An ImmunoPEGliposome for Targeted Antimalarial Combination Therapy at the Nanoscale

 , and
, and 
Abstract
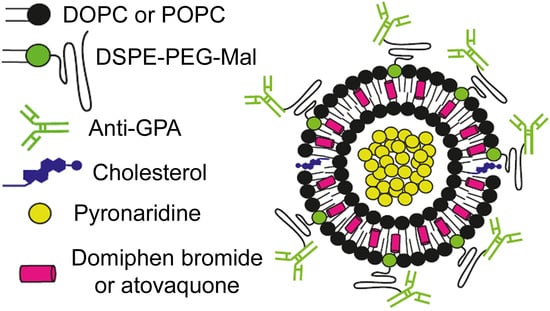
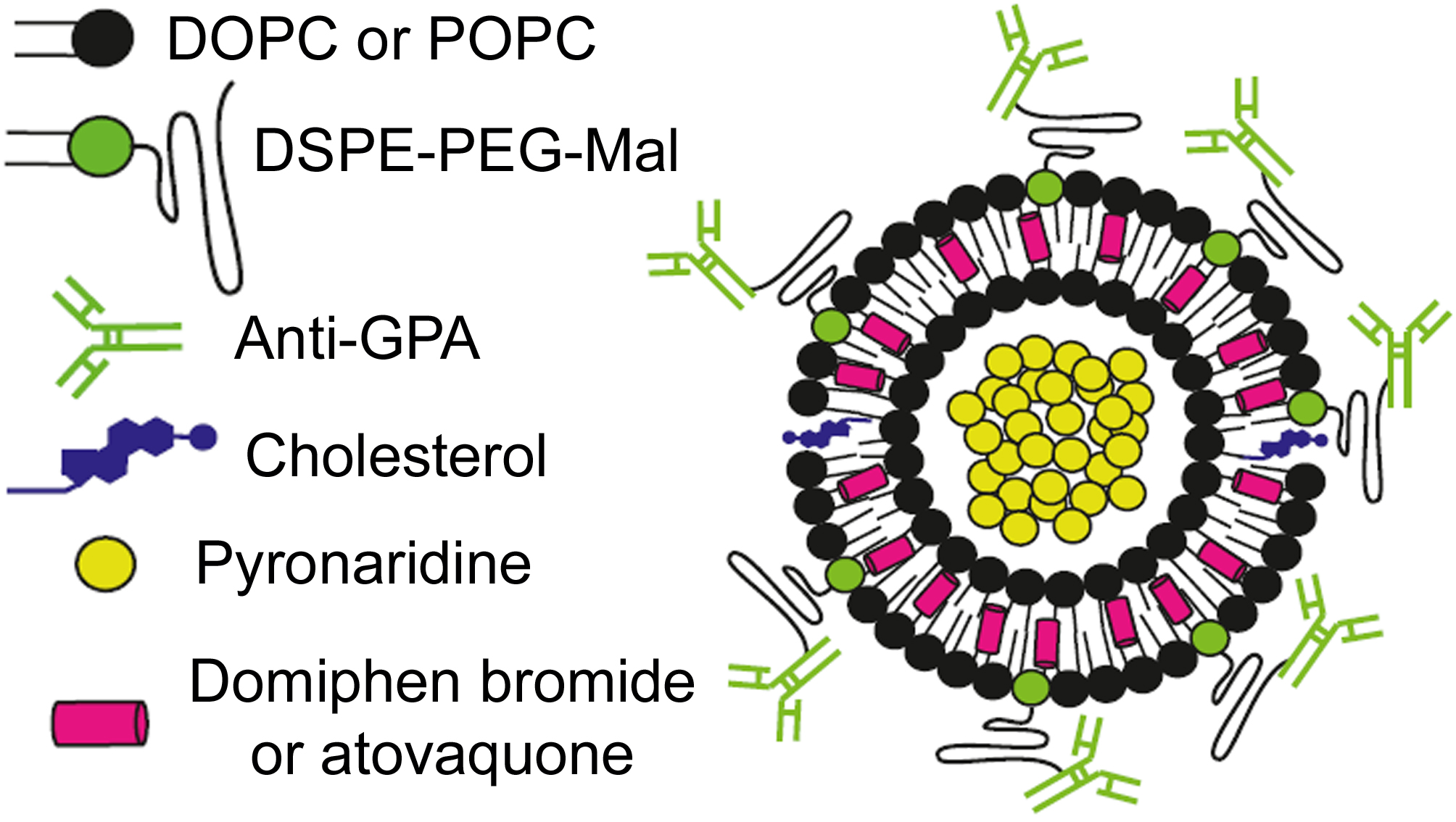
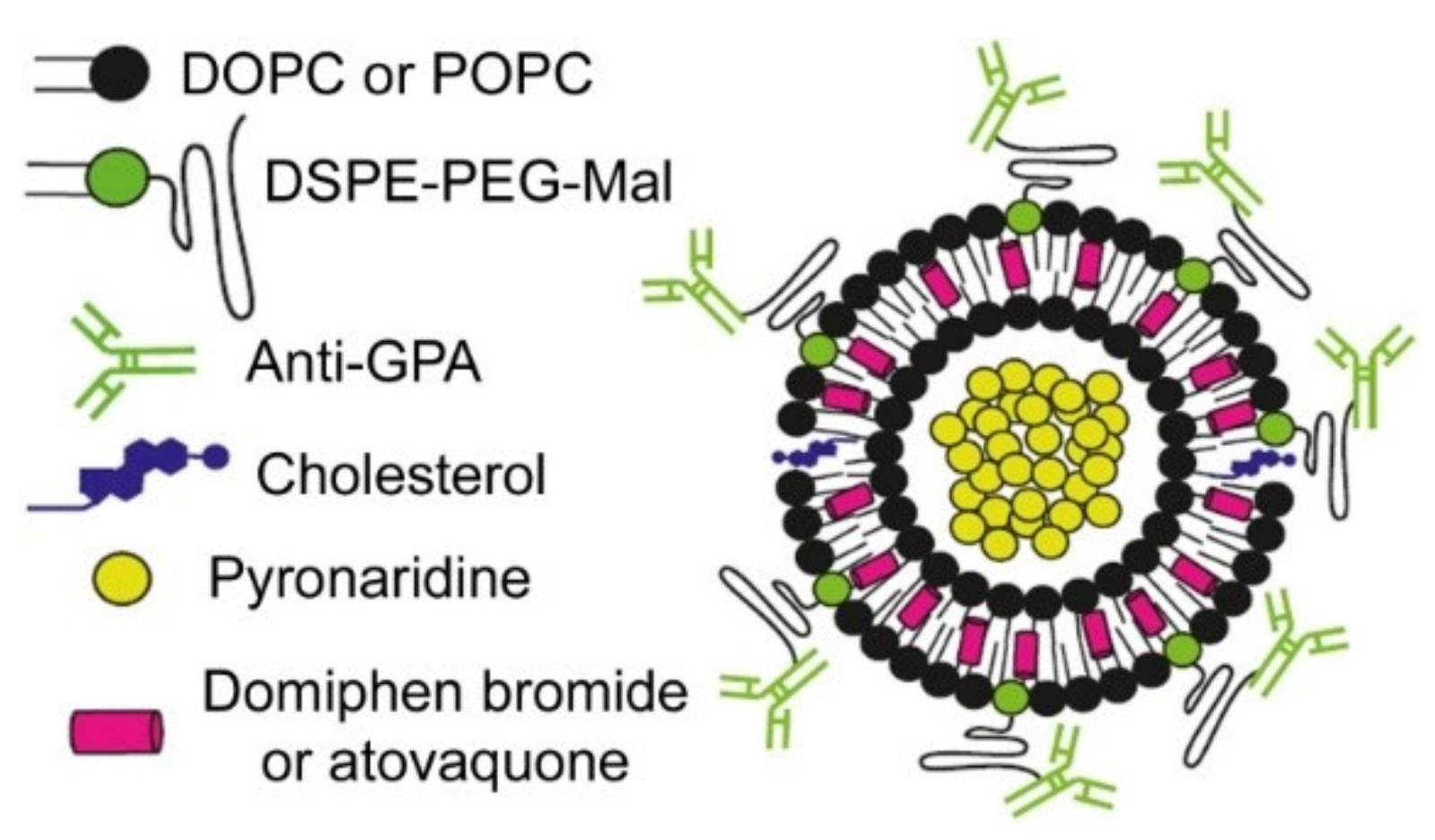
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Generation of Liposomes
2.3. Encapsulation of Drugs in Liposomes
2.4. Generation of Immunoliposomes
2.5. Quantification of Encapsulated Drugs
2.6. P. falciparum Cultures
2.7. Growth Inhibition Assays of P. falciparum Blood Stages
2.8. Cell Binding Assays
2.9. Liposome Stability and Drug Release Assays
