Rational Development of Liposomal Hydrogels: A Strategy for Topical Vaginal Antiretroviral Drug Delivery in the Context of HIV Prevention


 ,
,  , ,
, ,  ,
,  and
and 
Abstract
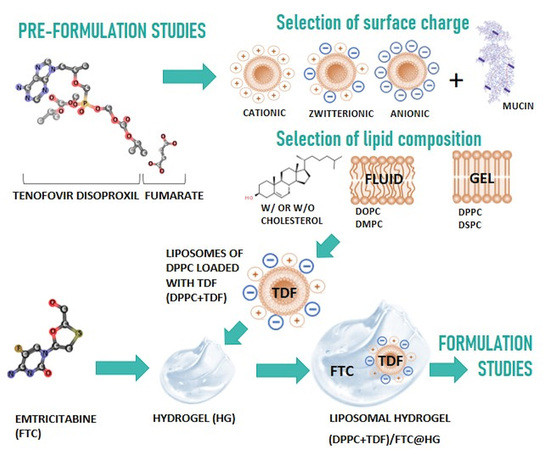
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. In Silico Studies
2.3. Preparation of Liposomes and Liposomal Hydrogels
2.4. Small-Angle and Wide-Angle X-ray Scattering (SAXS/WAXS)
2.5. Attenuated Total Reflection–Fourier Transform Infrared (ATR-FTIR)
2.6. Dynamic Light Scattering (DLS) Applied to Microviscosity Studies
2.7. Mucoadhesion Studies
2.8. Encapsulation Efficiency
2.9. Liposomal Size, Polydispersity Index, and Zeta Potential
2.10. In Vitro Drug Release Studies
2.11. In Vitro Permeation Studies
