Eradication of Human Immunodeficiency Virus Type-1 (HIV-1)-Infected Cells



{kind=link}
{kind=link}
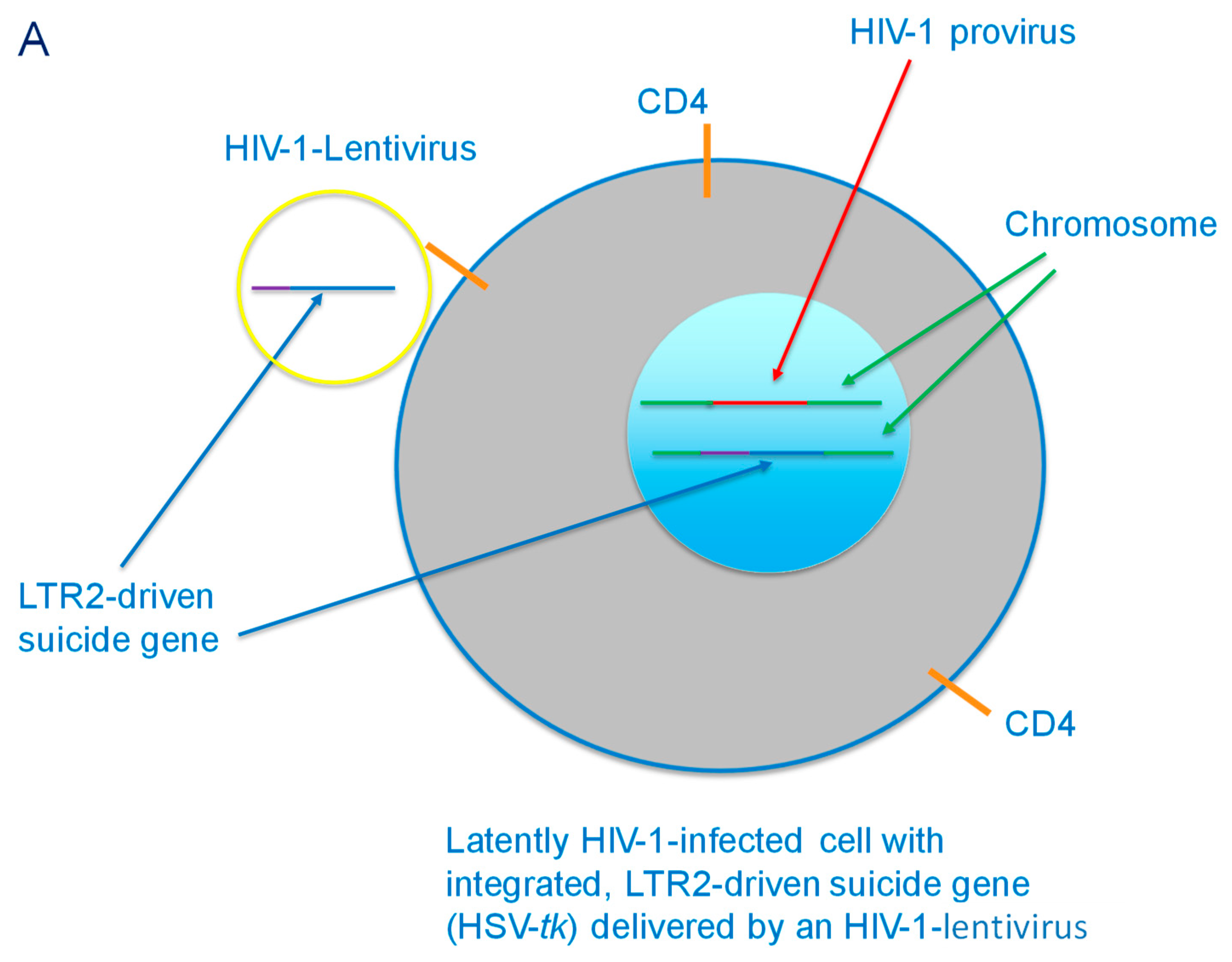{kind=link}
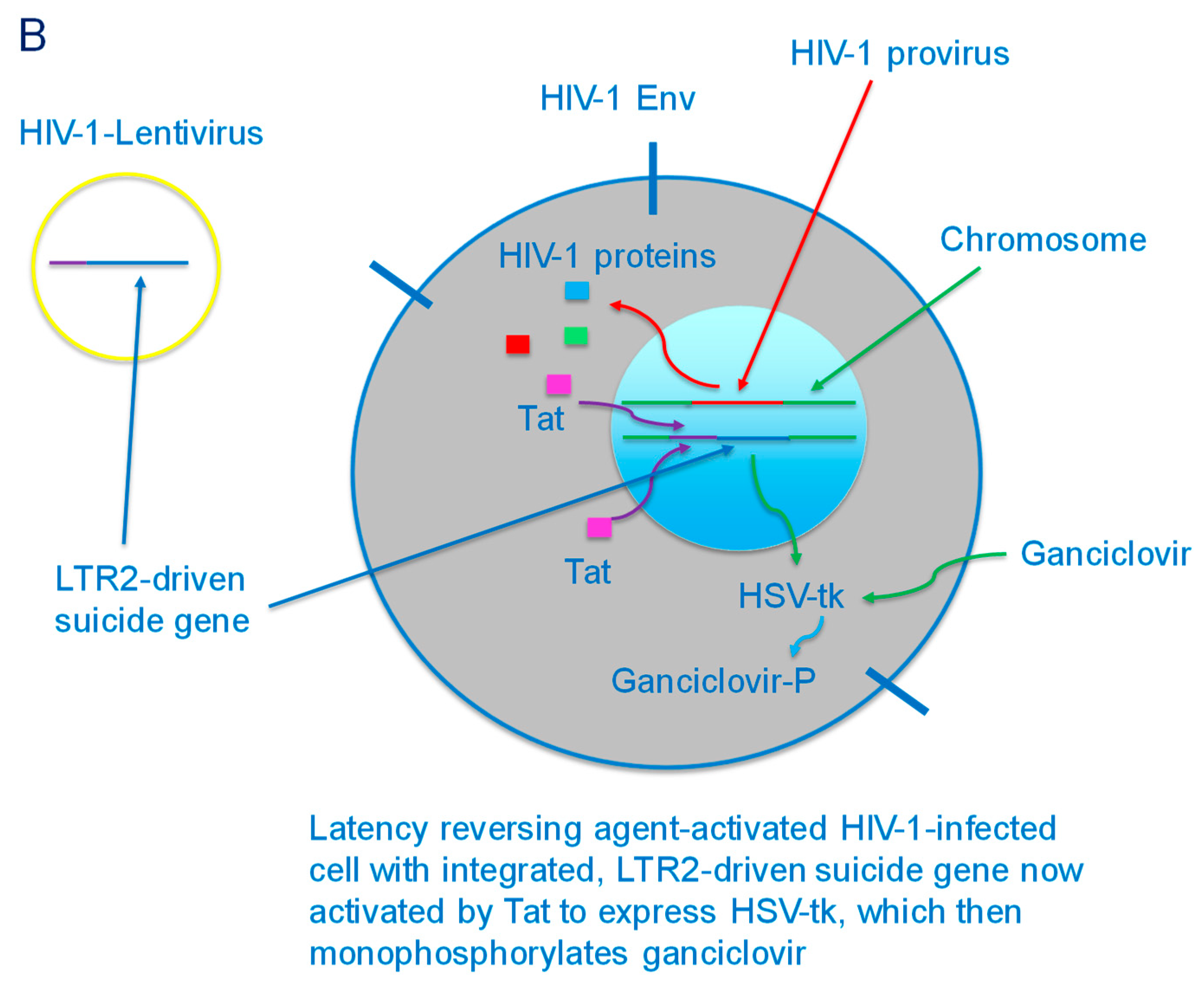{kind=link}
{kind=link}
Abstract
:1. Antiretroviral Therapy
2. HIV-1 Latency
3. Latency Reversal
4. Suicide Gene Therapy
5. Excision of Chromosome-Integrated HIV-1 DNA
6. Cytotoxic Liposomes Targeted to HIV-1-Infected Cells
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sundquist, W.I.; Kräusslich, H.-G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef] [PubMed]
- Baumgärtel, V.; Müller, B.; Lamb, D.C. Quantitative live-cell imaging of human immunodeficiency virus (HIV-1) assembly. Viruses 2012, 4, 777–799. [Google Scholar] [CrossRef]
- Perelson, A.S.; Essunger, P.; Cao, Y.; Vesanen, M.; Hurley, A.; Saksela, K.; Markowitz, M.; Ho, D.D. Decay characteristics of HIV-1-infected compartments during combination therapy. Nat. Cell Boil. 1997, 387, 188–191. [Google Scholar] [CrossRef]
- Kulkosky, J.; Sullivan, J.; Xu, Y.; Souder, E.; Hamer, D.H.; Pomerantz, R.J. Expression of latent HAART-persistent HIV Type 1 induced by novel cellular activating agents. AIDS Hum. Retrovir. 2004, 20, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Shehu-Xhilaga, M.; Tachedjian, G.; Crowe, S.; Kedzierska, K. Antiretroviral compounds: Mechanisms underlying failure of HAART to eradicate HIV-1. Med. Chem. 2005, 12, 1705–1719. [Google Scholar] [CrossRef]
- Chun, T.W.; Engel, D.; Berrey, M.M.; Shea, T.; Corey, L.; Fauci, A.S. Early establishment of a pool of latently infected, resting CD4+ T cells during primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 1998, 95, 8869–8873. [Google Scholar] [CrossRef]
- Blankson, J.N.; Persaud, D.; Siliciano, R.F. The challenge of viral reservoirs in HIV-1 infection. Annu. Med. 2002, 53, 557–593. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Siliciano, R.F. A long-term latent reservoir for HIV-1: Discovery and clinical implications. J. Antimicrob. Chemother. 2004, 54, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Richman, D.D.; Margolis, D.M.; Delaney, M.; Greene, W.C.; Hazuda, D.; Pomerantz, R.J. The challenge of finding a cure for HIV infection. Science 2009, 323, 1304–1307. [Google Scholar] [CrossRef]
- Murray, A.J.; Kwon, K.J.; Farber, D.L.; Siliciano, R.F. The latent reservoir for HIV-1: How immunologic memory and clonal expansion contribute to HIV-1 persistence. J. Immunol. 2016, 197, 407–417. [Google Scholar] [CrossRef]
- Siliciano, R.F.; Greene, W.C. HIV latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef]