2.10. Flow Cytometry
2.11. Fluorescence Microscopy
2.12. Cryogenic Transmission Electron Microscopy (cryoTEM)
3. Results
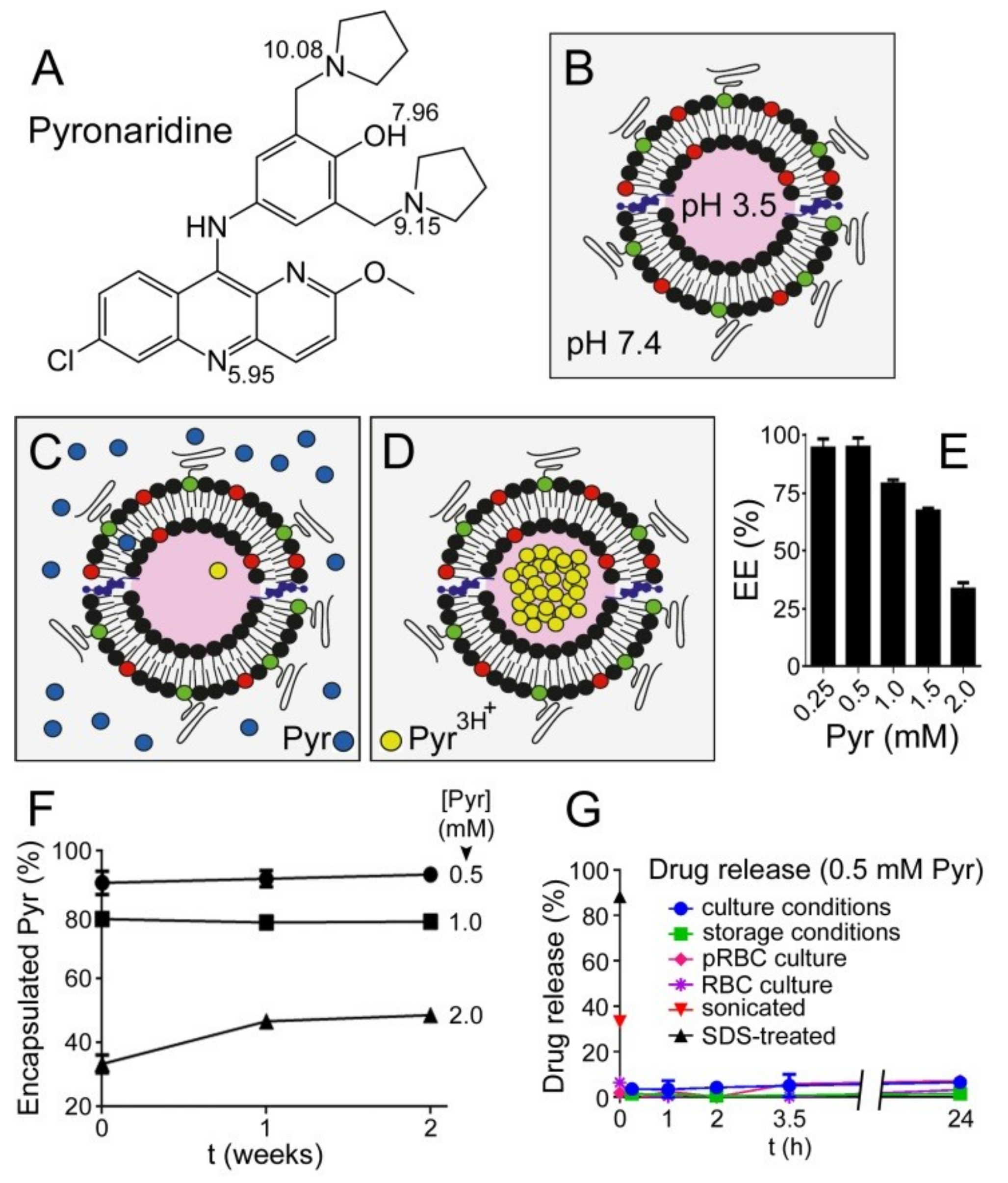
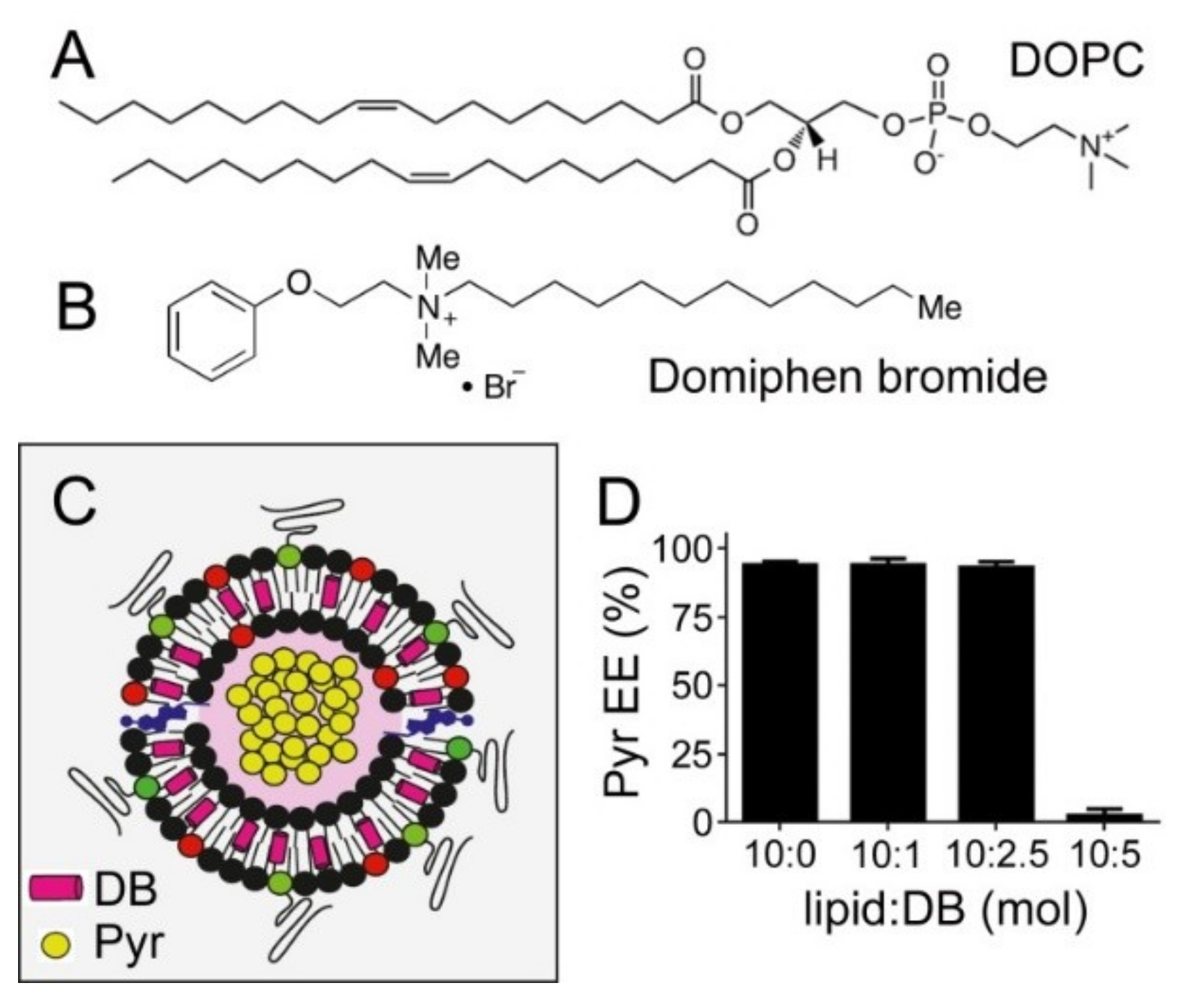
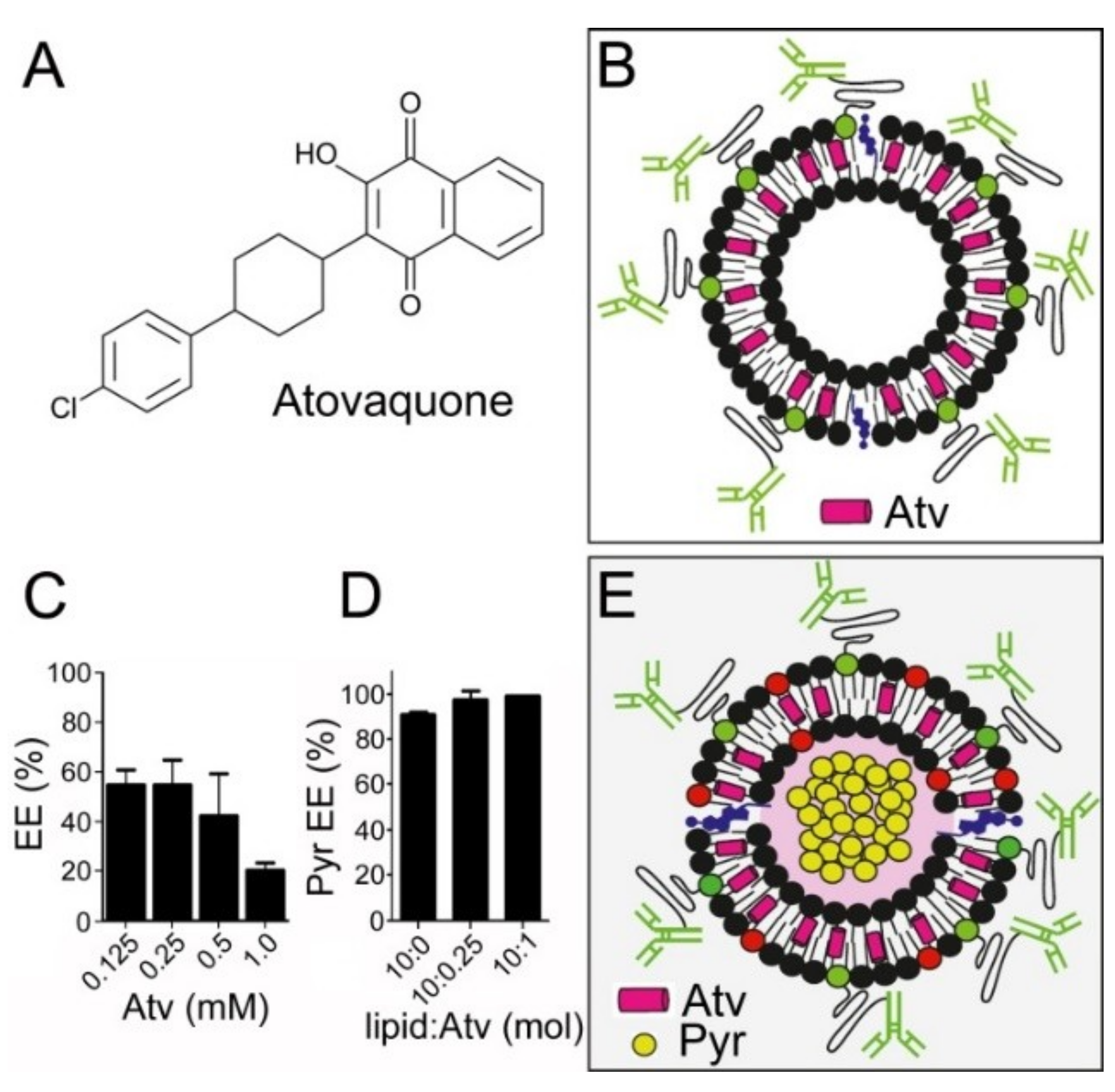
3.1. Liposome Encapsulation of Pyronaridine
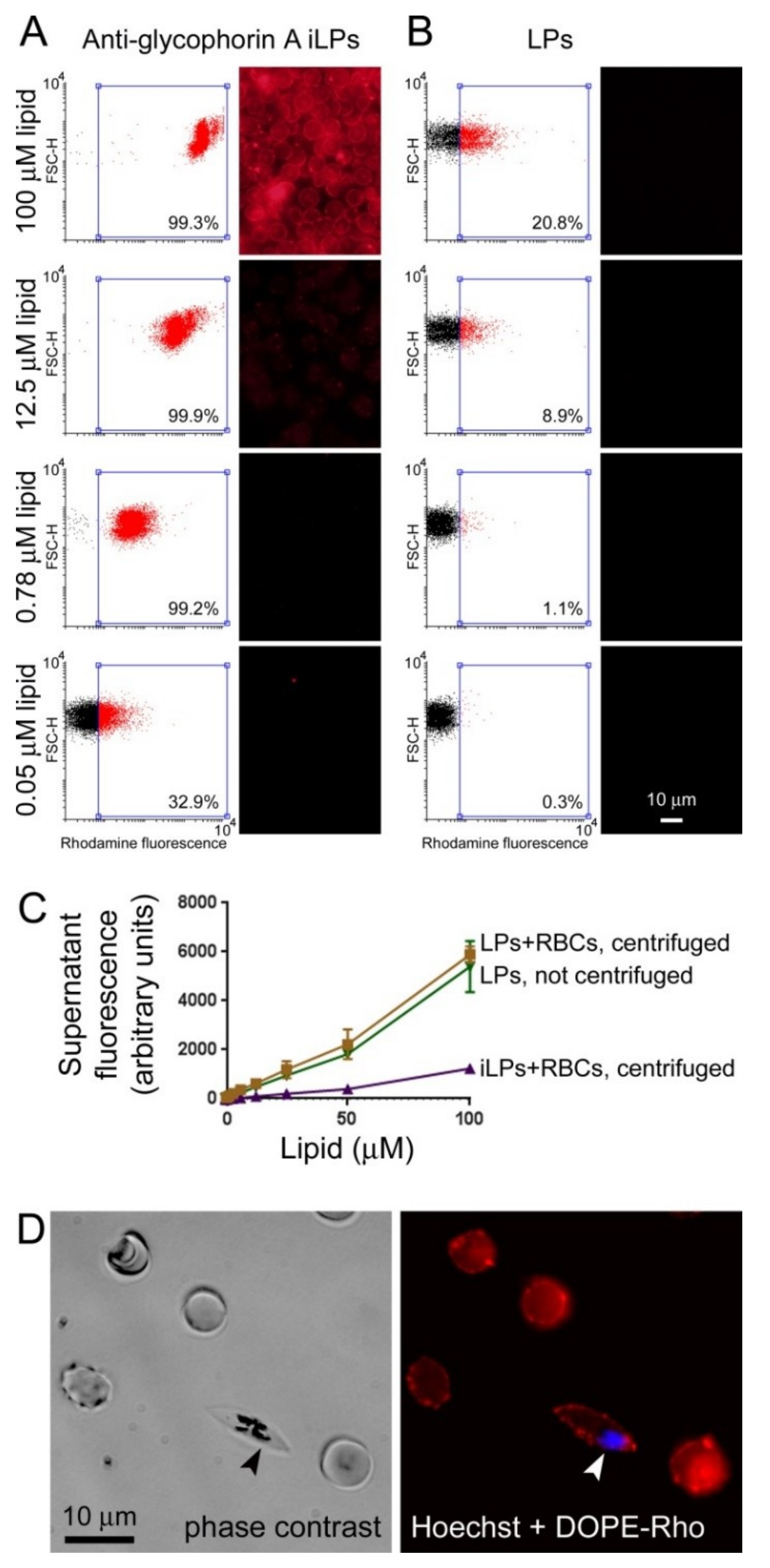
3.2. Immunoliposome Targeting
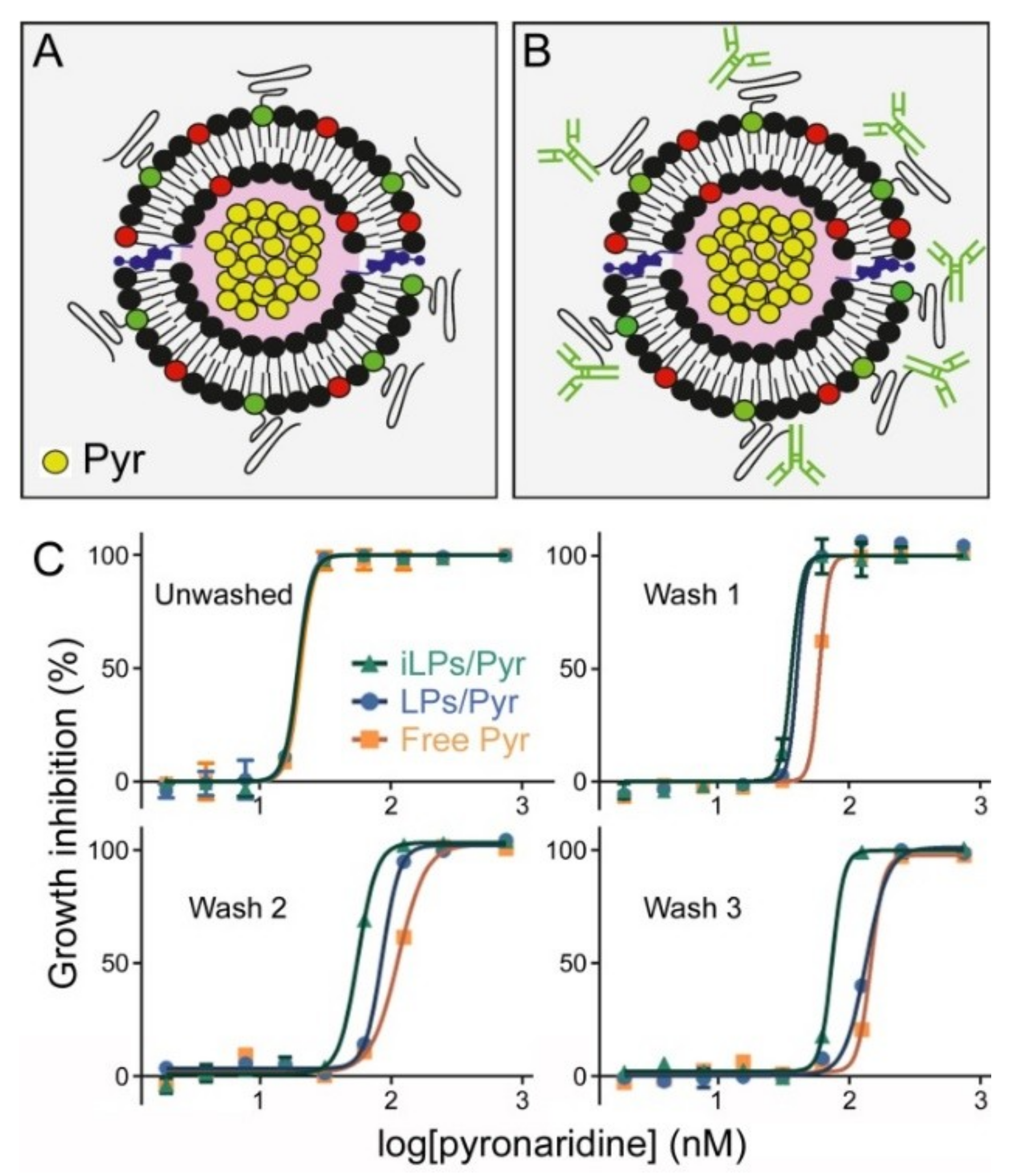
3.3. In Vitro Inhibition of P. falciparum Growth by iLPs Encapsulating Pyronaridine
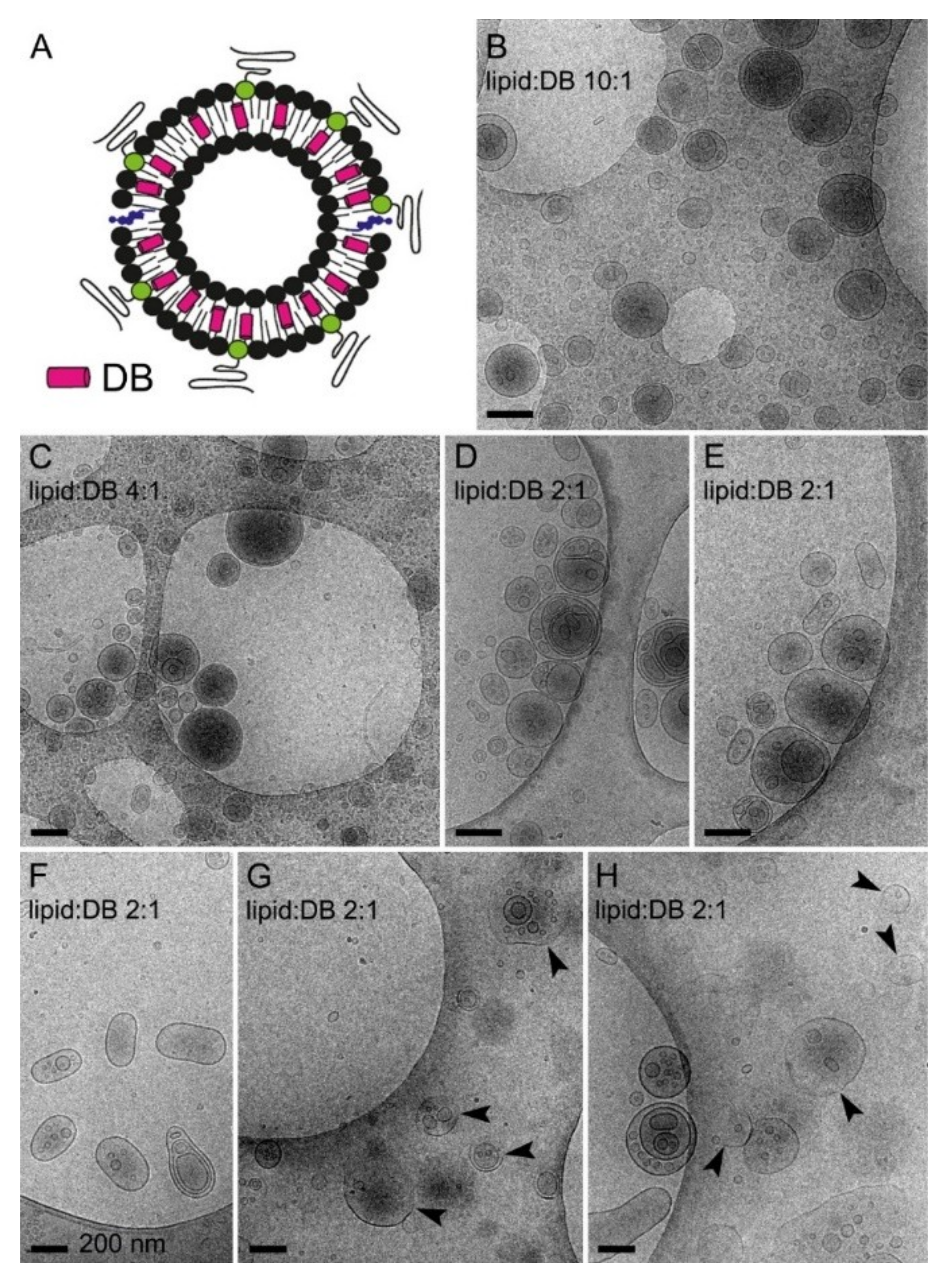
3.4. Simultaneous Encapsulation in Liposomes of Pyronaridine and DB