2.12. Rheological Behavior of Hydrogels
2.13. Cytotoxicity Studies
3. Results
3.1. Pre-Formulation Studies
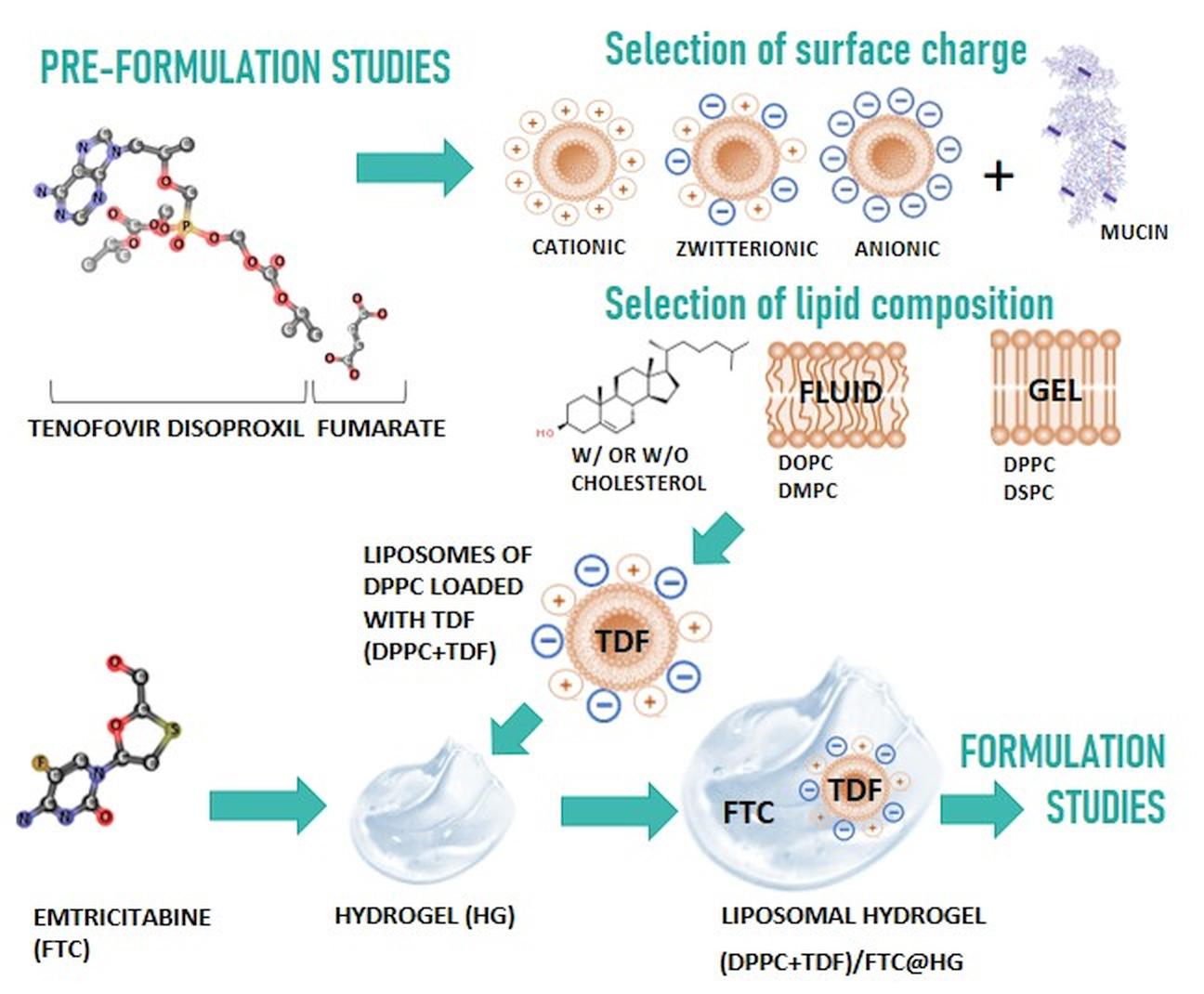
3.1.1. In Silico Studies
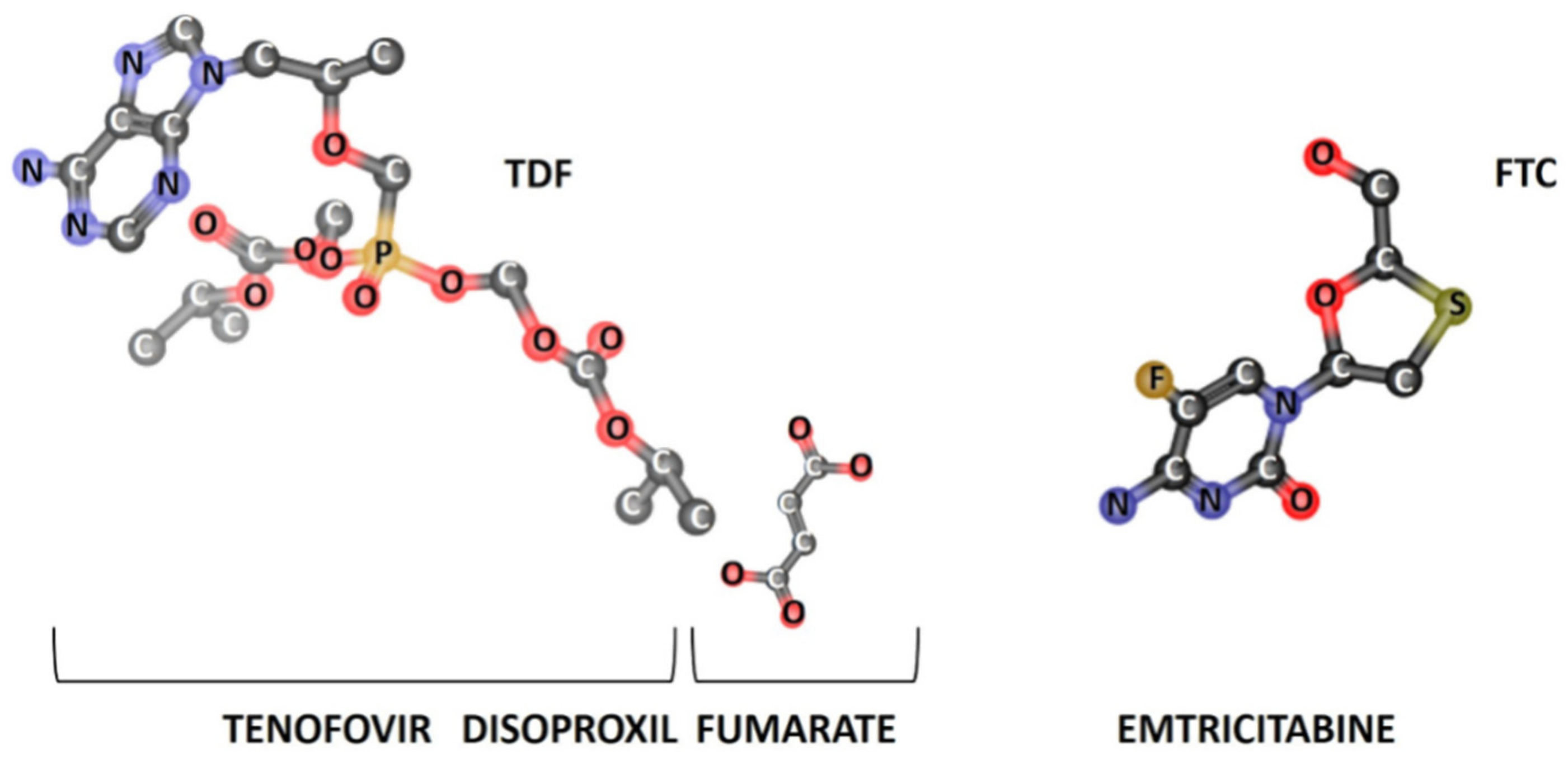
3.1.2. Drug–Membrane Interaction Studies
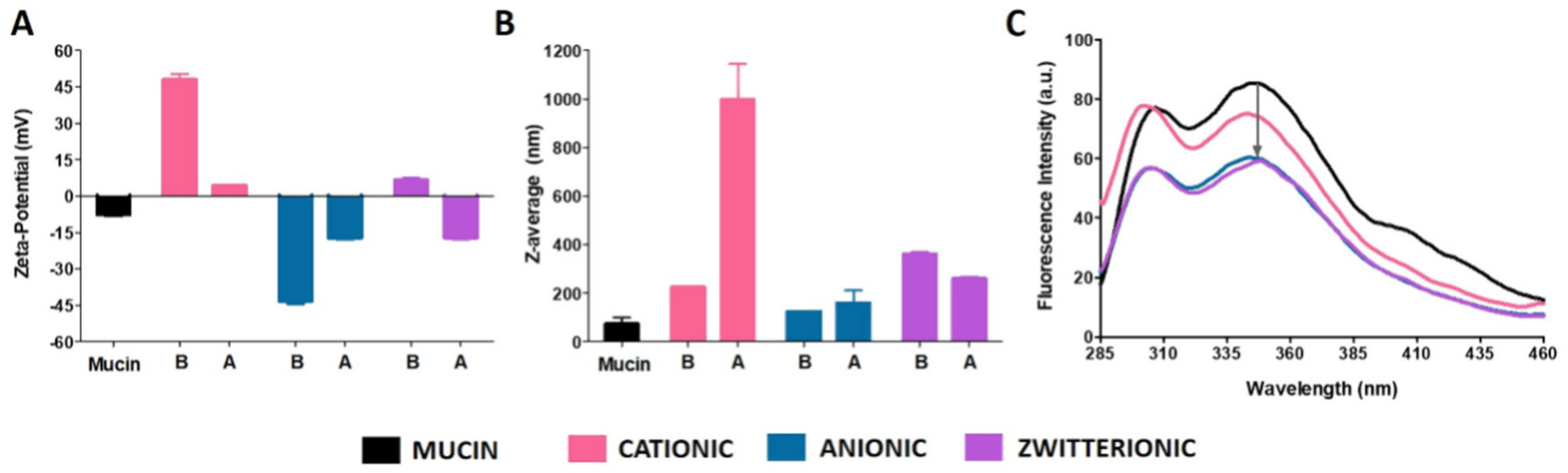
3.1.3. Mucoadhesive Properties of Liposomes with Different Surface Charges
3.2. Formulation Studies