- Siliciano, R.F. Scientific rationale for antiretroviral therapy in 2005: Viral reservoirs and resistance evolution. Top. HIV Med. Publ. Int. AIDS Soc. USA 2005, 13, 96–100. [Google Scholar]
- Ruelas, D.S.; Greene, W.C. An integrated overview of HIV-1 latency. Cell 2013, 155, 519–529. [Google Scholar] [CrossRef]
- Lehrman, G.; Ylisastigui, L.; Bosch, R.J.; Margolis, D.M. Interleukin-7 induces HIV type 1 outgrowth from peripheral resting CD4+ T cells. JAIDS J. Acquir. Immune Defic. Syndr. 2004, 36, 1103–1104. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Cheema, M.; Parker, D.; Wiegand, A.; Bosch, R.J.; Coffin, J.M.; Eron, J.; Cohen, M.; Margolis, D.M. Antiretroviral intensification and valproic acid lack sustained effect on residual HIV-1 viremia or resting CD4+ cell infection. PLoS ONE 2010, 5, e9390. [Google Scholar] [CrossRef] [PubMed]
- Margolis, D.M.; Garcia, J.V.; Hazuda, D.J.; Haynes, B.F. Latency reversal and viral clearance to cure HIV-1. Science 2016, 353, aaf6517. [Google Scholar] [CrossRef]
- Sengupta, S.; Siliciano, R.F. Targeting the latent reservoir for HIV-1. Immunity 2018, 48, 872–895. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Bateson, R.; Tripathy, M.K.; Crooks, A.M.; Yang, K.H.; Dahl, N.P.; Kearney, M.F.; Anderson, E.M.; Coffin, J.M.; Strain, M.C.; et al. HIV-1 expression within resting CD4+ T cells after multiple doses of vorinostat. J. Infect. Dis. 2014, 210, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Martinez, J.P.; Zorita, E.; Meyerhans, A.; Filion, G.J. Position effects influence HIV latency reversal. Nat. Struct. Mol. Biol. 2017, 24, 47–54. [Google Scholar] [CrossRef]
- Lehrman, G.; Hogue, I.B.; Palmer, S.; Jennings, C.; A Spina, C.; Wiegand, A.; Landay, A.L.; Coombs, R.W.; Richman, D.D.; Mellors, J.W.; et al. Depletion of latent HIV-1 infection in vivo: A proof-of-concept study. Lancet 2005, 366, 549–555. [Google Scholar] [CrossRef]
- Hamer, D.H.; Bocklandt, S.; McHugh, L.; Chun, T.-W.; Blumberg, P.M.; Sigano, D.M.; Marquez, V.E. Rational design of drugs that induce Human Immunodeficiency Virus replication. J. Virol. 2003, 77, 10227–10236. [Google Scholar] [CrossRef] [PubMed]
- Bocklandt, S.; Blumberg, P.M.; Hamer, D.H. Activation of latent HIV-1 expression by the potent anti-tumor promoter 12-deoxyphorbol 13-phenylacetate. Antivir. Res. 2003, 59, 89–98. [Google Scholar] [CrossRef]
- Demonte, D.; Quivy, V.; Colette, Y.; Van Lint, C. Administration of HDAC inhibitors to reactivate HIV-1 expression in latent cellular reservoirs: Implications for the development of therapeutic strategies. Biochem. Pharmacol. 2004, 68, 1231–1238. [Google Scholar] [CrossRef]