3.5. In Vitro Inhibition of P. falciparum Growth by iLPs Encapsulating DB
3.6. In Vitro Inhibition of P. falciparum Growth by iLPs Co-Encapsulating Pyronaridine and Atovaquone
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moorthy, V.S.; Newman, R.D.; Duclos, P.; Okwo-Bele, J.M.; Smith, P.G. Assessment of the RTS,S/AS01 malaria vaccine. Lancet Infect. Dis. 2013, 13, 280–282. [Google Scholar] [CrossRef]
- Mbengue, A.; Bhattacharjee, S.; Pandharkar, T.; Liu, H.; Estiu, G.; Stahelin, R.V.; Rizk, S.S.; Njimoh, D.L.; Ryan, Y.; Chotivanich, K.; et al. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 2015, 520, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Vangapandu, S.; Jain, M.; Kaur, K.; Patil, P.; Patel, S.R.; Jain, R. Recent advances in antimalarial drug development. Med. Res. Rev. 2007, 27, 65–107. [Google Scholar] [CrossRef] [PubMed]
- White, N. Antimalarial drug resistance and combination chemotherapy. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 739–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thu, A.M.; Phyo, A.P.; Landier, J.; Parker, D.M.; Nosten, F.H. Combating multidrug-resistant Plasmodium falciparum malaria. FEBS J. 2017, 284, 2569–2578. [Google Scholar] [CrossRef] [PubMed]
- Burrows, J.; Hooft van Huijsduijnen, R.; Möhrle, J.; Oeuvray, C.; Wells, T. Designing the next generation of medicines for malaria control and eradication. Malar. J. 2013, 12, 187. [Google Scholar] [CrossRef]
- World Health Organization. Guidelines for the Treatment of Malaria, 3rd ed.; World Health Organization: Geneva, Switzerland, 2015; Available online: http://apps.who.int/iris/bitstream/10665/162441/1/9789241549127_eng.pdf (accessed on 16 July 2019).