3.2.1. Physicochemical Characterization
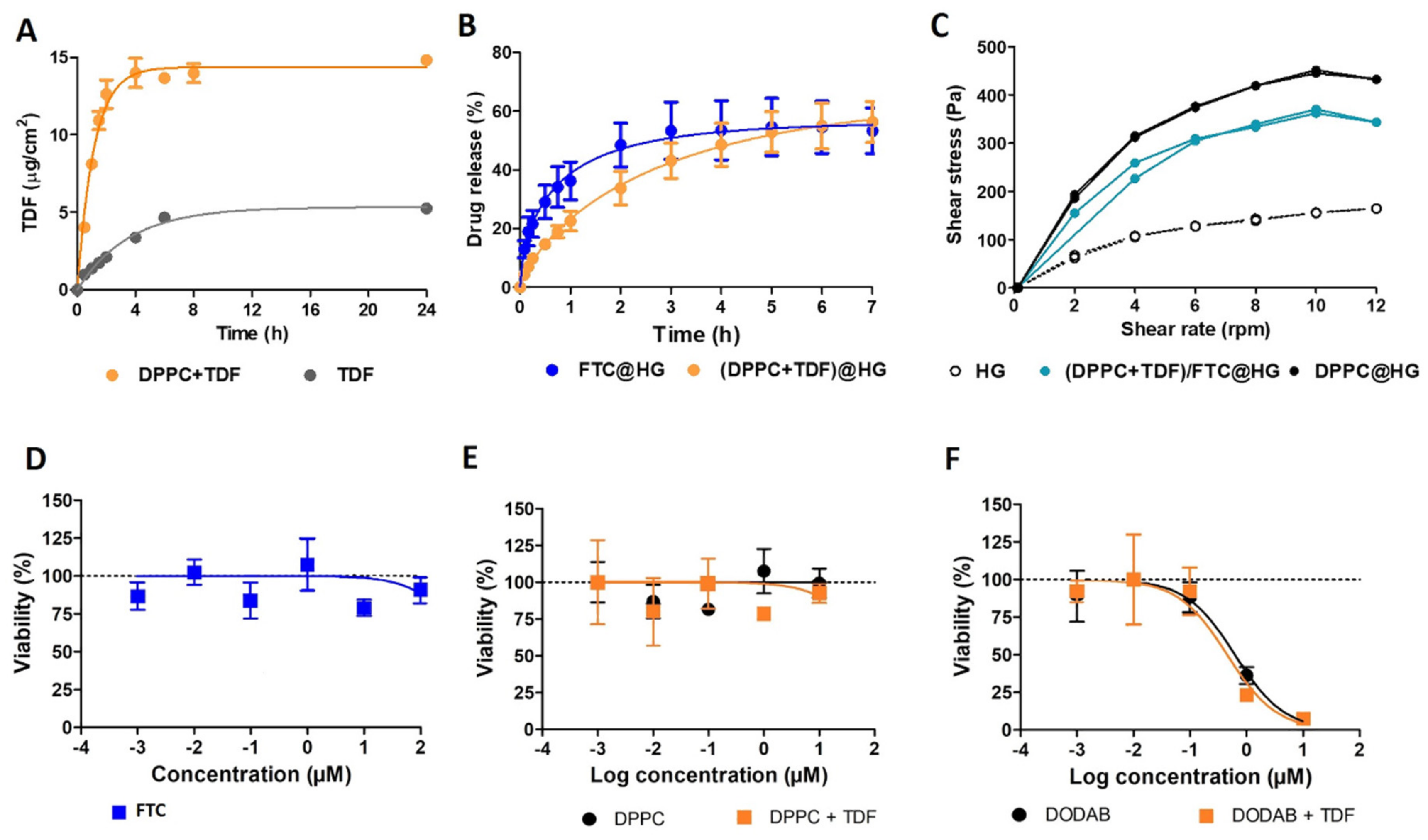
3.2.2. Pharmaceutical Performance of the Selected Formulation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- UNAIDS. UNAIDS Data 2018; UNAIDS: Geneva, Switzerland, 2018; Available online: http://www.unaids.org/en/resources/documents/2018/unaids-data-2018 (accessed on 18 March 2019).