- Macedo, A.B.; Novis, C.L.; De Assis, C.M.; Sorensen, E.S.; Moszczynski, P.; Huang, S.-H.; Ren, Y.; Spivak, A.M.; Jones, R.B.; Planelles, V.; et al. Dual TLR2 and TLR7 agonists as HIV latency-reversing agents. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Marsden, M.D.; Wu, X.; Navab, S.M.; Loy, B.A.; Schrier, A.J.; DeChristopher, B.A.; Shimizu, A.J.; Hardman, C.T.; Ho, S.; Ramirez, C.M.; et al. Characterization of designed, synthetically accessible bryostatin analog HIV latency reversing agents. Virology 2018, 520, 83–93. [Google Scholar] [CrossRef]
- Spina, C.A.; Anderson, J.; Archin, N.M.; Bosque, A.; Chan, J.; Famiglietti, M.; Greene, W.C.; Kashuba, A.; Lewin, S.R.; Margolis, D.M.; et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. 2013, 9, e1003834. [Google Scholar] [CrossRef] [PubMed]
- Beliakova-Bethell, N.; Hezareh, M.; Wong, J.K.; Strain, M.C.; Lewinski, M.K.; Richman, D.D.; Spina, C.A. Relative efficacy of T cell stimuli as inducers of productive HIV-1 replication in latently infected CD4 lymphocytes from patients on suppressive cART. Virology 2017, 508, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Richard, K.; Williams, D.E.; De Silva, E.D.; Brockman, M.A.; Brumme, Z.L.; Andersen, R.J.; Tietjen, I. Identification of novel HIV-1 latency-reversing agents from a Library of Marine Natural Products. Viruses 2018, 10, 348. [Google Scholar] [CrossRef] [PubMed]
- Ukah, O.B.; Puray-Chavez, M.; Tedbury, P.R.; Herschhorn, A.; Sodroski, J.G.; Sarafianos, S.G. Visualization of HIV-1 RNA Transcription from Integrated HIV-1 DNA in Reactivated Latently Infected Cells. Viruses 2018, 10, 534. [Google Scholar] [CrossRef]
- Bialek, J.K.; Dunay, G.A.; Voges, M.; Schäfer, C.; Spohn, M.; Stucka, R.; Hauber, J.; Lange, U.C. Targeted HIV-1 latency reversal using CRISPR/Cas9-derived transcriptional activator systems. PLoS ONE 2016, 11, e0158294. [Google Scholar] [CrossRef]
- Saayman, S.M.; Lazar, D.C.; Scott, T.A.; Hart, J.R.; Takahashi, M.; Burnett, J.C.; Planelles, V.; Morris, K.V.; Weinberg, M.S. Potent and targeted activation of latent HIV-1 using the CRISPR/dCas9 activator complex. Mol. Ther. 2016, 24, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Walker-Sperling, V.E.; Pohlmeyer, C.W.; Tarwater, P.M.; Blankson, J.N. The effect of latency reversal agents on primary CD8+ T cells: Implications for shock and kill strategies for human immunodeficiency virus eradication. EBioMedicine 2016, 8, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Clutton, G.T.; Jones, R.B. Diverse impacts of HIV latency-reversing agents on CD8+ T-cell function: Implications for HIV cure. Front. Immunol. 2018, 9, 1452. [Google Scholar] [CrossRef]
