Hogarth, J. On the solubility of solids in gases. Proc. R. Soc. London 1879, 30, 178–188. [Google Scholar] [CrossRef]
- Riekes, M.K.; Caon, T.; da Silva, J.; Sordi, R.; Kuminek, G.; Bernardi, L.S.; Rambo, C.R.; de Campos, C.E.M.; Fernandes, D.; Stulzer, H.K. Enhanced hypotensive effect of nimodipine solid dispersions produced by supercritical CO2 drying. Powder Technol. 2015, 278, 204–210. [Google Scholar] [CrossRef]
- Jun, S.W.; Kim, M.-S.; Jo, G.H.; Lee, S.; Woo, J.S.; Park, J.-S.; Hwang, S.-J. Cefuroxime axetil solid dispersions prepared using solution enhanced dispersion by supercritical fluids. J. Pharm. Pharmacol. 2005, 57, 1529–1537. [Google Scholar] [CrossRef]
- Abuzar, S.M.; Hyun, S.-M.; Kim, J.-H.; Park, H.J.; Kim, M.-S.; Park, J.-S.; Hwang, S.-J. Enhancing the solubility and bioavailability of poorly water-soluble drugs using supercritical antisolvent (SAS) process. Int. J. Pharm. 2018, 538, 1–13. [Google Scholar] [CrossRef]
- Singh, A.; Van den Mooter, G. Spray drying formulation of amorphous solid dispersions. Adv. Drug Deliv. Rev. 2016, 100, 27–50. [Google Scholar] [CrossRef]
- World cancer factsheet. Available online: https://www.cancerresearchuk.org/sites/default/files/cs_report_world.pdf (accessed on 15 January 2019).
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA. Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Park, J.-H.; Yan, Y.-D.; Chi, S.-C.; Hwang, D.H.; Shanmugam, S.; Lyoo, W.S.; Woo, J.S.; Yong, C.S.; Choi, H.-G. Preparation and evaluation of cremophor-free paclitaxel solid dispersion by a supercritical antisolvent process. J. Pharm. Pharmacol. 2011, 63, 491–499. [Google Scholar] [CrossRef]
- Thanki, K.; Gangwal, R.P.; Sangamwar, A.T.; Jain, S. Oral delivery of anticancer drugs: Challenges and opportunities. J. Control. Release 2013, 170, 15–40. [Google Scholar] [CrossRef]
- Sawicki, E.; Schellens, J.H.M.; Beijnen, J.H.; Nuijen, B. Inventory of oral anticancer agents: Pharmaceutical formulation aspects with focus on the solid dispersion technique. Cancer Treat. Rev. 2016, 50, 247–263. [Google Scholar] [CrossRef]
- Bennett, R.C.; Brough, C.; Miller, D.A.; O’Donnell, K.P.; Keen, J.M.; Hughey, J.R.; Williams, R.O.; McGinity, J.W. Preparation of amorphous solid dispersions by rotary evaporation and KinetiSol dispersing: Approaches to enhance solubility of a poorly water-soluble gum extract. Drug Dev. Ind. Pharm. 2015, 41, 382–397. [Google Scholar] [CrossRef] [PubMed]
- Keen, J.M.; LaFountaine, J.S.; Hughey, J.R.; Miller, D.A.; McGinity, J.W. Development of itraconazole tablets containing viscous KinetiSol solid dispersions: In vitro and in vivo analysis in dogs. AAPS PharmSciTech 2018, 19, 1998–2008. [Google Scholar] [CrossRef] [PubMed]
- DiNunzio, J.C.; Brough, C.; Miller, D.A.; Williams, R.O.; McGinity, J.W. Fusion processing of itraconazole solid dispersions by Kinetisol® dispersing: A comparative study to hot melt extrusion. J. Pharm. Sci. 2010, 99, 1239–1253. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Drugs |
|---|---|
| Melting/fusion method | Sulfathiazole [39], clotrimazole [43], albendazole [54], tacrolimus [61], fenofibrate [75], furosemide [85], paclitaxel [86], manidipine [88], olanzapine [89], diacerein [90] |
| Solvent evaporation method | Dutasteride [23], tadalafil [50], glimepiride [53], nimodipine [59], diclofenac [68], azithromycin [91], tectorigenin [92], flurbiprofen [93], cilostazol [94], ticagrelor [95], piroxicam [96], indomethacin [97], loratadine [98], abietic acid [99], efavirenz [100], repagnilide [101], prednisolone [102] |
| Hot-melt extrusion method | Ritonavir [37], naproxen [46], oleanolic acid [103], efavirenz [104], tamoxifen [105], lafutidine [106], disulfiram [107], bicalutamide [108], itraconazole [109], miconazole [110], glyburide [111] |