- Urbán, P.; Valle-Delgado, J.J.; Moles, E.; Marques, J.; Díez, C.; Fernàndez-Busquets, X. Nanotools for the delivery of antimicrobial peptides. Curr. Drug Targets 2012, 13, 1158–1172. [Google Scholar] [CrossRef]
- Kuntworbe, N.; Martini, N.; Shaw, J.; Al-Kassas, R. Malaria intervention policies and pharmaceutical nanotechnology as a potential tool for malaria management. Drug Dev. Res. 2012, 73, 167–184. [Google Scholar] [CrossRef]
- Urbán, P.; Fernàndez-Busquets, X. Nanomedicine against malaria. Curr. Med. Chem. 2014, 21, 605–629. [Google Scholar] [CrossRef]
- Beytia, E.D.; Porter, J.W. Biochemistry of polyisoprenoid biosynthesis. Annu. Rev. Biochem. 1976, 45, 113–142. [Google Scholar] [CrossRef]
- Rohmer, M.; Knani, M.; Simonin, P.; Sutter, B.; Sahm, H. Isoprenoid biosynthesis in bacteria: A novel pathway for the early steps leading to isopentenyl diphosphate. Biochem. J. 1993, 295, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Eisenreich, W.; Rohdich, F.; Bacher, A. Deoxyxylulose phosphate pathway to terpenoids. Trends Plant Sci. 2001, 6, 78–84. [Google Scholar] [CrossRef]
- Rohdich, F.; Kis, K.; Bacher, A.; Eisenreich, W. The non-mevalonate pathway of isoprenoids: Genes, enzymes and intermediates. Curr. Opin. Chem. Biol. 2001, 5, 535–540. [Google Scholar] [CrossRef]
- Takahashi, S.; Kuzuyama, T.; Watanabe, H.; Seto, H. A 1-deoxy-D-xylulose 5-phosphate reductoisomerase catalyzing the formation of 2-C-methyl-D-erythritol 4-phosphate in an alternative nonmevalonate pathway for terpenoid biosynthesis. Proc. Natl. Acad. Sci. USA 1998, 95, 9879–9884. [Google Scholar] [CrossRef]
- Jomaa, H.; Wiesner, J.; Sanderbrand, S.; Altincicek, B.; Weidemeyer, C.; Hintz, M.; Turbachova, I.; Eberl, M.; Zeidler, J.; Lichtenthaler, H.K.; et al. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science 1999, 285, 1573–1576. [Google Scholar] [CrossRef]
- Ralph, S.A.; D’Ombrain, M.C.; McFadden, G.I. The apicoplast as an antimalarial drug target. Drug Resist. Update 2001, 4, 145–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrmann, S.; Lundgren, I.; Oyakhirome, S.; Impouma, B.; Matsiegui, P.B.; Adegnika, A.A.; Issifou, S.; Kun, J.F.J.; Hutchinson, D.; Wiesner, J.; et al. Fosmidomycin plus clindamycin for treatment of pediatric patients aged 1 to 14 years with Plasmodium falciparum malaria. Antimicrob. Agents Chemother. 2006, 50, 2713–2718. [Google Scholar] [CrossRef]
- Wiesner, J.; Borrmann, S.; Jomaa, H. Fosmidomycin for the treatment of malaria. Parasitol. Res. 2003, 90 (Suppl. S2), S71–S76. [Google Scholar] [CrossRef]
- Altincicek, B.; Hintz, M.; Sanderbrand, S.; Wiesner, J.; Beck, E.; Jomaa, H. Tools for discovery of inhibitors of the 1-deoxy-D-xylulose 5-phosphate (DXP) synthase and DXP reductoisomerase: An approach with enzymes from the pathogenic bacterium Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2000, 190, 329–333. [Google Scholar] [CrossRef]
- Sisquella, X.; de Pourcq, K.; Alguacil, J.; Robles, J.; Sanz, F.; Anselmetti, D.; Imperial, S.; Fernàndez-Busquets, X. A single-molecule force spectroscopy nanosensor for the identification of new antibiotics and antimalarials. FASEB J. 2010, 24, 4203–4217. [Google Scholar] [CrossRef]
- Ghavami, M.; Merino, E.F.; Yao, Z.K.; Elahi, R.; Simpson, M.E.; Fernández-Murga, M.L.; Butler, J.H.; Casasanta, M.A.; Krai, P.M.; Totrov, M.M.; et al. Biological studies and target engagement of the 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase (IspD)-targeting antimalarial agent (1R,3S)-MMV008138 and analogs. ACS Infect. Dis. 2018, 4, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Yang, Y.; Xiao, C.; Liu, Y.; Gan, M.; Guan, Y.; Hao, X.; Meng, J.; Zhou, S.; Chen, X.; et al. Identification and validation of a novel lead compound targeting 4-diphosphocytidyl-2-C-methylerythritol synthetase (IspD) of mycobacteria. Eur. J. Pharmacol. 2012, 694, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Saggu, G.S.; Garg, S.; Pala, Z.R.; Kochar, S.K.; Saxena, V. Deciphering the role of IspD (2-C-methyl-D-erythritol 4-phosphate cytidyltransferase) enzyme as a potential therapeutic drug target against Plasmodium vivax. Gene 2018, 675, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.; Valle-Delgado, J.J.; Urbán, P.; Baró, E.; Prohens, R.; Mayor, A.; Cisteró, P.; Delves, M.; Sinden, R.E.; Grandfils, C.; et al. Adaptation of targeted nanocarriers to changing requirements in antimalarial drug delivery. Nanomed. NBM 2017, 13, 515–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, S.L.; Duparc, S.; Arbe-Barnes, S.J.; Craft, J.C.; Shin, C.S.; Fleckenstein, L.; Borghini-Fuhrer, I.; Rim, H.J. Review of pyronaridine anti-malarial properties and product characteristics. Malar. J. 2012, 11, 270. [Google Scholar] [CrossRef] [PubMed]