- Patel, P.; Borkowf, C.B.; Brooks, J.T.; Lasry, A.; Lansky, A.; Mermin, J. Estimating per-act HIV transmission risk: A systematic review. AIDS 2014, 28, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Piot, P.; Karim, S.S.; Hecht, R.; Legido-Quigley, H.; Buse, K.; Stover, J.; Resch, S.; Ryckman, T.; Møgedal, S.; Dybul, M. UNAIDS-Lancet Commission; et al. Defeating AIDS–advancing global health. Lancet 2015, 386, 171–218. [Google Scholar] [CrossRef]
- Baeten, J.; Celum, C. Systemic and topical drugs for the prevention of HIV infection: Antiretroviral pre-exposure prophylaxis. Annu. Rev. Med. 2013, 64, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Riddell, J.T.; Amico, K.R.; Mayer, K.H. HIV preexposure prophylaxis: A review. JAMA 2018, 319, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Sosnik, A.; Augustine, R. Challenges in oral drug delivery of antiretrovirals and the innovative strategies to overcome them. Adv. Drug Deliv. Rev. 2016, 103, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Beymer, M.R.; Holloway, I.W.; Pulsipher, C.; Landovitz, R.J. Current and future PrEP medications and modalities: On-demand, injectables, and topicals. Curr. HIV/AIDS Rep. 2019, 16, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Karim, Q.A.; Abdool Karim, S.S.; Frohlich, J.A.; Grobler, A.C.; Baxter, C.; Mansoor, L.E.; Kharsany, A.B.; Sibeko, S.; Mlisana, K.P.; Omar, Z.; et al. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science 2010, 329, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Baeten, J.M.; Palanee-Phillips, T.; Brown, E.R.; Schwartz, K.; Soto-Torres, L.E.; Govender, V.; Mgodi, N.M.; Matovu Kiweewa, F.; Nair, G.; MTN-Aspire Study Team; et al. Use of a vaginal ring containing dapivirine for HIV-1 prevention in women. N. Engl. J. Med. 2016, 375, 2121–2132. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.; van Niekerk, N.; Kapiga, S.; Bekker, L.G.; Gama, C.; Gill, K.; Kamali, A.; Kotze, P.; Louw, C.; Ring Study Team; et al. Safety and efficacy of a dapivirine vaginal ring for HIV prevention in women. N. Engl. J. Med. 2016, 375, 2133–2143. [Google Scholar] [CrossRef] [PubMed]
- Das Neves, J.; Nunes, R.; Rodrigues, F.; Sarmento, B. Nanomedicine in the development of anti-HIV microbicides. Adv. Drug Deliv. Rev. 2016, 103, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, C.; Sarmento, B.; das Neves, J. Targeted microbicides for preventing sexual HIV transmission. J. Control Release 2017, 266, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Andersen, T.; Vanić, Ž.; Flaten, G.E.; Mattsson, S.; Tho, I.; Škalko-Basnet, N. Pectosomes and chitosomes as delivery ystems for metronidazole: The one-pot preparation method. Pharmaceutics 2013, 5, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Jøraholmen, M.W.; Vanić, Ž.; Tho, I.; Škalko-Basnet, N. Chitosan-coated liposomes for topical vaginal therapy: Assuring localized drug effect. Int. J. Pharm. 2014, 472, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, M.; Guo, Y.; Pan, H.; Chen, X.; Gu, Z. Preparation, in vitro and in vivo evaluation of liposomal/niosomal gel delivery systems for clotrimazole. Drug Dev. Ind. Pharm. 2005, 31, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Foldvari, M.; Moreland, A. Clinical observations with topical liposome-encapsulated interferon alpha for the treatment of genital papillomavirus infections. J. Liposome Res. 1997, 7, 115–126. [Google Scholar] [CrossRef]
