Reverse Transcriptase Inhibitors Nanosystems Designed for Drug Stability and Controlled Delivery

 ,
, 

 ,
,  ,
, 

Abstract
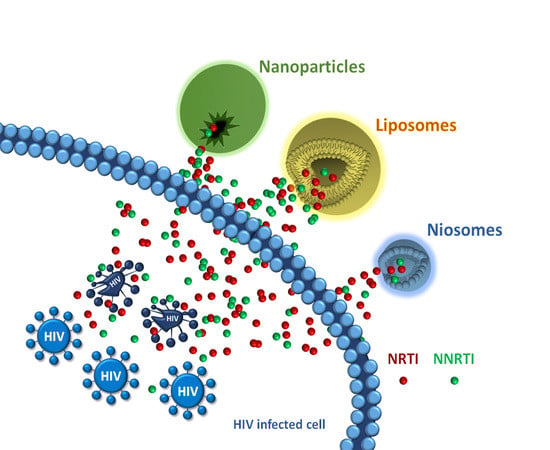
:
1. Introduction
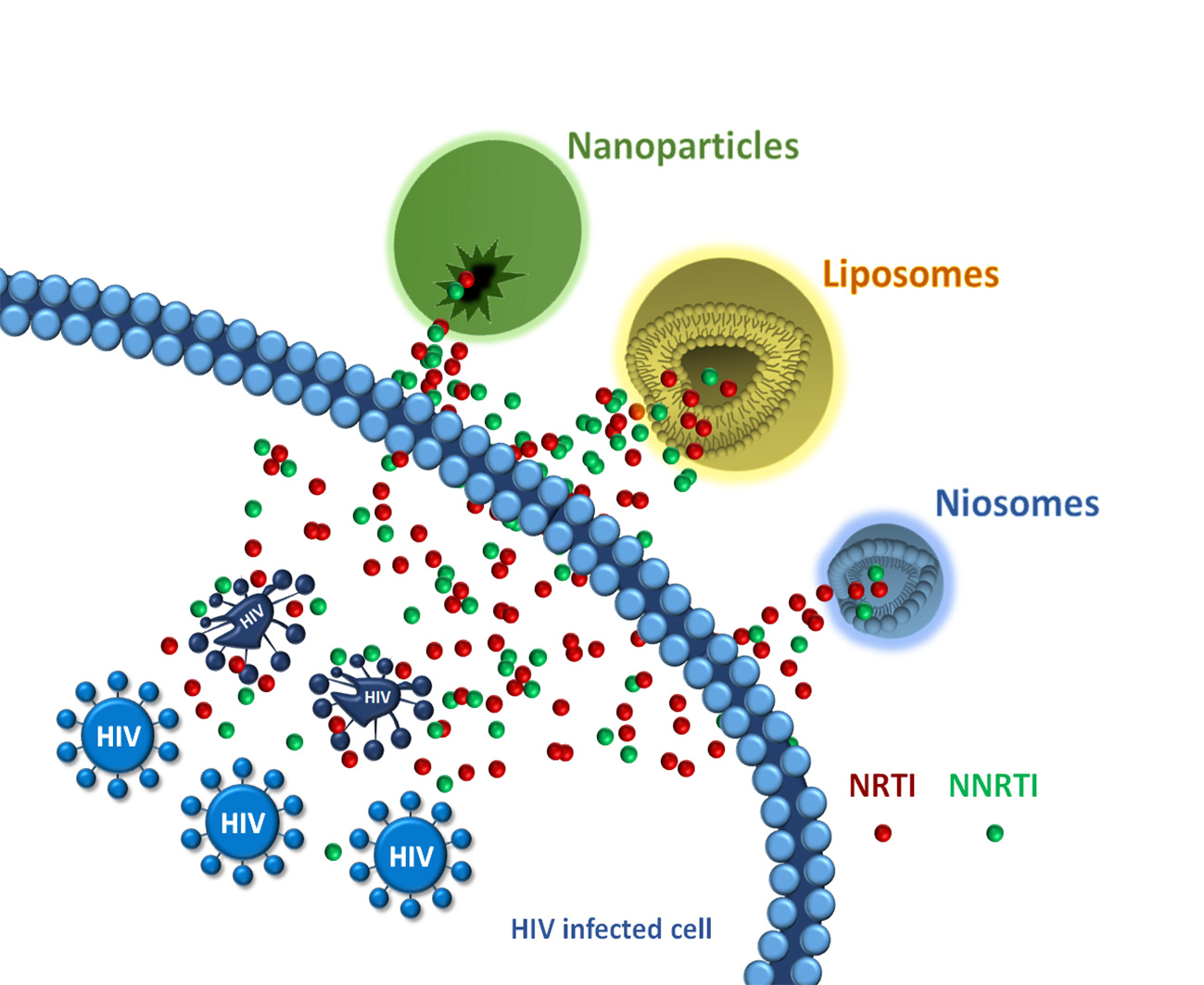
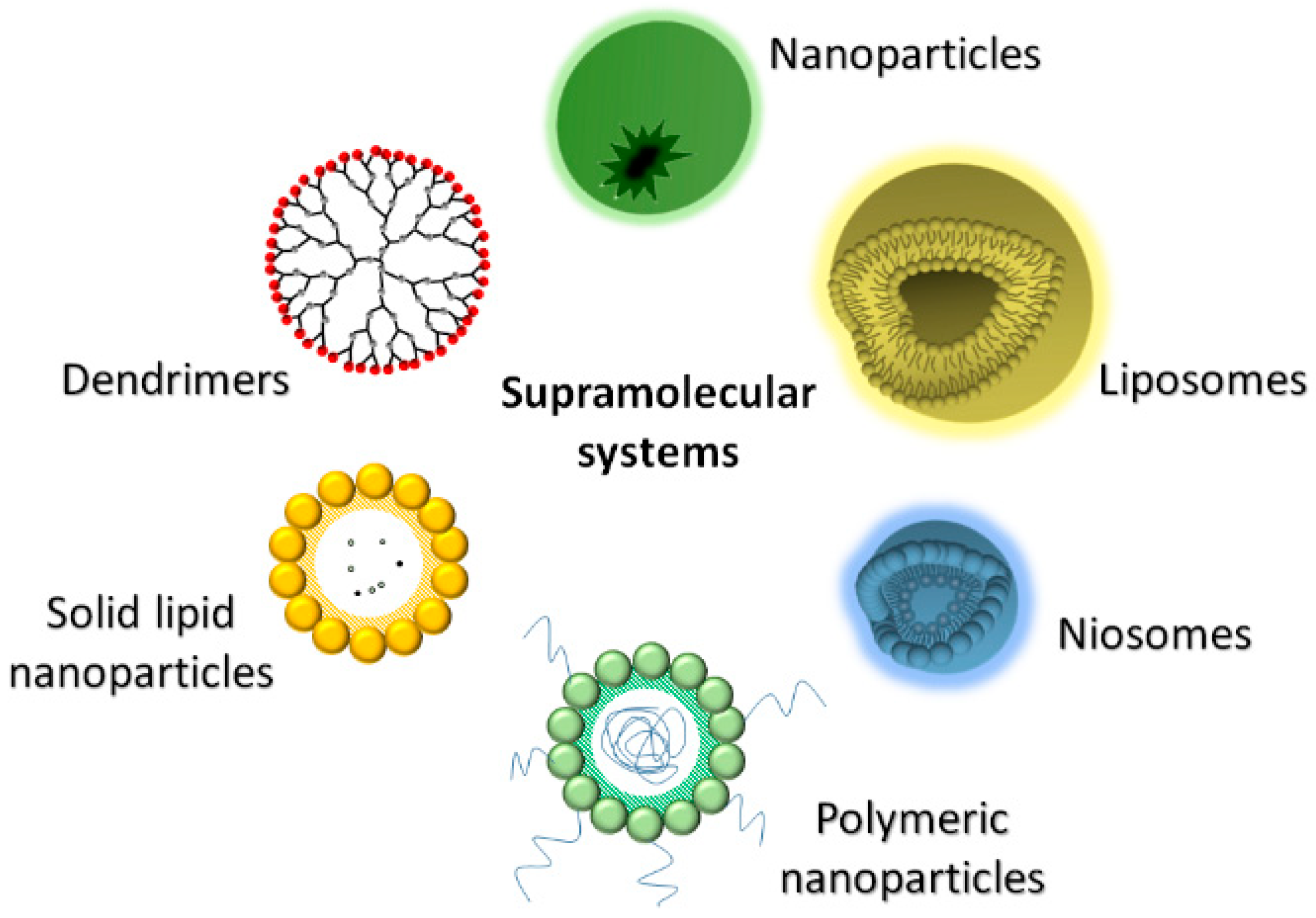
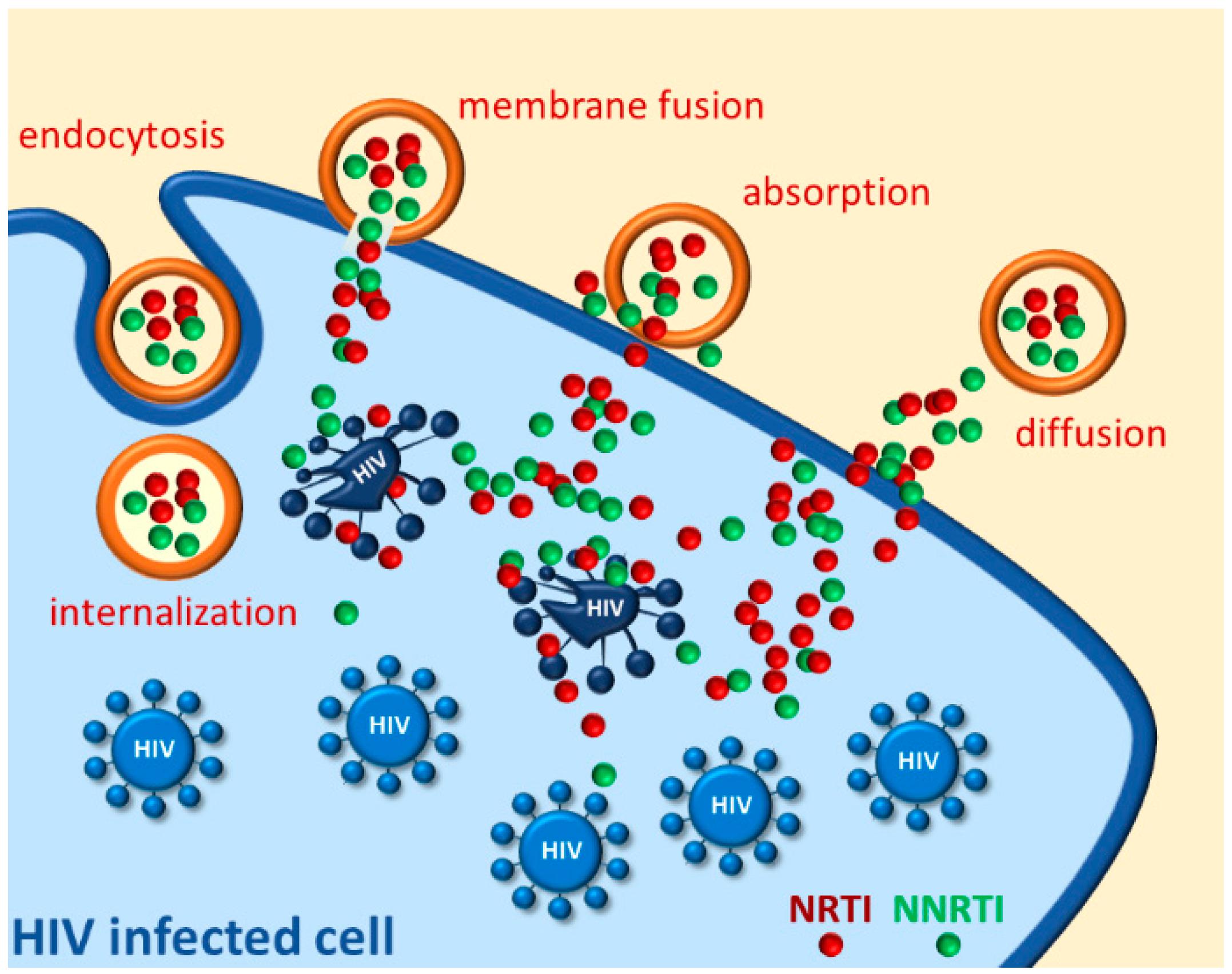
2. Drug Protection Nanosystems
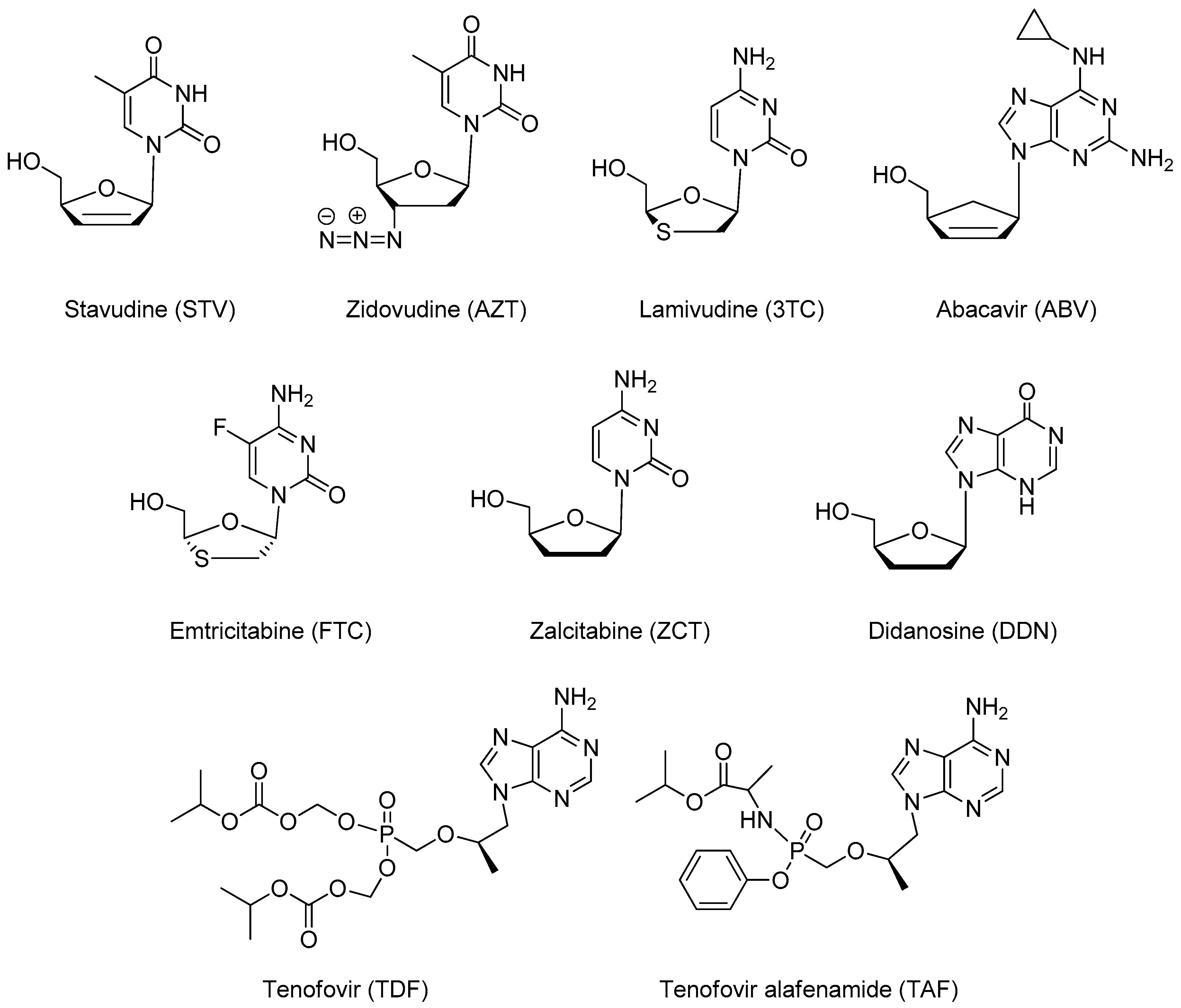
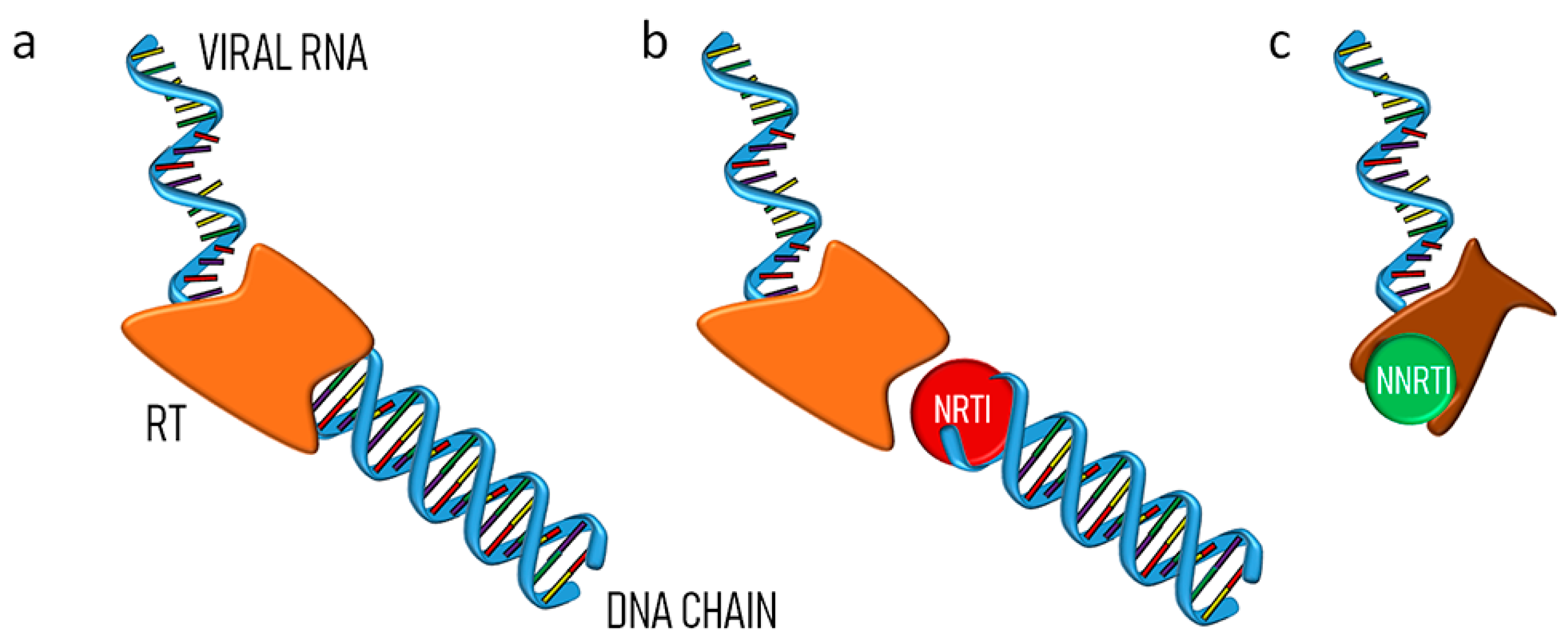
3. Nucleoside Reverse Transcriptase Inhibitors Nanosystems
3.1. Stavudine
3.2. Zidovudine
3.3. Lamivudine
3.4. Abacavir
3.5. Emtricitabine
3.6. Tenofovir
3.6.1. Tenofovir SLN
3.6.2. Tenofovir/Dendrimers Complexes
3.6.3. Chitosan based TDF Nanoparticles
3.6.4. Alternative Polymeric TDF NPs
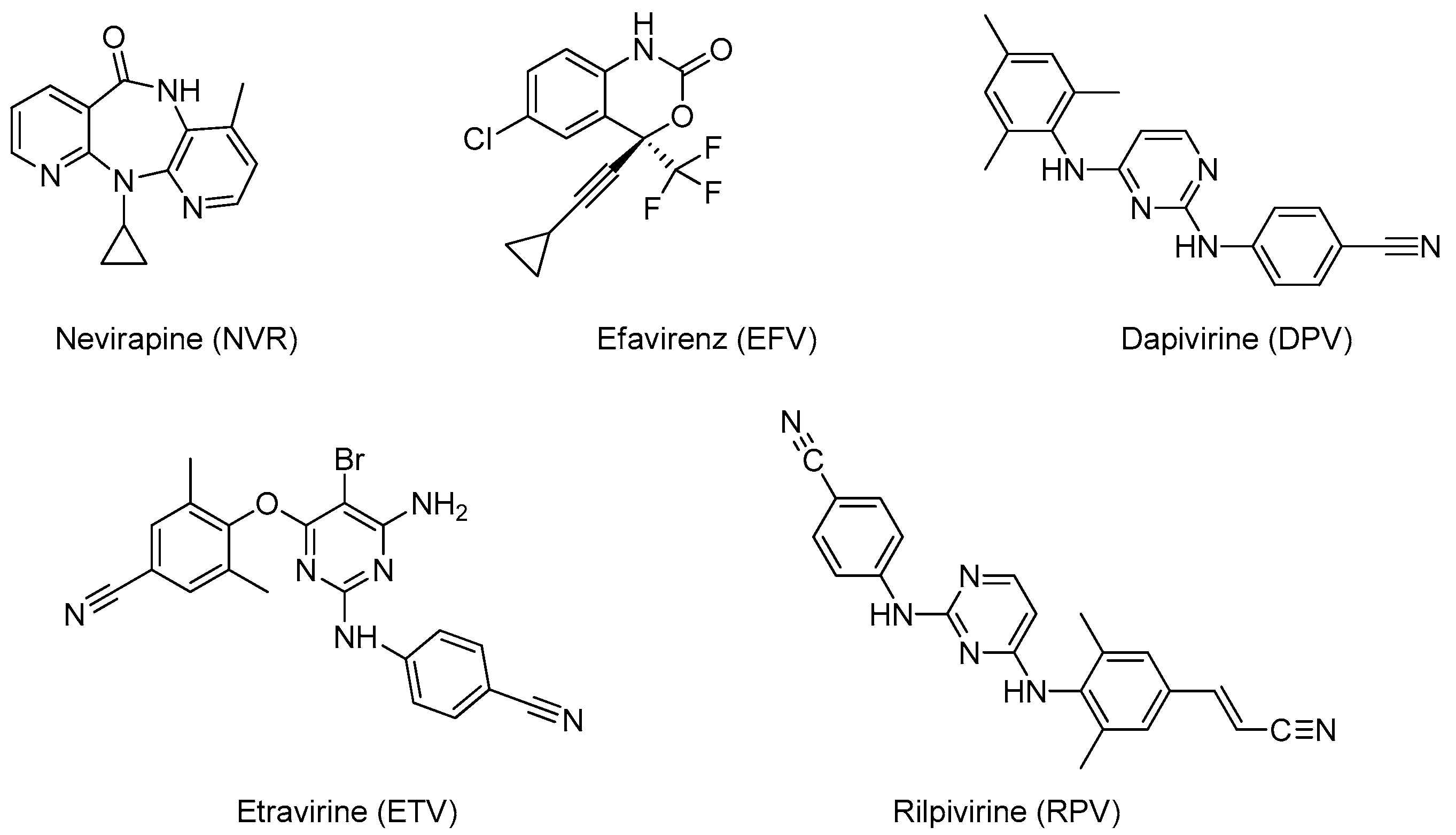
4. Non-Nucleoside Reverse Transcriptase Inhibitors Nanosystems
4.1. Nevirapine
4.2. Efavirenz
4.2.1. Efavirenz SLN
4.2.2. Polymeric EFV NPs
4.2.3. Efavirenz/Dendrimer Complexes
4.2.4. Alternative Supramolecular EFV NPs
4.3. Dapivirine
4.4. Etravirine
4.5. Rilpivirine
5. Conclusion
Author Contributions
Funding
Conflicts of Interest
References
- Vandamme, A.M.; Van Vaerenbergh, K.; De Clercq, E. Anti-human immunodeficiency virus drug combination strategies. Antivir. Chem. Chemoth. 1998, 9, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Lisziewicz, J.; Toke, E.R. Nanomedicine applications towards the cure of HIV. Nanomed-Nanotechnol. 2013, 9, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Bangsberg, D.R.; Hecht, F.M.; Charlebois, E.D.; Zolopa, A.R.; Holodniy, M.; Sheiner, L.; Bamberger, J.D.; Chesney, M.A.; Moss, A. Adherence to protease inhibitors, HIV-1 viral load, and development of drug resistance in an indigent population. Aids 2000, 14, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrager, L.K.; D’Souza, M.P. Cellular and anatomical reservoirs of HIV-1 in patients receiving potent antiretroviral combination