- Alonso, P.L.; Brown, G.; Arevalo-Herrera, M.; Binka, F.; Chitnis, C.; Collins, F.; Doumbo, O.K.; Greenwood, B.; Hall, B.F.; Levine, M.M.; et al. A research agenda to underpin malaria eradication. PLoS Med. 2011, 8, e1000406. [Google Scholar] [CrossRef] [PubMed]
- Delves, M.J.; Ramakrishnan, C.; Blagborough, A.M.; Leroy, D.; Wells, T.N.C.; Sinden, R.E. A high-throughput assay for the identification of malarial transmission-blocking drugs and vaccines. Int. J. Parasitol. 2012, 42, 999–1006. [Google Scholar] [CrossRef]
- Adjalley, S.H.; Johnston, G.L.; Li, T.; Eastman, R.T.; Ekland, E.H.; Eappen, A.G.; Richman, A.; Sim, B.K.L.; Lee, M.C.S.; Hoffman, S.L.; et al. Quantitative assessment of Plasmodium falciparum sexual development reveals potent transmission-blocking activity by methylene blue. Proc. Natl. Acad. Sci. USA 2011, 108, E1214–E1223. [Google Scholar] [CrossRef]
- Delves, M.J.; Ruecker, A.; Straschil, U.; Lelièvre, J.; Marques, S.; López-Barragán, M.J.; Herreros, E.; Sinden, R.E. Male and female Plasmodium falciparum mature gametocytes show different responses to antimalarial drugs. Antimicrob. Agents Chemother. 2013, 57, 3268–3274. [Google Scholar] [CrossRef]
- Ruecker, A.; Mathias, D.K.; Straschil, U.; Churcher, T.S.; Dinglasan, R.R.; Leroy, D.; Sinden, R.E.; Delves, M.J. A male and female gametocyte functional viability assay to identify biologically relevant malaria transmission-blocking drugs. Antimicrob. Agents Chemother. 2014, 58, 7292–7302. [Google Scholar] [CrossRef]
- Moles, E.; Urbán, P.; Jiménez-Díaz, M.B.; Viera-Morilla, S.; Angulo-Barturen, I.; Busquets, M.A.; Fernàndez-Busquets, X. Immunoliposome-mediated drug delivery to Plasmodium-infected and non-infected red blood cells as a dual therapeutic/prophylactic antimalarial strategy. J. Control. Release 2015, 210, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Moles, E.; Fernàndez-Busquets, X. Loading antimalarial drugs into noninfected red blood cells: An undesirable roommate for Plasmodium. Future Med. Chem. 2015, 7, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Moles, E.; Galiano, S.; Gomes, A.; Quiliano, M.; Teixeira, C.; Aldana, I.; Gomes, P.; Fernàndez-Busquets, X. ImmunoPEGliposomes for the targeted delivery of novel lipophilic drugs to red blood cells in a falciparum malaria murine model. Biomaterials 2017, 145 (Suppl. SC), 178–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basco, L.K.; Ramiliarisoa, O.; Le Bras, J. In vitro activity of atovaquone against the African isolates and clones of Plasmodium falciparum. Am. J. Trop. Med. Hyg. 1995, 53, 388–391. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, R.C.; MacDonald, R.I.; Menco, B.P.; Takeshita, K.; Subbarao, N.K.; Hu, L.R. Small-volume extrusion apparatus for preparation of large, unilamellar vesicles. Biochim. Biophys. Acta 1991, 1061, 297–303. [Google Scholar] [CrossRef]
- Moll, K.; Ljungström, I.; Perlmann, H.; Scherf, A.; Wahlgren, M. Methods in Malaria Research, 5th ed.; Malaria Research and Reference Reagent Resource Center (MR4): Manassas, VA, USA, 2008. [Google Scholar]
- Lambros, C.; Vanderberg, J.P. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 1979, 65, 418–420. [Google Scholar] [CrossRef] [PubMed]
- ChemAxon. 2016. Available online: http://www.chemaxon.com (accessed on 16 July 2019).
- Roy, B.; Guha, P.; Bhattarai, R.; Nahak, P.; Karmakar, G.; Chettri, P.; Panda, A.K. Influence of lipid composition, pH, and temperature on physicochemical properties of liposomes with curcumin as model drug. J. Oleo Sci. 2016, 65, 399–411. [Google Scholar] [CrossRef]
- Lichtenberg, D.; Ahyayauch, H.; Goñi, F.M. The mechanism of detergent solubilization of lipid bilayers. Biophys. J. 2013, 105, 289–299. [Google Scholar] [CrossRef]
- Munn, L.L.; Dupin, M.M. Blood cell interactions and segregation in flow. Ann. Biomed. Eng. 2008, 36, 534–544. [Google Scholar] [CrossRef]
- Feachem, R.G.; Phillips, A.A.; Targett, G.A.; Snow, R.W. Call to action: Priorities for malaria elimination. Lancet 2010, 376, 1517–1521. [Google Scholar] [CrossRef]
- Alonso, P.L. Malaria: Deploying a candidate vaccine (RTS,S/AS02A) for an old scourge of humankind. Int. Microbiol. 2006, 9, 83–93. [Google Scholar] [PubMed]
- Daily, J.P. Antimalarial drug therapy: The role of parasite biology and drug resistance. J. Clin. Pharmacol. 2006, 46, 1487–1497. [Google Scholar] [CrossRef] [PubMed]
- Saltzman, M.; Desai, T. Drug delivery in the BME curricula. Ann. Biomed. Eng. 2006, 34, 270–275. [Google Scholar] [CrossRef] [PubMed]
- European Science Fundation: ESF Forward Look on Nanomedicine 2005. Available online: http://archives.esf.org/fileadmin/Public_documents/Publications/Nanomedicine.pdf (accessed on 16 July 2019).