- Naderkhani, E.; Erber, A.; Škalko-Basnet, N.; Flaten, G.E. Improved permeability of acyclovir: Optimization of mucoadhesive liposomes using the phospholipid vesicle-based permeation assay. J. Pharm. Sci. 2014, 103, 661–668. [Google Scholar] [CrossRef]
- Caron, M.; Besson, G.; Etenna, S.L.; Mintsa-Ndong, A.; Mourtas, S.; Radaelli, A.; de Morghen, C.G.; Loddo, R.; La Colla, P.; Antimisiaris, S.G.; et al. Protective properties of non-nucleoside reverse transcriptase inhibitor (MC1220) incorporated into liposome against intravaginal challenge of Rhesus macaques with RT-SHIV. Virology 2010, 405, 225–233. [Google Scholar] [CrossRef]
- Alukda, D.; Sturgis, T.; Youan, B.B. Formulation of tenofovir-loaded functionalized solid lipid nanoparticles intended for HIV prevention. J. Pharm. Sci. 2011, 100, 3345–3356. [Google Scholar] [CrossRef]
- Malavia, N.K.; Zurakowski, D.; Schroeder, A.; Princiotto, A.M.; Laury, A.R.; Barash, H.E.; Sodroski, J.; Langer, R.; Madani, N.; Kohane, D.S. Liposomes for HIV prophylaxis. Biomaterials 2011, 32, 8663–8668. [Google Scholar] [CrossRef] [Green Version]
- Thomson, K.A.; Baeten, J.M.; Mugo, N.R.; Bekker, L.G.; Celum, C.L.; Heffron, R. Tenofovir-based oral preexposure prophylaxis prevents HIV infection among women. Curr. Opin. HIV AIDS 2016, 11, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Moss, J.A.; Srinivasan, P.; Smith, T.J.; Butkyavichene, I.; Lopez, G.; Brooks, A.A.; Martin, A.; Dinh, C.T.; Smith, J.M.; Baum, M.M. Pharmacokinetics and preliminary safety study of pod-intravaginal rings delivering antiretroviral combinations for HIV prophylaxis in a macaque model. Antimicrob. Agents Chemother. 2014, 58, 5125–5135. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, P.; Moss, J.A.; Gunawardana, M.; Churchman, S.A.; Yang, F.; Dinh, C.T.; Mitchell, J.M.; Zhang, J.; Fanter, R.; Miller, C.S.; et al. Topical delivery of tenofovir disoproxil fumarate and emtricitabine from pod-intravaginal rings protects macaques from multiple SHIV exposures. PLoS ONE 2016, 11, e0157061. [Google Scholar] [CrossRef] [PubMed]
- Vincent, K.L.; Moss, J.A.; Marzinke, M.A.; Hendrix, C.W.; Anton, P.A.; Gunawardana, M.; Dawson, L.N.; Olive, T.J.; Pyles, R.B.; Baum, M.M. Phase I trial of pod-intravaginal rings delivering antiretroviral agents for HIV-1 prevention: Rectal drug exposure from vaginal dosing with tenofovir disoproxil fumarate, emtricitabine, and maraviroc. PLoS ONE 2018, 13, e0201952. [Google Scholar] [CrossRef] [PubMed]
- Das Neves, J.; Bahia, M.F. Gels as vaginal drug delivery systems. Int. J. Pharm. 2006, 318, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lúcio, M.; Ferreira, H.; Lima, J.L.F.C.; Matos, C.; de Castro, B.; Reis, S. Influence of some anti-inflammatory drugs in membrane fluidity studied by fluorescence anisotropy measurements. Phys. Chem. Chem. Phys. 2004, 6, 1493–1498. [Google Scholar] [CrossRef]
- Pereira-Leite, C.; Nunes, C.; Lima, J.L.; Reis, S.; Lúcio, M. Interaction of celecoxib with membranes: The role of membrane biophysics on its therapeutic and toxic effects. J. Phys. Chem. B 2012, 116, 13608–13617. [Google Scholar] [CrossRef] [PubMed]
- Lúcio, M.; Lima, J.L.F.C.; Reis, S. Drug-membrane interactions: Molecular mechanisms underlying therapeutic and toxic effects of drugs. In Ideas in Chemistry and Molecular Sciences: Where Chemistry Meets Life; Pignataro, B., Ed.; Wiley-VCH: Weinheim, Germany, 2010; pp. 191–214. [Google Scholar]