therapy. JAMA 1998, 280, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Richman, D.D. HIV chemotherapy. Nature 2001, 410, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, A.; Price, R.W.; Gisslen, M. Antiretroviral drug treatment of CNS HIV-1 infection. J. Antimicrob. Chemother. 2012, 67, 299–311. [Google Scholar] [CrossRef]
- Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Available online: http://aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-treatment-guidelines/0 (accessed on 10 April 2019).
- Rohit, S.; Ramesh, J.; Karan, G.; Raman, K.; Anil, K.S. Nanotechnological interventions in HIV drug delivery and therapeutics. Biointerface Res. Appl. Chem. 2014, 4, 820–831. [Google Scholar]
- Harrigan, P.R.; Hogg, R.S.; Dong, W.W.; Yip, B.; Wynhoven, B.; Woodward, J.; Brumme, C.J.; Brumme, Z.L.; Mo, T.; et al. Predictors of HIV drug-resistance mutations in a large antiretroviral-naive cohort initiating triple antiretroviral therapy. J. Infect. Dis. 2005, 191, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Davey, R.T., Jr.; Engel, D.; Lane, H.C.; Fauci, A.S. Re-emergence of HIV after stopping therapy. Nature 1999, 401, 874–875. [Google Scholar] [CrossRef] [PubMed]
- Marsden, M.D.; Zack, J.A. Eradication of HIV: current challenges and new directions. J. Antimicrob. Chemother. 2009, 63, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Xing, Y.; Kim, G.J.; Simons, J.W. Nanotechnology applications in cancer. Annu. Rev. Biomed. Eng. 2007, 9, 257–288. [Google Scholar] [CrossRef] [PubMed]
- Mazzuca, P.; Caruso, A.; Caccuri, F. HIV-1 infection, microenvironment and endothelial cell dysfunction. New. Microbiol. 2016, 39, 163–173. [Google Scholar] [PubMed]