| Lyophilization/Freeze-drying | Nifedipine and sulfamethoxazole [112], celecoxib [113], meloxicam [114], docetaxel [115] |
| Co-precipitation method | Silymarin [116], celecoxib [117], GDC-0810 [118] |
| Supercritical fluid method | Ketoprofen [66], irbesartan [119], apigenin [120], carbamazepine [121], glibenclamide [122], carvedilol [123] |
| Spray-drying method | Nilotinib [124], spironolactone [125], valsartan [126], rebamipide [127], artemether [128], naproxen [129] |
| Kneading method | Cefixime [67], efavirenz [100], domperidone [130] |
| Products | Drugs | Polymers | Company |
|---|---|---|---|
| Afeditab® | Nifedipine | Poloxamer or PVP | Elan Corp, Ireland |
| Cesamet® | Nabilone | PVP | Lilly, USA |
| Cesamet® | Nabilone | PVP | Valeant Pharmaceuticals, Canada |
| Certican® | Everolimus | HPMC | Novartis, Switzweland |
| Gris-PEG® | Griseofulvin | PEG | Novartis, Switzweland |
| Gris-PEG® | Griseofulvin | PVP | VIP Pharma, Denmark |
| Fenoglide® | Fenofibrate | PEG | LifeCycle Pharma, Denmark |
| Nivadil® | Nivaldipine | HPC/HPMC | Fujisawa Pharmaceuticals Co., Ltd |
| Nimotop® | Nimodipine | PEG | Bayer |
| Torcetrapib® | Torcetrapib | HPMC AS | Pfizer, USA |
| Ibuprofen® | Ibuprofen | Various | Soliqs, Germany |
| Incivek® | Telaprevir | HPMC AS | Vertex |
| Sporanox® | Itraconazole | HPMC | Janssen Pharmaceutica, Belgium |
| Onmel® | Itraconazole | HPMC | Stiefel |
| Prograf® | Tacrolimus | HPMC | Fujisawa Pharmaceuticals Co., Ltd |
| Cymbalta® | Duloxetine | HPMC AS | Lilly, USA |
| Noxafil® | Posaconazole | HPMC AS | Merck |
| LCP-Tacro® | Tacrolimus | HPMC | LifeCycle Pharma, Denmark |
| Intelence® | Etravirine | HPMC | Tibotec, Yardley, PA |
| Incivo® | Etravirine | HPMC | Janssen Pharmaceutica, Belgium |
| Rezulin® | Troglitazone | PVP | Pfizer, USA |
| Isoptin SRE-240® | Verapamil | Various | Soliqs, Germany |
| Isoptin SR-E® | Verapamil | HPC/HPMC | Abbott Laboratories, USA |
| Crestor® | Rosuvastatin | HPMC | AstraZeneca |
| Zelboraf® | Vemurafenib | HPMC AS | Roche |
| Zortress® | Everolimus | HPMC | Novartis, Switzweland |
| Kalydeco® | Ivacaflor | HPMC AS | Vertex |
| Kaletra® | Lopinavir and Ritonavir | PVP/polyvinyl acetate | Abbott Laboratories, USA |
| Anticancer Drugs | Carriers | Methods | Attributes of Modified Anticancer Drugs | Reference | Years |
|---|---|---|---|---|---|
| Bicalutamide | PVP K30 | Solvent evaporation | Using PVP K30 as carrier, SD showed the highest cumulative released percentage (about 98% during the initial 10 min) and stability after 6 months | [134] | 2006 |
| Docetaxel | HPMC, PEG | Solvent evaporation | The solubility and dissolution of emulsified SD of docetaxel at 2 h were 34.2- and 12.7-fold higher, respectively, compared to the pure conventional drug | [76] | 2011 |
| Docetaxel | Poloxamer F68/P85 | Freeze-drying | A combination of poloxamer F68 and P85 in the preparation of docetaxel SD not only enhanced solubility, but also improved intestinal permeation | [135] | 2016 |
| Etoposide | PEG | Fusion method | The solubility and dissolution of etoposide in SD were higher in comparison with etoposide alone | [136] | 1993 |
| Everolimus | HPMC | Co-precipitation | At a ratio of drug to HPMC (1:15), drug release from SD was 75% after 30 min, thereby improving oral absorption of everolimus | [137] | 2014 |
| Exemestane | Lipoid® E80S/sodium deoxycholate | Freeze-drying | The exemestane SD showed 4-6-fold increase in absorptive transport compared to the pure drug. In addition, AUC0-72h of exemestane SD was 2.3-fold higher in comparison with that of drug alone | [138] | 2017 |