- Huang, S.H.; Ren, Y.; Thomas, A.S.; Chan, D.; Mueller, S.; Ward, A.R.; Patel, S.; Bollard, C.M.; Cruz, C.R.; Karandish, S.; et al. Latent HIV reservoirs exhibit inherent resistance to elimination by CD8+ T cells. J. Clin. Investig. 2018, 128, 876–889. [Google Scholar] [CrossRef] [PubMed]
- Harrison, G.S.; Maxwell, F.; Long, C.J.; Rosen, C.A.; Glode, L.M.; Maxwell, I.H. Activation of a Diphtheria Toxin A gene by expression of human immunodeficiency virus-1 Tat and Rev proteins in transfected cells. Hum. Gene Ther. 1991, 2, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Harrison, G.S.; Long, C.J.; Curiel, T.J.; Maxwell, F.; Maxwell, I.H. Inhibition of Human Immunodeficiency Virus-1 production resulting from transduction with a retrovirus containing an HIV-regulated diphtheria toxin A chain gene. Hum. Gene Ther. 1992, 3, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Cook, D.R.; Wang, Y.; Hahn, B.H.; Ghosh, S.K.; Harrison, G.S. Long-Term inhibition of clinical and laboratory human immunodeficiency virus strains in human T-cell lines containing an HIV-regulated diphtheria toxin A chain gene. Hum. Gene Ther. 1993, 4, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Konopka, K.; Harrison, G.S.; Felgner, P.L.; Düzgüneş, N. Cationic liposome-mediated expression of HIV-regulated luciferase and diphtheria toxin genes in HeLa cells infected with or expressing HIV. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 1997, 1356, 185–197. [Google Scholar] [CrossRef]
- Gebremedhin, S.; Au, A.; Konopka, K.; Milnes, M.; Düzgüneş, N. A gene therapy approach to eliminate HIV-1-infected cells. J. Calif. Dent. Assoc. 2012, 40, 402–406. [Google Scholar]
- Young, M.; Overlid, N.; Konopka, K.; Düzgüneş, N. Gene therapy for oral cancer: Efficient delivery of a ’suicide gene’ to murine oral cancer cells in physiological milieu. J. Calif. Dent. Assoc. 2005, 33, 967–971. [Google Scholar]
- Gebremedhin, S.; Singh, A.; Koons, S.; Bernt, W.; Konopka, K.; Düzgüneş, N. Gene delivery to carcinoma cells via novel non-viral vectors: Nanoparticle tracking analysis and suicide gene therapy. Eur. J. Pharm. Sci. 2014, 60, 72–79. [Google Scholar] [CrossRef]
- Düzgüneş, N.; Cheung, J.; Konopka, K. Non-viral suicide gene therapy in cervical, oral and pharyngeal carcinoma cells with CMV- and EEV-plasmids. J. Gene Med. 2018, 20, e3054. [Google Scholar] [CrossRef] [PubMed]
- Düzgüneş, N.; Cheung, J.; Konopka, K. Suicide gene therapy of oral squamous cell carcinoma and cervical carcinoma in vitro. Methods Mol. Biol. 2019, 1895, 177–184. [Google Scholar]