- Mahajan, S.D.; Aalinkeel, R.; Law, W.C.; Reynolds, J.L.; Nair, B.B.; Sykes, D.E.; Yong, K.T.; Roy, I.; Prasad, P.N.; Schwartz, S.A. Anti-HIV-1 nanotherapeutics: promises and challenges for the future. Int. J. Nanomed. 2012, 7, 5301–5314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, T.K.; Shah, L.; Amiji, M.M. Nanoparticulate drug carriers for delivery of HIV/AIDS therapy to viral reservoir sites. Expert Opin. Drug Deliv. 2006, 3, 613–628. [Google Scholar] [CrossRef]
- Amiji, M.M.; Vyas, T.K.; Shah, L.K. Role of nanotechnology in HIV/AIDS treatment: potential to overcome the viral reservoir challenge. Discov. Med. 2006, 6, 157–162. [Google Scholar]
- Ferrari, M. Cancer nanotechnology: opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Farokhzad, O.C. Nanotechnology for drug delivery: the perfect partnership. Expert Opin. Drug Deliv. 2008, 5, 927–929. [Google Scholar] [CrossRef] [PubMed]
- Farokhzad, O.C.; Langer, R. Impact of nanotechnology on drug delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Dunge, A.; Sharda, N.; Singh, B.; Singh, S. Establishment of inherent stability of stavudine and development of a validated stability-indicating HPLC assay method. J. Pharm. Biomed. Anal. 2005, 37, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- Devrukhakar, P.S.; Shiva Shankar, M.; Shankar, G.; Srinivas, R. A stability-indicating LC-MS/MS method for zidovudine: Identification, characterization and toxicity prediction of two major acid degradation products. J. Pharm. Biomed. Anal. 2017, 7, 231–236. [Google Scholar] [CrossRef]
- Shegokar, R.; Singh, K.K.; Muller, R.H. Production & stability of stavudine solid lipid nanoparticles--from lab to industrial scale. Int. J. Pharm. 2011, 416, 461–470. [Google Scholar] [PubMed]
- Nowacek, A.; Gendelman, H.E. NanoART, neuroAIDS and CNS drug delivery. Nanomedicine (Lond) 2009, 4, 557–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ICH Q1A(R2)- Stability Testing of New Drug Substances and Products; International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH): Geneve, Switzerland, 2003.