| Flutamide | PVP K30, PEG, Pluronic F127 | Lyophilization | The dissolution of flutamide was higher (81.64%) than the drug alone (13.45%) using poloxamer 407 as a carrier | [77] | 2010 |
| Lapatinib | Soluplus, poloxamer 188 | Solvent evaporation, hot-melt extrusion | Solubility and dissolution of lapatinib SD were enhanced compared to the drug alone. After 15 min, the drug in SD was released at 92%compared to the drug alone (48%) | [78] | 2018 |
| Letrozole | CO2-menthol | Supercritical fluid | Solubility of letrozole SD using supercritical fluid is 7.1 times higher compared to that of the conventional drug | [139] | 2018 |
| Megestrol acetate | HPMC, Ryoto sugar ester L1695 | Supercritical fluid | The SD with drug: HPMC: Ryoto sugar ester L1695 ratio of 1:2:1 showed over 95% rapid dissolution within 30 min. In addition, AUC and Cmax (0-24h) of drug in SD were 4.0- and 5.5-fold higher, respectively, compared to those in pure drug | [140] | 2015 |
| Oridonin | PVP K17 | Supercritical fluid | The dissolution of oridonin SD significantly increased compared to the original drug. In addition, the absorption of oridonin in SD showed 26.4-fold improvement in BA | [141] | 2011 |
| Paclitaxel | Poloxamer 188, PEG | Fusion method | Paclitaxel SD was successfully prepared, and the drug release from SD was higher than that of the drug alone | [86] | 2013 |
| Paclitaxel | HPMC AS | Solvent method | The solubility and permeability of paclitaxel were not increased simultaneously through supersaturation in vivo | [133] | 2018 |
| Prednisolone | HP-β-CD, PEG, PVP, PEG 4000, MNT, SMP, Cremophor | Solvent evaporation, melting method, kneading method | The in vitro dissolution of prednisolone SD was improved compared with the pure drug | [87] | 2011 |
| Raloxifene | PVP K30 | Spray-drying | The absorption of raloxifene from SD showed 2.6-fold enhanced BA in comparison with the conventional drug | [142] | 2013 |
| Sorafenib | Soluplus | Spray-drying | The Cmax and AUC0-48h of sorafenib in SD formulation increased 1.5- and 1.8-fold, resocetuvely, compared with the pure drug | [143] | 2015 |
| Tamoxifen | Soluplus | Hot-melt extrusion | The dissolution and BA of tamoxifen in SD were improved compared with the drug alone | [105] | 2018 |
| Vemurafenib | HPMC AS | Solvent-controlled precipitation | The BA of vemurafenib in SD was improved 4~5-fold compared to the conventional drug | [144] | 2013 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, P.; Pyo, Y.-C.; Kim, D.-H.; Lee, S.-E.; Kim, J.-K.; Park, J.-S. Overview of the Manufacturing Methods of Solid Dispersion Technology for Improving the Solubility of Poorly Water-Soluble Drugs and Application to Anticancer Drugs. Pharmaceutics 2019, 11, 132. https://doi.org/10.3390/pharmaceutics11030132
Tran P, Pyo Y-C, Kim D-H, Lee S-E, Kim J-K, Park J-S. Overview of the Manufacturing Methods of Solid Dispersion Technology for Improving the Solubility of Poorly Water-Soluble Drugs and Application to Anticancer Drugs. Pharmaceutics. 2019; 11(3):132. https://doi.org/10.3390/pharmaceutics11030132
Chicago/Turabian StyleTran, Phuong, Yong-Chul Pyo, Dong-Hyun Kim, Sang-Eun Lee, Jin-Ki Kim, and Jeong-Sook Park. 2019. "Overview of the Manufacturing Methods of Solid Dispersion Technology for Improving the Solubility of Poorly Water-Soluble Drugs and Application to Anticancer Drugs" Pharmaceutics 11, no. 3: 132. https://doi.org/10.3390/pharmaceutics11030132
APA StyleTran, P., Pyo, Y. -C., Kim, D. -H., Lee, S. -E., Kim, J. -K., & Park, J. -S. (2019). Overview of the Manufacturing Methods of Solid Dispersion Technology for Improving the Solubility of Poorly Water-Soluble Drugs and Application to Anticancer Drugs. Pharmaceutics, 11(3), 132. https://doi.org/10.3390/pharmaceutics11030132