- Neves, S.; Faneca, H.; Bertin, S.; Konopka, K.; Düzgüneş, N.; Pierrefite-Carle, V.; Simoes, S.; De Lima, M.P. Transferrin lipoplex-mediated suicide gene therapy of oral squamous cell carcinoma in an immunocompetent murine model and mechanisms involved in the antitumoral response. Cancer Gene Ther. 2009, 16, 91–101. [Google Scholar] [CrossRef]
- Faneca, H.; Düzgüneş, N.; Pedroso de Lima, M.C. Suicide gene therapy for oral squamous cell carcinoma. Suicide Gene Ther. 2019, 1895, 43–55. [Google Scholar]
- Garg, H.; Joshi, A. Conditional cytotoxic anti-HIV gene therapy for selectable cell modification. Hum. Gene Ther. 2016, 27, 400–415. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Esvelt, K.M.; Church, G.M. Cas9 as a versatile tool for engineering biology. Nat. Methods 2013, 10, 957–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3, 2510. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Pan, Q.; Gendron, P.; Zhu, W.; Guo, F.; Cen, S.; Wainberg, M.A.; Liang, C. CRISPR/Cas9-Derived mutations both inhibit HIV-1 replication and accelerate viral escape. Cell Rep. 2016, 15, 481–489. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas based antiviral strategies against HIV-1. Virus Res. 2018, 244, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Ebina, H.; Kanemura, Y.; Misawa, N.; Sakuma, T.; Kobayashi, T.; Yamamoto, T.; Koyanagi, Y. A high excision potential of TALENs for integrated DNA of HIV-based lentiviral vector. PLoS ONE 2015, 10, e0120047. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Kaminski, R.; Yang, F.; Zhang, Y.; Cosentino, L.; Li, F.; Luo, B.; Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Karn, J.; et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 11461–11466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminski, R.; Chen, Y.; Fischer, T.; Tedaldi, E.; Napoli, A.; Zhang, Y.; Karn, J.; Hu, W.; Khalili, K. Elimination of HIV-1 genomes from human T-lymphoid cells by CRISPR/Cas9 gene editing. Sci. Rep. 2016, 6, 22555. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, R.; Bella, R.; Yin, C.; Otte, J.; Ferrante, P.; Gendelman, H.E.; Li, H.; Booze, R.; Gordon, J.; Hu, W.; et al. Excision of HIV-1 DNA by gene editing: A proof-of-concept in vivo study. Gene Ther. 2016, 23, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.M.; Phogat, S.K.; Chan-Hui, P.-Y.; Wagner, D.; Phung, P.; Goss, J.L.; Wrin, T.; Simek, M.D.; Fling, S.; Mitcham, J.L.; et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 2009, 326, 285–289. [Google Scholar] [CrossRef]