- Tonnesen, H.H. Photostability of Drugs and Drug Formulations; CRC Press: New York, NY, USA, 2004. [Google Scholar]
- De Luca, M.; Ioele, G.; Spatari, C.; Ragno, G. Photostabilization studies of antihypertensive 1,4-dihydropyridines using polymeric containers. Int. J. Pharm. 2016, 505, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Asthana, A.; Agashe, H.B.; Agrawal, G.P.; Jain, N.K. Stavudine-loaded mannosylated liposomes: in-vitro anti-HIV-I activity, tissue distribution and pharmacokinetics. J. Pharm. Pharmacol. 2006, 58, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Dutta, T.; Jain, N.K. Reduced hepatic toxicity, enhanced cellular uptake and altered pharmacokinetics of stavudine loaded galactosylated liposomes. Eur. J. Pharm. Biopharm. 2007, 67, 76–85. [Google Scholar] [CrossRef]
- Nayak, D.; Boxi, A.; Ashe, S.; Thathapudi, N.C.; Nayak, B. Stavudine loaded gelatin liposomes for HIV therapy: Preparation, characterization and in vitro cytotoxic evaluation. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 73, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Ramana, L.N.; Sethuraman, S.; Ranga, U.; Krishnan, U.M. Development of a liposomal nanodelivery system for nevirapine. Int. J. Biomed. Sci. 2010, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Malik, T.; Chauhan, G.; Rath, G.; Kesarkar, R.N.; Chowdhary, A.S.; Goyal, A.K. Efaverinz and nano-gold-loaded mannosylated niosomes: a host cell-targeted topical HIV-1 prophylaxis via thermogel system. Artif. Cells Nanomed. Biotechnol. 2018, 46, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.R.P.; Babrekar, L.S. Liposomal Drug Delivery for Solubility and Bioavailability Enhancement of Efavirenz. Indian J. Pharm. Sci. 2018, 80, 1115–1124. [Google Scholar]
- Abdelkader, H.; Alani, A.W.; Alany, R.G. Recent advances in non-ionic surfactant vesicles (niosomes): self-assembly, fabrication, characterization, drug delivery applications and limitations. Drug Deliv. 2014, 21, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Irchhaiya, R. Niosomes: a potential tool for novel drug delivery. J. Pharm. Investig. 2016, 46, 195–204. [Google Scholar] [CrossRef]
- Naseri, N.; Valizadeh, H.; Zakeri-Milani, P. Solid Lipid Nanoparticles and Nanostructured Lipid Carriers: Structure, Preparation and Application. Adv. Pharm. Bull. 2015, 5, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Joshy, K.S.; Chandra, P.S.; Nandakumar, K.; Sandeep, K.; Sabu, T.; Laly, A.P. Evaluation of in-vitro cytotoxicity and cellular uptake efficiency of zidovudine-loaded solid lipid nanoparticles modified with Aloe Vera in glioma cells. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 66, 40–50. [Google Scholar]
- Freeling, J.P.; Koehn, J.; Shu, C.; Sun, J.; Ho, R.J. Anti-HIV drug-combination nanoparticles enhance plasma drug exposure duration as well as triple-drug combination levels in cells within lymph nodes and blood in primates. AIDS Res. Hum. Retroviruses 2015, 31, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Mishra, M.; Padh, H. Formulation Development and Optimization of Efavirenz Loaded SLNs and NLCs using Plackett- Burman Design and its Statistical Elucidation. Int. J. Pharm. Res. Health Sci. 2018, 6, 2379–2388. [Google Scholar]
- Gupta, S.; Kesarla, R.; Chotai, N.; Misra, A.; Omri, A. Systematic Approach for the Formulation and Optimization of Solid Lipid Nanoparticles of Efavirenz by High Pressure Homogenization Using Design of Experiments for Brain Targeting and Enhanced Bioavailability. Biomed. Res. Int. 2017, 2017, 5984014. [Google Scholar] [CrossRef] [PubMed]
- Madhusudhan, A.; Bhagavanth, R.G.; Venkatesham, M.; Veerabhadram, G. Design and Evaluation of Efavirenz loaded Solid Lipid Nanoparticles to Improve the Oral Bioavailability. Int. J. Pharm. Pharm. Sci. 2012, 2, 84–89. [Google Scholar]
- Pardeshi, C.; Rajput, P.; Belgamwar, V.; Tekade, A.; Patil, G.; Chaudhary, K.; Sonje, A. Solid lipid based nanocarriers: an overview. Acta Pharm. 2012, 62, 433–472. [Google Scholar] [CrossRef]
- Date, A.A.; Destache, C.J. A review of nanotechnological approaches for the prophylaxis of HIV/AIDS. Biomaterials 2013, 34, 6202–6228. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yu, B.; Zhong, Z.; Guo, B.H.; Huang, Y. Nevirapine-polycaprolactone crystalline inclusion complex as a potential long-acting injectable solid form. Int. J. Pharm. 2018, 543, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Woodrow, K.A. Nanotechnology approaches to eradicating HIV reservoirs. Eur. J. Pharm. Biopharm. 2018, 138, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.; Kruger, H.G.; Maguire, G.E.M.; Govender, T.; Parboosing, R. The role of nanotechnology in the treatment of viral infections. Therap. Adv. Infect. Dis. 2017, 4, 105–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parboosing, R.; Maguire, G.E.; Govender, P.; Kruger, H.G. Nanotechnology and the treatment of HIV infection. Viruses 2012, 4, 488–520. [Google Scholar] [CrossRef] [PubMed]
- das Neves, J.; Amiji, M.M.; Bahia, M.F.; Sarmento, B. Nanotechnology-based systems for the treatment and prevention of HIV/AIDS. Adv. Drug Deliv. Rev. 2010, 62, 458–477. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.K.; Parikh, R.H.; Patel, N. Targeted delivery of mannosylated-PLGA nanoparticles of antiretroviral drug to brain. Int. J. Nanomed. 2018, 13, 97–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, T.O.; Giardiello, M.; Martin, P.; Siccardi, M.; Liptrott, N.J.; Smith, D.; Roberts, P.; Curley, P.; Schipani, A.; Khoo, S.H.; et al. Antiretroviral solid drug nanoparticles with enhanced oral bioavailability: production, characterization, and in vitro-in vivo correlation. Adv. Healthc. Mater. 2014, 3, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Almaraz, M.T.; Gref, R.; Agostoni, V.; Kreuz, C.; Clayette, P.; Serre, C.; Couvreurb, P.; Horcajada, P. Towards improved HIV-microbicide activity through the co-encapsulation of NRTI drugs in biocompatible metal organic framework nanocarriers. J. Mater. Chem. B 2017, 5, 8563–8569. [Google Scholar] [CrossRef]
- Chiodo, F.; Marradi, M.; Calvo, J.; Yuste, E.; Penades, S. Glycosystems in nanotechnology: Gold glyconanoparticles as carrier for anti-HIV prodrugs. Beilstein J. Org. Chem. 2014, 10, 1339–1346. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Wu, Z.; Qi, X.; Chen, Y.; Li, X. Dendrimers as potential therapeutic tools in HIV inhibition. Molecules 2013, 18, 7912–7929. [Google Scholar] [CrossRef]
- Ionov, M.; Ciepluch, K.; Klajnert, B.; Glinska, S.; Gomez-Ramirez, R.; de la Mata, F.J.; Munoz-Fernandez, M.A.; Bryszewska, M. Complexation of HIV derived peptides with carbosilane dendrimers. Colloids Surf. B Biointerfaces 2013, 101, 236–242. [Google Scholar] [CrossRef]
- Garcia-Broncano, P.; Cena-Diez, R.; de la Mata, F.J.; Gomez, R.; Resino, S.; Munoz-Fernandez, M.A. Efficacy of carbosilane dendrimers with an antiretroviral combination against HIV-1 in the presence of semen-derived enhancer of viral infection. Eur. J. Pharmacol. 2017, 811, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Cena-Diez, R.; Vacas-Cordoba, E.; Garcia-Broncano, P.; de la Mata, F.J.; Gomez, R.; Maly, M.; Munoz-Fernandez, M.A. Prevention of vaginal and rectal herpes simplex virus type 2 transmission in mice: mechanism of antiviral action. Int. J. Nanomed. 2016, 11, 2147–2162. [Google Scholar] [PubMed] [Green Version]
- das Neves, J.; Amiji, M.; Bahia, M.F.; Sarmento, B. Assessing the physical-chemical properties and stability of dapivirine-loaded polymeric nanoparticles. Int. J. Pharm. 2013, 456, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Nucleoside Reverse Transcriptase Inhibitors (NRTIs or nukes). Available online: https://www.hiv.va.gov/patient/treat/nrtis.asp (accessed on 10 April 2019).