- Velasques, K.; Maciel, T.R.; de Castro Dal Forno, A.H.; Teixeira, F.E.G.; da Fonseca, A.L.; Varotti, F.d.P.; Fajardo, A.R.; Ávila, D.S.d.; Haas, S.E. Co-nanoencapsulation of antimalarial drugs increases their in vitro efficacy against Plasmodium falciparum and decreases their toxicity to Caenorhabditis elegans. Eur. J. Pharm. Sci. 2018, 118, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Oyeyemi, O.; Morenkeji, O.; Afolayan, F.; Dauda, K.; Busari, Z.; Meena, J.; Panda, A. Curcumin-artesunate based polymeric nanoparticle; antiplasmodial and toxicological evaluation in murine model. Front. Pharmacol. 2018, 9, 562. [Google Scholar] [CrossRef] [PubMed]
- Mott, B.T.; Eastman, R.T.; Guha, R.; Sherlach, K.S.; Siriwardana, A.; Shinn, P.; McKnight, C.; Michael, S.; Lacerda-Queiroz, N.; Patel, P.R.; et al. High-throughput matrix screening identifies synergistic and antagonistic antimalarial drug combinations. Sci. Rep. 2015, 5, 13891. [Google Scholar] [CrossRef] [PubMed]
- Griffith, K.S.; Lewis, L.S.; Mali, S.; Parise, M.E. Treatment of malaria in the United States: A systematic review. JAMA 2007, 297, 2264–2277. [Google Scholar] [CrossRef]
- Delves, M.; Plouffe, D.; Scheurer, C.; Meister, S.; Wittlin, S.; Winzeler, E.; Sinden, R.E.; Leroy, D. The activities of current antimalarial drugs on the life cycle stages of Plasmodium: A comparative study with human and rodent parasites. PLoS Med. 2012, 9, e1001169. [Google Scholar] [CrossRef]
- Sinden, R.; Carter, R.; Drakeley, C.; Leroy, D. The biology of sexual development of Plasmodium: The design and implementation of transmission-blocking strategies. Malar. J. 2012, 11, 70. [Google Scholar] [CrossRef]
- Delves, M.J. Plasmodium cell biology should inform strategies used in the development of antimalarial transmission-blocking drugs. Future Med. Chem. 2012, 4, 2251–2263. [Google Scholar] [CrossRef]
- Yeh, E.; DeRisi, J.L. Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol. 2011, 9, e1001138. [Google Scholar] [CrossRef] [PubMed]
- Gisselberg, J.E.; Dellibovi-Ragheb, T.A.; Matthews, K.A.; Bosch, G.; Prigge, S.T. The Suf iron-sulfur cluster synthesis pathway is required for apicoplast maintenance in malaria parasites. PLoS Pathog. 2013, 9, e1003655. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.D.; Merino, E.F.; Brooks, C.F.; Striepen, B.; Carlier, P.R.; Cassera, M.B. Antiapicoplast and gametocytocidal screening to identify the mechanisms of action of compounds within the Malaria Box. Antimicrob. Agents Chemother. 2014, 58, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Blasco, B.; Leroy, D.; Fidock, D.A. Antimalarial drug resistance: Linking Plasmodium falciparum parasite biology to the clinic. Nat. Med. 2017, 23, 917–928. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No Wash | Wash 1 | Wash 2 | Wash 3 | |
|---|---|---|---|---|
| Free Pyr | 20.6 (18.8–22.5) | 57.2 (55.0–59.6) | 113.6 (106.2–121.5) | 146.4 (121.3–176.7) |
| LPs/Pyr | 19.7 (17.7–21.9) | 43.1 (40.0–46.3) | 85.5 (80.7–90.7) | 134.8 (128.1–141.9) |
| iLPs/Pyr | 19.7 (18.5–21.0) | 36.2 (17.4–75.2) | 55.8 (52.4–59.4) | 74.2 (62.0–88.8) |
| No Wash | Wash 1 | Wash 2 | Wash 3 | |
|---|---|---|---|---|
| Free DB | 1.1 (1.0–1.3) | 4.1 (3.6–4.8) | 6.0 (5.3–6.8) | 8.3 (7.0–9.7) |
| LPs/DB | 1.7 (1.5–1.9) | 12.0 (9.6–15.0) | 28.9 (18.5–45.3) | 34.9 (16.2–74.9) |
| iLPs/DB | 1.0 (0.8–1.2) | 2.0 (1.7–2.2) | 2.4 (2.1–2.7) | 3.0 (2.7–3.4) |
| % Inhibition at Drug IC50 of Immunoliposome Three-Wash Sample | |||
|---|---|---|---|
| Immunoliposomized Drug | Liposomized Drug | Free Drug | |
| Pyronaridine | 50.0 | 16.2 | 2.0 |
| Domiphen bromide | 50.0 | 0.2 | 3.5 |
| Atovaquone | 50.0 | 17.3 | 0.0 |
| Atovaquone/pyronaridine | 50.0 | 28.4 | 0.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biosca, A.; Dirscherl, L.; Moles, E.; Imperial, S.; Fernàndez-Busquets, X. An ImmunoPEGliposome for Targeted Antimalarial Combination Therapy at the Nanoscale. Pharmaceutics 2019, 11, 341. https://doi.org/10.3390/pharmaceutics11070341
Biosca A, Dirscherl L, Moles E, Imperial S, Fernàndez-Busquets X. An ImmunoPEGliposome for Targeted Antimalarial Combination Therapy at the Nanoscale. Pharmaceutics. 2019; 11(7):341. https://doi.org/10.3390/pharmaceutics11070341
Chicago/Turabian StyleBiosca, Arnau, Lorin Dirscherl, Ernest Moles, Santiago Imperial, and Xavier Fernàndez-Busquets. 2019. "An ImmunoPEGliposome for Targeted Antimalarial Combination Therapy at the Nanoscale" Pharmaceutics 11, no. 7: 341. https://doi.org/10.3390/pharmaceutics11070341
APA StyleBiosca, A., Dirscherl, L., Moles, E., Imperial, S., & Fernàndez-Busquets, X. (2019). An ImmunoPEGliposome for Targeted Antimalarial Combination Therapy at the Nanoscale. Pharmaceutics, 11(7), 341. https://doi.org/10.3390/pharmaceutics11070341