- Yee, M.; Konopka, K.; Balzarini, J.; Düzgüneş, N. Inhibition of HIV-1 Env-mediated cell-cell fusion by lectins, peptide T-20, and neutralizing antibodies. Open Virol. J. 2011, 5, 44–51. [Google Scholar] [CrossRef]
- Stephenson, K.E.; Barouch, D.H. Broadly neutralizing antibodies for HIV eradication. HIV/AIDS Rep. 2016, 13, 31–37. [Google Scholar] [CrossRef]
- Flasher, D.; Konopka, K.; Chamow, S.M.; Dazin, P.; Ashkenazi, A.; Pretzer, E.; Düzgüneş, N. Liposome targeting to human immunodeficiency virus type 1-infected cells via recombinant soluble CD4 and CD4 immunoadhesin (CD4-IgG). Biochim. et Biophys. Acta (BBA) Biomembr. 1994, 1194, 185–196. [Google Scholar] [CrossRef]
- Slepushkin, V.A.; Salem, I.I.; Andreev, S.M.; Dazin, P.; Düzgüneş, N. Targeting of liposomes to HIV-1-infected cells by peptides derived from the CD4 receptor. Biochem. Biophys. Commun. 1996, 227, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kirpotin, D.; Hong, K.; Shalaby, R.; Shao, Y.; Nielsen, U.; Marks, J.; Papahadjopoulos, D.; Benz, C. Tumor targeting using anti-her2 immunoliposomes. J. Control. Release 2001, 74, 95–113. [Google Scholar] [CrossRef]
- Eliaz, R.E.; Nir, S.; Marty, C.; Szoka, F.C., Jr. Determination and modeling of kinetics of cancer cell killing by doxorubicin and doxorubicin encapsulated in targeted liposomes. Cancer Res. 2004, 64, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Hu, S.; Chang, Y.; Zhang, Z.; Zha, Z.; Huang, H.; Shen, G.; Liu, J.; Song, L.; Wei, W.; et al. Development and characterization of a humanized anti-HER2 antibody HuA21 with potent anti-tumor properties in breast cancer cells. Int. J. Mol. Sci. 2016, 17, 563. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.A.; Patil, Y.; La-Beck, N.M. New insights and evolving role of pegylated liposomal doxorubicin in cancer therapy. Drug Resist. Updat. 2016, 29, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Oussoren, C.; Storm, G. Liposomes to target the lymphatics by subcutaneous administration. Adv. Drug Deliv. Rev. 2001, 50, 143–156. [Google Scholar] [CrossRef]
- Allen, T.M.; Hansen, C.B.; Guo, L.S.S. Subcutaneous administration of liposomes: A comparison with the intravenous and intraperitoneal routes of injection. Biochim. Biophys. Acta (BBA) Biomembr. 1993, 1150, 9–16. [Google Scholar] [CrossRef]
- Pantaleo, G.; Graziosi, C.; Butini, L.; Pizzo, P.A.; Schnittman, S.M.; Kotler, D.P.; Fauci, A.S. Lymphoid organs function as major reservoirs for human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 1991, 88, 9838–9842. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, G.; Graziosi, C.; Demarest, J.F.; Butini, L.; Montroni, M.; Fox, C.H.; Orenstein, J.M.; Kotler, D.P.; Fauci, A.S. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nat. Cell Boil. 1993, 362, 355–358. [Google Scholar] [CrossRef]