- Shegokar, R.; Singh, K.K. Stavudine entrapped lipid nanoparticles for targeting lymphatic HIV reservoirs. Pharmazie 2011, 66, 264–271. [Google Scholar] [PubMed]
- Kaur, C.D.; Nahar, M.; Jain, N.K. Lymphatic targeting of zidovudine using surface-engineered liposomes. J. Drug Target 2008, 16, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Weibull, W. A statistical distribution function of wide applicability. J. Appl. Mech. 1951, 18, 293–297. [Google Scholar]
- Carvalho, F.C.; Sarmento, V.H.; Chiavacci, L.A.; Barbi, M.S.; Gremiao, M.P. Development and in vitro evaluation of surfactant systems for controlled release of zidovudine. J. Appl. Mech. 2010, 99, 2367–2374. [Google Scholar] [CrossRef]
- Mainardes, R.M.; Gremiao, M.P.; Brunetti, I.L.; da Fonseca, L.M.; Khalil, N.M. Zidovudine-loaded PLA and PLA-PEG blend nanoparticles: influence of polymer type on phagocytic uptake by polymorphonuclear cells. J. Pharm. Sci. 2009, 98, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Mainardes, R.M.; Gremiao, M.P. Nanoencapsulation and characterization of zidovudine on poly(l-lactide) and poly(l -lactide)-poly(ethylene glycol)-blend nanoparticles. J. Nanosci. Nanotechnol. 2012, 12, 8513–8521. [Google Scholar] [CrossRef] [PubMed]
- Joshy, K.S.; Susan, M.A.; Snigdha, S.; Nandakumar, K.; Laly, A.P.; Sabu, T. Encapsulation of zidovudine in PF-68 coated alginate conjugate nanoparticles for anti-HIV drug delivery. Int. J. Biol. Macromol. 2018, 107, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Dunge, A.; Sharda, N.; Singh, B.; Singh, S. Validated specific HPLC method for determination of zidovudine during stability studies. J. Pharm. Biomed. Anal. 2005, 37, 1109–1114. [Google Scholar] [CrossRef]
- Schenfeld, E.M.; Ribone, S.R.; Quevedo, M.A. Stability and plasmatic protein binding of novel zidovudine prodrugs: Targeting site ii of human serum albumin. Eur. J. Pharm. Sci. 2018, 115, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.M.; Faulds, D. Lamivudine. A review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy in the management of HIV infection. Drugs 1997, 53, 657–680. [Google Scholar] [CrossRef]
- Vogenthaler, N.S. Lamivudine and second-line antiretroviral regimens. Clin. Infect. Dis. 2007, 44, 1387. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, S.V.; Poluektova, L.Y.; Makarov, E.; Gerson, T.; Senanayake, M.T. Nano-NRTIs: efficient inhibitors of HIV type-1 in macrophages with a reduced mitochondrial toxicity. Antivir. Chem. Chemother. 2010, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bedse, G.; Kumar, V.; Singh, S. Study of forced decomposition behavior of lamivudine using LC, LC-MS/TOF and MS(n). J. Pharm. Biomed. Anal. 2009, 49, 55–63. [Google Scholar] [CrossRef]
- Konari, N.S.; Jacob, J.T. Stability indicating validated UPLC technique for the simultaneous analysis of raltegravir and lamivudine in pharmaceutical dosage forms. HIV AIDS Rev. 2016, 15, 161–169. [Google Scholar] [CrossRef]
- Kurmi, M.; Sahu, A.; Singh, D.K.; Singh, I.P.; Singh, S. Stability behaviour of antiretroviral drugs and their combinations. 8: Characterization and in-silico toxicity prediction of degradation products of efavirenz. J. Pharm. Biomed. Anal. 2018, 148, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Tamizhrasi, S.; Shukla, A.; Shivkumar, T.; Rathi, V.; Rathi, J.C. Formulation and evaluation of lamivudine loaded polymethacrylic acid nanoparticles. Int. J. Pharmtech Res. 2009, 1, 411–415. [Google Scholar]
- Bing, W.; Guan Qun, C.; Zheng Wei, M.; Yu Ying, Z.; Da Hai, Y.; Chang You, G. Preparation and cellular uptake of PLGA particles loaded with lamivudine. Chin. Sci. Bull. 2012, 57, 3985–3993. [Google Scholar] [Green Version]
- Rajca, A.; Li, Q.; Date, A.; Belshan, M.; Destache, C. Thermosensitive Vaginal Gel Containing PLGA-NRTI conjugated nanoparticles for HIV prophylaxis. NSTI-Nanotech 2013, 3, 293–296. [Google Scholar]
- Ramadan, E. Transdermal microneedle–mediated delivery of polymeric lamivudine loaded nanoparticles. J. Pharm. Technol. Drug Res. 2016, 5. [Google Scholar] [CrossRef]
- Hatami, E.; Mu, Y.; Shields, D.N.; Chauhan, S.C.; Kumar, S.; Cory, T.J.; Yallapu, M.M. Mannose-decorated hybrid nanoparticles for enhanced macrophage targeting. Biochem. Biophys. Rep. 2019, 17, 197–207. [Google Scholar] [CrossRef]
- Dev, A.; Binulal, N.S.; Anitha, A.; Nair, S.V.; Furuike, T.; Tamura, H.; Jayakumar, R. Preparation of poly(lactic acid)/chitosan nanoparticles for anti-HIV drug delivery applications. Carbohydr. Polym. 2010, 80, 833–838. [Google Scholar] [CrossRef] [Green Version]
- Nesalin, J.A.J.; Smith, A.A. Stability study of chitosan nanoparticles containing some antiretroviral drugs. Res. J. Pharm. Biol. Chem. Sci. 2014, 5, 193–203. [Google Scholar]
- Deepak, M.; Nivrati, J.; Vaibhav, R.; Ashish, K.J. Glycyrrhizin conjugated chitosan nanoparticles for hepatocyte-targeted delivery of lamivudine. J. Pharm. Pharmacol. 2014, 66, 1082–1093. [Google Scholar]
- Tshweu, L.; Katata, L.; Kalombo, L.; Swai, H. Nanoencapsulation of water-soluble drug, lamivudine, using a double emulsion spray-drying technique for improving HIV treatment. J. Nanopart. Res. 2013, 15. [Google Scholar] [CrossRef]
- Oluwaseun, O.; Namita, K.; Kahli, A.S.; Oleg, B.; Simeon, A.; Ayele, G.; Winston, A.A.; Sergei, N.; Emmanuel, O.A. Antiretroviral Drugs-Loaded Nanoparticles Fabricated by Dispersion Polymerization with Potential for HIV/AIDS Treatment. Inf. Dis. Res. Treat. 2016, 9, 21–32. [Google Scholar]
- Pravalika, R.P.; Madhava, R.B.; Prakash, D.J.; Latha, K.; Prashanthi, D. Formulation and characterisation of chitosan based lamivudine nanoparticles. Eur. J. Pharm. Med. Res. 2017, 4, 377–383. [Google Scholar]
- Kumar, P.; Lakshmi, Y.S.; C, B.; Golla, K.; Kondapi, A.K. Improved Safety, Bioavailability and Pharmacokinetics of Zidovudine through Lactoferrin Nanoparticles during Oral Administration in Rats. PLoS ONE 2015, 10, e0140399. [Google Scholar] [CrossRef]
- Kumar, P.; Lakshmi, Y.S.; Kondapi, A.K. Triple Drug Combination of Zidovudine, Efavirenz and Lamivudine Loaded Lactoferrin Nanoparticles: an Effective Nano First-Line Regimen for HIV Therapy. Pharm. Res. 2017, 34, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Shahabadia, N.; Khorshidia, A.; Zhalehc, H.; Kashaniand, S. Synthesis, characterization, cytotoxicity and DNA binding studies of Fe3O4@ SiO2 nanoparticles coated by an antiviral drug lamivudine. J. Drug Deliv. Sci. Technol. 2018, 46, 55–65. [Google Scholar] [CrossRef]
- Vasilyeva, S.V.; Shtil, A.A.; Petrova, A.S.; Balakhnin, S.M.; Achigecheva, P.Y.; Stetsenko, D.A.; Silnikov, V.N. Conjugates of phosphorylated zalcitabine and lamivudine with SiO2 nanoparticles: Synthesis by CuAAC click chemistry and preliminary assessment of anti-HIV and antiproliferative activity. Bioorg. Med. Chem. 2017, 25, 1696–1702. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.A.; Kumar, A.N.; Arnab, D.; Debmalya, M.; Amalesh, S. Development of lamivudine containing multiple emulsions stabilizedby gum odina. Future J. Pharm. Sci. 2018, 4, 71–79. [Google Scholar]
- Suma, U.S.; Parthiban, S.; Senthil, K.G.P.; Tamiz, M.T. Effect of span-80 in the formulation lamivudine niosomal gel. Asian J. Res. Biol. Pharm. Sci. 2015, 4, 35–45. [Google Scholar]
- Godbole, M.D.; Mathur, V. Selection of phospholipid and method of formulation for optimum entrapment and release of lamivudine from liposome. J. Drug Deliv. Ther. 2018, 8, 175–183. [Google Scholar] [CrossRef]
- Wilson, B.; Paladugu, L.; Priyadarshini, S.R.; Jenita, J.J. Development of albumin-based nanoparticles for the delivery of abacavir. Int. J. Biol. Macromol. 2015, 81, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Corbo, C.; Molinaro, R.; Parodi, A.; Toledano Furman, N.E.; Salvatore, F.; Tasciotti, E. The impact of nanoparticle protein corona on cytotoxicity, immunotoxicity and target drug delivery. Nanomedicine (Lond.) 2016, 11, 81–100. [Google Scholar] [CrossRef]
- Lin, Z.; Gautam, N.; Alnouti, Y.; McMillan, J.; Bade, A.N.; Gendelman, H.E.; Edagwa, B. ProTide generated long-acting abacavir nanoformulations. Chem. Commun. 2018, 54, 8371–8374. [Google Scholar] [CrossRef]
- Singh, G.; Pai, R.S. Pharmacokinetics and in vivo biodistribution of optimized PLGA nanoparticulate drug delivery system for controlled release of emtricitabine. Drug Deliv. 2014, 21, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Pai, R.S. Optimization (central composite design) and validation of HPLC method for investigation of emtricitabine loaded poly(lactic-co-glycolic acid) nanoparticles: in vitro drug release and in vivo pharmacokinetic studies. Sci. World J. 2014, 2014, 583090. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Belshan, M.; Holec, A.; Zhou, Y.; Destache, C.J. An Enhanced Emtricitabine-Loaded Long-Acting Nanoformulation for Prevention or Treatment of HIV Infection. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Rao, R.N.; Vali, R.M.; Ramachandra, B.; Raju, S.S. Separation and characterization of forced degradation products of abacavir sulphate by LC-MS/MS. J. Pharm. Biomed. Anal. 2011, 54, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Kurmi, M.; Sahu, A.; Singh, S. Stability behaviour of antiretroviral drugs and their combinations. 5: Characterization of novel degradation products of abacavir sulfate by mass and nuclear magnetic resonance spectrometry. J. Pharm. Biomed. Anal. 2017, 134, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Youa, J.; Yub, F. Study on the thermal decomposition of emtricitabine. J. Anal. Appl. Pyrolysis 2015, 115, 344–352. [Google Scholar] [CrossRef]
- Kurmi, M.; Singh, S. Stability behavior of antiretroviral drugs and their combinations. 7: Comparative degradation pathways of lamivudine and emtricitabine and explanation to their differential degradation behavior by density functional theory. J.Pharm. Biomed. Anal. 2017, 142, 155–161. [Google Scholar] [CrossRef]
- Duan, J.; Freeling, J.P.; Koehn, J.; Shu, C.; Ho, R.J. Evaluation of atazanavir and darunavir interactions with lipids for developing pH-responsive anti-HIV drug combination nanoparticles. J. Pharm. Sci. 2014, 103, 2520–2529. [Google Scholar] [CrossRef] [PubMed]
- Freeling, J.P.; Koehn, J.; Shu, C.; Sun, J.; Ho, R.J. Long-acting three-drug combination anti-HIV nanoparticles enhance drug exposure in primate plasma and cells within lymph nodes and blood. Aids 2014, 28, 2625–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, A.I.; Naidu, E.C.; Akang, E.; Ogedengbe, O.O.; Offor, U.; Rambharose, S.; Kalhapure, R.; Chuturgoon, A.; Govender, T.; Azu, O.O. Investigating Organ Toxicity Profile of Tenofovir and Tenofovir Nanoparticle on the Liver and Kidney: Experimental Animal Study. Toxicol. Res. 2018, 34, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Pokharkar, V.B.; Jolly, M.R.; Kumbhar, D.D. Engineering of a hybrid polymer-lipid nanocarrier for the nasal delivery of tenofovir disoproxil fumarate: physicochemical, molecular, microstructural, and stability evaluation. Eur. J. Pharm. Sci. 2015, 71, 99–111. [Google Scholar] [CrossRef]
- Ramanathan, R.; Jiang, Y.; Read, B.; Golan-Paz, S.; Woodrow, K.A. Biophysical characterization of small molecule antiviral-loaded nanolipogels for HIV-1 chemoprophylaxis and topical mucosal application. Acta Biomater. 2016, 36, 122–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayant, R.D.; Atluri, V.S.; Agudelo, M.; Sagar, V.; Kaushik, A.; Nair, M. Sustained-release nanoART formulation for the treatment of neuroAIDS. Int. J. Nanomed. 2015, 10, 1077–1093. [Google Scholar] [CrossRef] [Green Version]
- Vacas-Cordoba, E.; Galan, M.; de la Mata, F.J.; Gomez, R.; Pion, M.; Munoz-Fernandez, M.A. Enhanced activity of carbosilane dendrimers against HIV when combined with reverse transcriptase inhibitor drugs: searching for more potent microbicides. Int. J. Nanomed. 2014, 9, 3591–3600. [Google Scholar] [Green Version]
- Briz, V.; Sepulveda-Crespo, D.; Diniz, A.R.; Borrego, P.; Rodes, B.; de la Mata, F.J.; Gomez, R.; Taveira, N.; Munoz-Fernandez, M.A. Development of water-soluble polyanionic carbosilane dendrimers as novel and highly potent topical anti-HIV-2 microbicides. Nanoscale 2015, 7, 14669–14683. [Google Scholar] [CrossRef] [Green Version]
- Sepulveda-Crespo, D.; Sanchez-Rodriguez, J.; Serramia, M.J.; Gomez, R.; De La Mata, F.J.; Jimenez, J.L.; Munoz-Fernandez, M.A. Triple combination of carbosilane dendrimers, tenofovir and maraviroc as potential microbicide to prevent HIV-1 sexual transmission. Nanomedicine (Lond) 2015, 10, 899–914. [Google Scholar] [CrossRef]
- Sepulveda-Crespo, D.; Gomez, R.; De La Mata, F.J.; Jimenez, J.L.; Munoz-Fernandez, M.A. Polyanionic carbosilane dendrimer-conjugated antiviral drugs as efficient microbicides: Recent trends and developments in HIV treatment/therapy. Nanomed.- Nanotechnol. Biol. Med. 2015, 11, 1481–1498. [Google Scholar] [CrossRef] [PubMed]
- Agrahari, V.; Zhang, C.; Zhang, T.; Li, W.; Gounev, T.K.; Oyler, N.A.; Youan, B.B. Hyaluronidase-sensitive nanoparticle templates for triggered release of HIV/AIDS microbicide in vitro. AAPS J. 2014, 16, 181–193. [Google Scholar] [CrossRef]
- Ngo, A.N.; Ezoulin, M.J.; Murowchick, J.B.; Gounev, A.D.; Youan, B.B. Sodium Acetate Coated Tenofovir-Loaded Chitosan Nanoparticles for Improved Physico-Chemical Properties. Pharm. Res. 2016, 33, 367–383. [Google Scholar] [CrossRef]
- Timur, S.S.; Sahin, A.; Aytekin, E.; Ozturk, N.; Polat, K.H.; Tezel, N.; Gursoy, R.N.; Calis, S. Design and in vitro evaluation of tenofovir-loaded vaginal gels for the prevention of HIV infections. Pharm. Dev. Technol. 2018, 23, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Shailender, J.; Ravi, P.R.; Reddy Sirukuri, M.; Dalvi, A.; Keerthi Priya, O. Chitosan nanoparticles for the oral delivery of tenofovir disoproxil fumarate: formulation optimization, characterization and ex vivo and in vivo evaluation for uptake mechanism in rats. Drug Dev. Ind. Pharm. 2018, 44, 1109–1119. [Google Scholar] [CrossRef]
- Shohani, S.; Mondanizadeh, M.; Abdoli, A.; Khansarinejad, B.; Salimi-Asl, M.; Ardestani, M.S.; Ghanbari, M.; Haj, M.S.; Zabihollahi, R. Trimethyl Chitosan Improves Anti-HIV Effects of Atripla as a New Nanoformulated Drug. Curr. HIV Res. 2017, 15, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Ensinas, A.; Verrier, B.; Primard, C.; Cuvillier, A.; Champier, G.; Paul, S.; Delair, T. Zinc-Stabilized Chitosan-Chondroitin Sulfate Nanocomplexes for HIV-1 Infection Inhibition Application. Mol. Pharm. 2016, 13, 3279–3291. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Agrahari, V.; Ezoulin, M.J.; Zhang, C.; Purohit, S.S.; Molteni, A.; Dim, D.; Oyler, N.A.; Youan, B.C. Tenofovir Containing Thiolated Chitosan Core/Shell Nanofibers: In Vitro and in Vivo Evaluations. Mol. Pharm. 2016, 13, 4129–4140. [Google Scholar] [CrossRef] [PubMed]
- Destache, C.J.; Mandal, S.; Yuan, Z.; Kang, G.; Date, A.A.; Lu, W.; Shibata, A.; Pham, R.; Bruck, P.; Rezich, M.; et al. Topical Tenofovir Disoproxil Fumarate Nanoparticles Prevent HIV-1 Vaginal Transmission in a Humanized Mouse Model. Antimicrob. Agents Chemother. 2016, 60, 3633–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, A.; Cunha-Reis, C.; Araujo, F.; Nunes, R.; Seabra, V.; Ferreira, D.; das Neves, J.; Sarmento, B. Development and in vivo safety assessment of tenofovir-loaded nanoparticles-in-film as a novel vaginal microbicide delivery system. Acta Biomater. 2016, 44, 332–340. [Google Scholar] [CrossRef]
- Cunha-Reis, C.; Machado, A.; Barreiros, L.; Araujo, F.; Nunes, R.; Seabra, V.; Ferreira, D.; Segundo, M.A.; Sarmento, B.; das Neves, J. Nanoparticles-in-film for the combined vaginal delivery of anti-HIV microbicide drugs. J. Control. Release 2016, 243, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Kang, G.; Prathipati, P.K.; Fan, W.; Li, Q.; Destache, C.J. Long-acting parenteral combination antiretroviral loaded nano-drug delivery system to treat chronic HIV-1 infection: A humanized mouse model study. Antiviral Res. 2018, 156, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Kang, G.; Prathipati, P.K.; Zhou, Y.; Fan, W.; Li, Q.; Destache, C.J. Nanoencapsulation introduces long-acting phenomenon to tenofovir alafenamide and emtricitabine drug combination: A comparative pre-exposure prophylaxis efficacy study against HIV-1 vaginal transmission. J. Control. Release 2019, 294, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Prathipati, P.K.; Kang, G.; Zhou, Y.; Yuan, Z.; Fan, W.; Li, Q.; Destache, C.J. Tenofovir alafenamide and elvitegravir loaded nanoparticles for long-acting prevention of HIV-1 vaginal transmission. Aids 2017, 31, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Prathipati, P.K.; Mandal, S.; Pon, G.; Vivekanandan, R.; Destache, C.J. Pharmacokinetic and Tissue Distribution Profile of Long Acting Tenofovir Alafenamide and Elvitegravir Loaded Nanoparticles in Humanized Mice Model. Pharm. Res. 2017, 34, 2749–2755. [Google Scholar] [CrossRef] [PubMed]