- Embretson, J.; Zupancic, M.; Ribas, J.L.; Burke, A.; Racz, P.; Tenner-Racz, K.; Haase, A.T. Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nat. Cell Boil. 1993, 362, 359–362. [Google Scholar] [CrossRef]
- Désormeaux, A.; Bergeron, M.G. Lymphoid tissue targeting of anti-HIV drugs using liposomes. Methods Enzymol. 2005, 391, 330–351. [Google Scholar] [PubMed]
- Kinman, L.; Brodie, S.J.; Tsai, C.C.; Bui, T.; Larsen, K.; Schmidt, A.; Anderson, D.; Morton, W.R.; Hu, S.-L.; Ho, R.J.Y. Lipid–drug association enhanced HIV-1 protease inhibitor Indinavir localization in lymphoid tissues and viral load reduction: A proof of concept study in HIV-2287-infected macaques. JAIDS J. Acquir. Immune Defic. Syndr. 2003, 34, 387–397. [Google Scholar] [CrossRef]
- Pretzer, E.; Flasher, D.; Düzgüneş, N. Inhibition of human immunodeficiency virus type-1 replication in macrophages and H9 cells by free or liposome-encapsulated L-689,502, an inhibitor of the viral protease. Antivir. Res. 1997, 34, 1–15. [Google Scholar] [CrossRef]
- Clayton, R.; Öhagen, Å.; Nicol, F.; Del Vecchio, A.M.; Jonckers, T.H.; Goethals, O.; Van Loock, M.; Michiels, L.; Grigsby, J.; Xu, Z.; et al. Sustained and specific in vitro inhibition of HIV-1 replication by a protease inhibitor encapsulated in gp120-targeted liposomes. Antivir. Res. 2009, 84, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Slepushkin, V.A.; Simões, S.; Dazin, P.; Newman, M.S.; Guo, L.S.; de Lima, M.C.P.; Düzgüneş, N. Sterically stabilized pH-sensitive liposomes. Intracellular delivery of aqueous contents and prolonged circulation in vivo. J. Biol. Chem. 1997, 272, 2382–2388. [Google Scholar] [CrossRef] [PubMed]
- Slepushkin, V.; Simões, S.; de Lima, M.C.; Düzgüneş, N. Sterically stabilized pH-sensitive liposomes. Methods Enzymol. 2004, 387, 134–147. [Google Scholar] [PubMed]
- Gabizon, A.; Shmeeda, H.; Grenader, T. Pharmacological basis of pegylated liposomal doxorubicin: Impact on cancer therapy. Eur. J. Pharm. Sci. 2012, 45, 388–398. [Google Scholar] [CrossRef]
- Phillips, W.T.; Medina, L.A.; Klipper, R.; Goins, B. A novel approach for the increased delivery of pharmaceutical agents to peritoneum and associated lymph nodes. J. Pharmacol. Exp. Ther. 2002, 303, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Bestman-Smith, J.; Gourde, P.; Désormeaux, A.; Tremblay, M.J.; Bergeron, M.G. Sterically stabilized liposomes bearing anti-HLA-DR antibodies for targeting the primary cellular reservoirs of HIV-1. Biochim. Biophys. Acta (BBA) Biomembr. 2000, 1468, 161–174. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Scheerer, S.; Geyer, M.A.; Howell, S.B. Direct cerebrospinal fluid delivery of an antiretroviral agent using multivesicular liposomes. J. Infect. Dis. 1990, 162, 750–752. [Google Scholar] [CrossRef]