- Shailender, J.; Ravi, P.R.; Saha, P.; Dalvi, A.; Myneni, S. Tenofovir disoproxil fumarate loaded PLGA nanoparticles for enhanced oral absorption: Effect of experimental variables and in vitro, ex vivo and in vivo evaluation. Colloids Surf. B Biointerfaces 2017, 158, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Cautela, M.P.; Moshe, H.; Sosnik, A.; Sarmento, B.; das Neves, J. Composite films for vaginal delivery of tenofovir disoproxil fumarate and emtricitabine. Eur. J. Pharm. Biopharm. 2018, 138, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Tomitaka, A.; Arami, H.; Huang, Z.; Raymond, A.; Rodriguez, E.; Cai, Y.; Febo, M.; Takemura, Y.; Nair, M. Hybrid magneto-plasmonic liposomes for multimodal image-guided and brain-targeted HIV treatment. Nanoscale 2017, 10, 184–194. [Google Scholar] [CrossRef]
- Kraft, J.C.; McConnachie, L.A.; Koehn, J.; Kinman, L.; Sun, J.; Collier, A.C.; Collins, C.; Shen, D.D.; Ho, R.J.Y. Mechanism-based pharmacokinetic (MBPK) models describe the complex plasma kinetics of three antiretrovirals delivered by a long-acting anti-HIV drug combination nanoparticle formulation. J. Control. Release 2018, 275, 229–241. [Google Scholar] [CrossRef]
- McConnachie, L.A.; Kinman, L.M.; Koehn, J.; Kraft, J.C.; Lane, S.; Lee, W.; Collier, A.C.; Ho, R.J.Y. Long-Acting Profile of 4 Drugs in 1 Anti-HIV Nanosuspension in Nonhuman Primates for 5 Weeks After a Single Subcutaneous Injection. J. Pharm. Sci. 2018, 107, 1787–1790. [Google Scholar] [CrossRef]
- Koehn, J.; Iwamoto, J.F.; Kraft, J.C.; McConnachie, L.A.; Collier, A.C.; Ho, R.J.Y. Extended cell and plasma drug levels after one dose of a three-in-one nanosuspension containing lopinavir, efavirenz, and tenofovir in nonhuman primates. Aids 2018, 32, 2463–2467. [Google Scholar] [PubMed]
- Perazzolo, S.; Shireman, L.M.; Koehn, J.; McConnachie, L.A.; Kraft, J.C.; Shen, D.D.; Ho, R.J.Y. Three HIV Drugs, Atazanavir, Ritonavir, and Tenofovir, Coformulated in Drug-Combination Nanoparticles Exhibit Long-Acting and Lymphocyte-Targeting Properties in Nonhuman Primates. J. Pharm. Sci. 2018, 107, 3153–3162. [Google Scholar] [CrossRef] [PubMed]
- Golla, V.M.; Kurmi, M.; Shaik, K.; Singh, S. Stability behaviour of antiretroviral drugs and their combinations. 4: Characterization of degradation products of tenofovir alafenamide fumarate and comparison of its degradation and stability behaviour with tenofovir disoproxil fumarate. J. Pharm. Biomed. Anal. 2016, 131, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.C.; Lin, P.I.; Wang, C.C. Targeting nevirapine delivery across human brain microvascular endothelial cells using transferrin-grafted poly(lactide-co-glycolide) nanoparticles. Nanomedicine (Lond) 2011, 6, 1011–1026. [Google Scholar] [CrossRef] [PubMed]
- Shegokar, R.; Singh, K.K. Nevirapine nanosuspensions for HIV reservoir targeting. Die Pharmazie 2011, 66, 408–415. [Google Scholar]
- Shegokar, R.; Singh, K.K. Surface modified nevirapine nanosuspensions for viral reservoir targeting: In vitro and in vivo evaluation. Int. J. Pharm. 2011, 421, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Reis, N.F.; de Assis, J.C.; Fialho, S.L.; Pianetti, G.A.; Fernandes, C. Stability-indicating UHPLC method for determination of nevirapine in its bulk form and tablets: identification of impurities and degradation kinetic study. J. Pharm. Biomed. Anal. 2016, 126, 103–108. [Google Scholar] [CrossRef]
- Aungst, B.J.; Nguyen, N.H.; Taylor, N.J.; Bindra, D.S. Formulation and food effects on the oral absorption of a poorly water soluble, highly permeable antiretroviral agent. J. Pharm. Sci. 2002, 91, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Csajka, C.; Marzolini, C.; Fattinger, K.; Decosterd, L.A.; Fellay, J.; Telenti, A.; Biollaz, J.; Buclin, T. Population pharmacokinetics and effects of efavirenz in patients with human immunodeficiency virus infection. Clin. Pharmacol. Ther. 2003, 73, 20–30. [Google Scholar] [CrossRef]
- Vyas, A.; Jain, A.; Hurkat, P.; Jain, A.; Jain, S.K. Targeting of AIDS related encephalopathy using phenylalanine anchored lipidic nanocarrier. Colloids Surf. B Biointerfaces 2015, 131, 155–161. [Google Scholar] [CrossRef]
- Makwana, V.; Jain, R.; Patel, K.; Nivsarkar, M.; Joshi, A. Solid lipid nanoparticles (SLN) of Efavirenz as lymph targeting drug delivery system: Elucidation of mechanism of uptake using chylomicron flow blocking approach. Int. J. Pharm. 2015, 495, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Gaur, P.K.; Mishra, S.; Bajpai, M.; Mishra, A. Enhanced Oral Bioavailability of Efavirenz by Solid Lipid Nanoparticles: In Vitro Drug Release and Pharmacokinetics Studies. BioMed Res. Int. 2014, 2014, 363404. [Google Scholar] [CrossRef]
- Vedha Hari, B.N.; Dhevendaran, K.; Narayanan, N. Development of Efavirenz nanoparticle for enhanced efficiency of anti-retroviral therapy against HIV and AIDS. In Proceedings of the First International Science Symposium on HIV and Infectious Diseases (HIV SCIENCE 2012), Chennai, India, 20–22 January 2012; Springer-Verlag GmbH: Heidelberg, Germany, 2012. [Google Scholar]
- Chaowanachan, T.; Krogstad, E.; Ball, C.; Woodrow, K.A. Drug synergy of tenofovir and nanoparticle-based antiretrovirals for HIV prophylaxis. PLoS ONE 2013, 8, e61416. [Google Scholar] [CrossRef]
- Date, A.A.; Shibata, A.; McMullen, E.; La Bruzzo, K.; Bruck, P.; Belshan, M.; Zhou, Y.; Destache, C.J. Thermosensitive Gel Containing Cellulose Acetate Phthalate-Efavirenz Combination Nanoparticles for Prevention of HIV-1 Infection. J. Biomed. Nanotechnol. 2015, 11, 416–427. [Google Scholar] [CrossRef]
- Roy, U.; Ding, H.; Pilakka-Kanthikeel, S.; Raymond, A.D.; Atluri, V.; Yndart, A.; Kaftanovskaya, E.M.; Batrakova, E.; Agudelo, M.; Nair, M. Preparation and characterization of anti-HIV nanodrug targeted to microfold cell of gut-associated lymphoid tissue. Int. J. Nanomed. 2015, 10, 5819–5835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haria, V.B.N.; Lu, C.L.; Narayananc, N.; Wang, R.R.; Zheng, Y.T. Engineered polymeric nanoparticles of Efavirenz: Dissolution enhancement through particle size reduction. Chem. Eng. Sci. 2016, 155, 366–375. [Google Scholar] [CrossRef]
- Hari, B.N.V.; Narayanan, N.; Dhevendaran, K.; Ramyadevi, D. Engineered nanoparticles of Efavirenz using methacrylate co-polymer (Eudragit-E100) and its biological effects in-vivo. Mater. Sci. Eng. C. Mater. Biol. Appl. 2016, 67, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Belgamwar, A.; Khan, S.; Yeole, P. Intranasal chitosan-g-HPbetaCD nanoparticles of efavirenz for the CNS targeting. Artif. Cells Nanomed. Biotechnol. 2018, 46, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Nunes, R.; Araujo, F.; Barreiros, L.; Bartolo, I.; Segundo, M.A.; Taveira, N.; Sarmento, B.; das Neves, J. Noncovalent PEG Coating of Nanoparticle Drug Carriers Improves the Local Pharmacokinetics of Rectal Anti-HIV Microbicides. ACS Appl. Mater. Interfaces 2018, 10, 34942–34953. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.; Araújo, F.; Gomes, M.J.; Fernandes, C.; Nunes, R.; Li, W.; Santos, H.A.; Borges, F.; Sarmento, B. Using microfluidic platforms to develop CNS-targeted polymeric nanoparticles for HIV therapy. Eur. J. Pharm. Biopharm. 2018, 138, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Lakshmi, Y.S.; Kondapi, A.K. An oral formulation of efavirenz-loaded lactoferrin nanoparticles with improved biodistribution and pharmacokinetic profile. HIV Med. 2017, 18, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, Y.S.; Kumar, P.; Kishore, G.; Bhaskar, C.; Kondapi, A.K. Triple combination MPT vaginal microbicide using curcumin and efavirenz loaded lactoferrin nanoparticles. Sci. Rep. 2016, 6, 25479. [Google Scholar] [CrossRef]
- Dutta, T.; Agashe, H.B.; Garg, M.; Balakrishnan, P.; Kabra, M.; Jain, N.K. Poly (propyleneimine) dendrimer based nanocontainers for targeting of efavirenz to human monocytes/macrophages in vitro. J. Drug Target. 2007, 15, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Dutta, T.; Garg, M.; Jain, N.K. Targeting of efavirenz loaded tuftsin conjugated poly(propyleneimine) dendrimers to HIV infected macrophages in vitro. Eur. J. Pharm. Sci. 2008, 34, 181–189. [Google Scholar] [CrossRef]
- Hong, X.; Long, L.; Guohong, F.; Xiangfeng, C. DFT study of nanotubes as the drug delivery vehicles of Efavirenz. Comput. Theor. Chem. 2018, 1131, 57–68. [Google Scholar]
- Suvarna, V.; Thorat, S.; Nayak, U.; Sherje, A.; Murahari, M. Host-guest interaction study of Efavirenz with hydroxypropyl-β-cyclodextrin and L-arginine by computational simulation studies: Preparation and characterization of supramolecular complexes. J. Mol. Liq. 2018, 259, 55–64. [Google Scholar] [CrossRef]
- Moura Ramos, J.J.; Piedade, M.F.M.; Diogo, H.P.; Viciosa, M.T. Thermal Behavior and Slow Relaxation Dynamics in Amorphous Efavirenz: A Study by DSC, XRPD, TSDC, and DRS. J. Pharm. Sci. 2019, 108, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- das Neves, J.; Sarmento, B.; Amiji, M.M.; Bahia, M.F. Development and validation of a rapid reversed-phase HPLC method for the determination of the non-nucleoside reverse transcriptase inhibitor dapivirine from polymeric nanoparticles. J. Pharm. Biomed. Anal. 2010, 52, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Akil, A.; Devlin, B.; Cost, M.; Rohan, L.C. Increased Dapivirine tissue accumulation through vaginal film codelivery of dapivirine and Tenofovir. Mol. Pharm. 2014, 11, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- das Neves, J.; Araújo, F.; Andrade, F.; Michiels, J.; Ariën, K.K.; Vanham, G.; Amiji, M.; Bahia, M.F.; Sarmento, B. In Vitro and Ex Vivo Evaluation of Polymeric Nanoparticles for Vaginal and Rectal Delivery of the Anti-HIV Drug Dapivirine. Mol. Pharm. 2013, 10, 2793–2807. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Cao, S.; Bright, D.K.; Bever, A.M.; Blakney, A.K.; Suydam, I.T.; Woodrow, K.A. Nanoparticle-Based ARV Drug Combinations for Synergistic Inhibition of Cell-Free and Cell-Cell HIV Transmission. Mol. Pharm. 2015, 12, 4363–4374. [Google Scholar] [CrossRef] [PubMed]
- Krogstad, E.A.; Ramanathan, R.; Nhan, C.; Kraft, J.C.; Blakney, A.K.; Cao, S.; Ho, R.J.Y.; Woodrow, K.A. Nanoparticle-releasing nanofiber composites for enhanced in vivo vaginal retention. Biomaterials 2017, 144, 1–16. [Google Scholar] [CrossRef]
- Goebel, F.; Yakovlev, A.; Pozniak, A.; Vinogradova, E.; Boogaerts, G.; Hoetelmans, R.; de Béthune, M.P.; Peeters, M.; Woodfall, B. Short-term antiviral activity of TMC278 – a novel NNRTI – in treatment-naive HIV-1-infected subjects. Aids 2006, 20, 1721–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, A.; McGowan, I. Long-acting rilpivirine for HIV prevention. Curr. Opin. HIV AIDS 2015, 10, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Viciana, P. Rilpivirine: The Key for Long-term Success. AIDS Rev. 2017, 19, 156–166. [Google Scholar] [CrossRef]
- Spreen, W.R.; Margolis, D.A.; Pottage, J.C. Long-acting injectable antiretrovirals for HIV treatment and prevention. Curr. Opin. HIV AIDS 2013, 8, 565–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolis, D.A.; Boffito, M. Long-acting antiviral agents for HIV treatment. Curr. Opin. HIV AIDS 2015, 10, 246–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferretti, F.; Boffito, M. Rilpivirine long-acting for the prevention and treatment of HIV infection. Curr. Opin. HIV AIDS 2018, 13, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Kovarova, M.; Council, O.D.; Date, A.A.; Long, J.M.; Nochi, T.; Belshan, M.; Shibata, A.; Vincent, H.; Baker, C.E.; Thayer, W.O.; et al. Nanoformulations of Rilpivirine for Topical Pericoital and Systemic Coitus-Independent Administration Efficiently Prevent HIV Transmission. PLoS Pathog. 2015, 11, e1005075. [Google Scholar]
- Ottemann, B.M.; Helmink, A.J.; Zhang, W.; Mukadam, I.; Woldstad, C.; Hilaire, J.R.; Liu, Y.; McMillan, J.M.; Edagwa, B.J.; Mosley, R.L.; et al. Bioimaging predictors of rilpivirine biodistribution and antiretroviral activities. Biomaterials 2018, 185, 174–193. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Class | Name (Acronym) | Year* | LS | OB (%) | t/2 (hours) | Side Effects |
|---|---|---|---|---|---|---|
| NRTIs | Stavudine (STV) | 1996 | low | 86 | 1.3–1.4 | Peripheral neuropathy, pancreatitis, asymptomatic acidosis, lipoatrophy, hepatic steatosis |
| Zidovudine (AZT) | 1986 | low | 60 | 0.5–3 | Neutropenia, anemia, nausea, vomiting, asthenia, headache, insomnia, skin hyperpigmentation, acidosis, hepatic steatosis | |
| Lamivudine (3TC) | 1995 | low | 86 | 5–7 | Cough, diarrhea, fatigue, headache, malaise, nasal symptoms, lactic acidosis, hepatic steatosis | |
| Abacavir (ABV) | 1998 | low | 83 | 0.8–1.5 | Systemic respiratory hypersensitivity, gastrointestinal symptoms, fever, tiredness, sore throat | |
| Emtricitabine (FTC) | 2006 | low | 93 | 8–10 | Headache, nausea, upset stomach, diarrhea, trouble sleeping, dizziness, skin rash, strange dreams, cough, runny nose | |
| Zalcitabine (ZCT) | 1992 | low | 85 | Peripheral neuropathy, stomatitis, esophageal ulcerations, acidosis, hepatic steatosis | ||
| Didanosine (DDN) | 1991 | low | 30 | 2 | Gastrointestinal intolerance, peripheral neuropathy, pancreatitis, asymptomatic acidosis, lipoatrophy, hepatic steatosis | |
| Tenofovir (TDF) | 2001 | low | 25-39 | 12–15 | Nausea, depression, confusion, headache, hitching, weakness, kidneys problems | |
| NNRTIs | Nevirapine (NVR) | 1996 | moderate | 92 | 25–30 | Rash, Stevens-Johnson syndrome, elevated transaminases blood level, hepatitis, severe hypersensitivity reaction |
| Efavirenz (EFV) | 1998 | high | 50 | 40–55 | Rash, Stevens-Johnson syndrome, sleep disturbances, dizziness, vertigo, depression, euphoria, difficulty concentrating, hallucination. | |
| Etravirine (ETV) | 2008 | high | -- | 30–40 | Rash, Stevens-Johnson syndrome, toxic epidermal necrosis and multiform erythema, hypersensitivity reactions, hepatic failure | |
| Rilpivirine (RPV) | 2011 | high | 50 | 19 | Rash, depression, liver problems, mood changes |
| DDS | TARGET | |
|---|---|---|
| Matrix | Surface | |
| Liposome NPs | Mannose | Liver, spleen, lung, brain, macrophages |
| Liposomes | Galactose | Liver |
| Chitosan NPs | Glycyrrhizin | Liver |
| NPs | Transferrin | Brain, endothelial cells |
| NPs | Serum albumin | Brain, liver, spleen |
| SLN | Phenylalanine | Blood brain barrier |
| Polymeric micelles | Anti-GP2 antibody | M-cell of gut-associated lymphoid tissue |
| Dendrimers | Tuftsin | Macrophages, monocytes, polymorph nuclear leukocytes |
| DDS | Advantages | Limitations |
|---|---|---|
| Liposomes | Co-delivery of hydrophilic and lipophilic drug Selective uptake by mononuclear phagocytes Surface modification with target moiety of virus reservoir | Low drug loading capacity Physical and chemical instability Drug leakage Difficulty in sterilization Short half-life Poor scale up |
| Niosomes | Chemical stability Protection of drug from degradation Large uptake by mononuclear phagocytes and localization in virus reservoir organs Less expensive respect liposomes Functionalization with target ligand | Physical instability during the storage Difficulty in sterilization Difficulty in large-scale production |
| Polymeric NPs | High drug loading capacity Co-delivery of different drug for anti-HIV combination therapy Selective uptake by lymphoid organ Prolonged circulation time Surface functionalization with target moiety | Fast burst release Limited safe correlated to polymer toxicity High cost production |
| SLN | Higher stability and biological compatibility than liposomes and polymeric NPs Increase the bioavailability of poorly water soluble drug Avoidance of organic solvent Slow uptake by the RES Feasible-large scale production and sterilization Less expensive than polymeric and surfactant carriers | Low drug solubility in lipid matrix and loading capacity Drug leakage Particle growth Unpredictable gelation tendency |
| Dendrimers | Uniform particle size Large surface functional groups for the conjugation with target moieties | Toxicity problems |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grande, F.; Ioele, G.; Occhiuzzi, M.A.; De Luca, M.; Mazzotta, E.; Ragno, G.; Garofalo, A.; Muzzalupo, R. Reverse Transcriptase Inhibitors Nanosystems Designed for Drug Stability and Controlled Delivery. Pharmaceutics 2019, 11, 197. https://doi.org/10.3390/pharmaceutics11050197
Grande F, Ioele G, Occhiuzzi MA, De Luca M, Mazzotta E, Ragno G, Garofalo A, Muzzalupo R. Reverse Transcriptase Inhibitors Nanosystems Designed for Drug Stability and Controlled Delivery. Pharmaceutics. 2019; 11(5):197. https://doi.org/10.3390/pharmaceutics11050197
Chicago/Turabian StyleGrande, Fedora, Giuseppina Ioele, Maria Antonietta Occhiuzzi, Michele De Luca, Elisabetta Mazzotta, Gaetano Ragno, Antonio Garofalo, and Rita Muzzalupo. 2019. "Reverse Transcriptase Inhibitors Nanosystems Designed for Drug Stability and Controlled Delivery" Pharmaceutics 11, no. 5: 197. https://doi.org/10.3390/pharmaceutics11050197
APA StyleGrande, F., Ioele, G., Occhiuzzi, M. A., De Luca, M., Mazzotta, E., Ragno, G., Garofalo, A., & Muzzalupo, R. (2019). Reverse Transcriptase Inhibitors Nanosystems Designed for Drug Stability and Controlled Delivery. Pharmaceutics, 11(5), 197. https://doi.org/10.3390/pharmaceutics11050197