- Bissel, S.J.; Wiley, C.A. Human immunodeficiency virus infection of the brain: Pitfalls in evaluating infected/affected cell populations. Brain Pathol. 2004, 14, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Hütter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Müßig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kücherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Abdul-Jawad, S.; McCoy, L.E.; Mok, H.P.; Peppa, D.; Salgado, M.; Martinez-Picado, J.; Nijhuis, M.; Wensing, A.M.J.; Lee, H.; et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nat. Cell Boil. 2019, 568, 1. [Google Scholar] [CrossRef] [PubMed]
- Corey, D.; Schultz, P. Generation of a hybrid sequence-specific single-stranded deoxyribonuclease. Science 1987, 238, 1401–1403. [Google Scholar] [CrossRef] [PubMed]
- Pei, D.; Corey, D.R.; Schultz, P.G. Site-specific cleavage of duplex DNA by a semisynthetic nuclease via triple-helix formation. Proc. Natl. Acad. Sci. USA 1990, 87, 9858–9862. [Google Scholar] [CrossRef]
- Guieysse, A.; Praseuth, D.; François, J.; Helene, C. Inhibition of replication initiation by triple helix-forming oligonucleotides. Biochem. Biophys. Commun. 1995, 217, 186–194. [Google Scholar] [CrossRef]
- Faria, M.; Wood, C.; Perrouault, L.; Nelson, J.S.; Winter, A.; White, M.R.H.; Helene, C.; Giovannangeli, C. Targeted inhibition of transcription elongation in cells mediated by triplex-forming oligonucleotides. Proc. Natl. Acad. Sci. USA 2000, 97, 3862–3867. [Google Scholar] [CrossRef] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Düzgüneş, N.; Konopka, K. Eradication of Human Immunodeficiency Virus Type-1 (HIV-1)-Infected Cells. Pharmaceutics 2019, 11, 255. https://doi.org/10.3390/pharmaceutics11060255
Düzgüneş N, Konopka K. Eradication of Human Immunodeficiency Virus Type-1 (HIV-1)-Infected Cells. Pharmaceutics. 2019; 11(6):255. https://doi.org/10.3390/pharmaceutics11060255
Chicago/Turabian StyleDüzgüneş, Nejat, and Krystyna Konopka. 2019. "Eradication of Human Immunodeficiency Virus Type-1 (HIV-1)-Infected Cells" Pharmaceutics 11, no. 6: 255. https://doi.org/10.3390/pharmaceutics11060255
APA StyleDüzgüneş, N., & Konopka, K. (2019). Eradication of Human Immunodeficiency Virus Type-1 (HIV-1)-Infected Cells. Pharmaceutics, 11(6), 255. https://doi.org/10.3390/pharmaceutics11060255