- Fernandes, E.; Soares, T.B.; Gonçalves, H.; Lúcio, M. Spectroscopic studies as a toolbox for biophysical and chemical characterization of lipid-based nanotherapeutics. Front. Chem. 2018, 6, 323. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, E.; Soares, T.B.; Gonçalves, H.; Bernstorff, S.; Real Oliveira, M.E.C.D.; Lopes, C.M.; Lúcio, M. A molecular biophysical approach to diclofenac topical gastrointestinal damage. Int. J. Mol. Sci. 2018, 19, 3411. [Google Scholar] [CrossRef]
- Michel, N.; Fabiano, A.S.; Polidori, A.; Jack, R.; Pucci, B. Determination of phase transition temperatures of lipids by light scattering. Chem. Phys. Lipids 2006, 139, 11–19. [Google Scholar] [CrossRef]
- Hiemenz, P.C.; Rajagopalan, R. Principles of Colloid and Surface Chemistry, 3rd ed.; Dekker: New York, NY, USA, 1997. [Google Scholar]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Yu, L.X.; Amidon, G.; Khan, M.A.; Hoag, S.W.; Polli, J.; Raju, G.K.; Woodcock, J. Understanding pharmaceutical quality by design. AAPS J. 2014, 16, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Lúcio, M.; Lima, J.L.; Reis, S. Drug-membrane interactions: Significance for medicinal chemistry. Curr. Med. Chem. 2010, 17, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Truvada (Emtricitabine and Tenofovir Disoproxil Fumarate) Tablets; Food and Drug Administration: Silver Spring, MD, USA, 2016. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/021752s000_TruvadaTOC.cfm (accessed on 22 August 2019).
- Das Neves, J.; Palmeira-de-Oliveira, R.; Palmeira-de-Oliveira, A.; Rodrigues, F.; Sarmento, B. Vaginal mucosa and drug delivery. In Mucoadhesive Materials and Drug Delivery Systems; Khutoryanskiy, V.V., Ed.; Wiley: Chichester, UK, 2014; pp. 99–131. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- European Medicines Agency. Emtricitabine/Tenofovir Disoproxil Product-Specific Bioequivalence Guidance; European Medicines Agency: London, UK, 2015; Available online: https://www.ema.europa.eu/en/emtricitabinetenofovir-disoproxil-product-specific-bioequivalence-guidance (accessed on 22 August 2019).
- Benet, L.Z.; Broccatelli, F.; Oprea, T.I. BDDCS applied to over 900 drugs. AAPS J. 2011, 13, 519–547. [Google Scholar] [CrossRef] [PubMed]
- Hladik, F.; McElrath, M.J. Setting the stage: Host invasion by HIV. Nat. Rev. Immunol. 2008, 8, 447–457. [Google Scholar] [CrossRef]
- Zakeri-Milani, P.; Tajerzadeh, H.; Islambolchilar, Z.; Barzegar, S.; Valizadeh, H. The relation between molecular properties of drugs and their transport across the intestinal membrane. DARU 2006, 14, 164–171. [Google Scholar]
- Avgidou, M.; Dimopoulou, M.; Mackie, A.R.; Rigby, N.M.; Ritzoulis, C.; Panayiotou, C. Physicochemical aspects of mucosa surface. RSC Adv. 2016, 6, 102634–102646. [Google Scholar] [CrossRef]
- Sigurdsson, H.H.; Kirch, J.; Lehr, C.M. Mucus as a barrier to lipophilic drugs. Int. J. Pharm. 2013, 453, 56–64. [Google Scholar] [CrossRef]
- Trezza, C.R.; Kashuba, A.D. Pharmacokinetics of antiretrovirals in genital secretions and anatomic sites of HIV transmission: Implications for HIV prevention. Clin. Pharm. 2014, 53, 611–624. [Google Scholar] [CrossRef]
- Shinitzky, M. Biomembranes: Physical Aspects; Wiley-VCH: Weinheim, Germany, 1993. [Google Scholar]
- Lúcio, M.; Bringezu, F.; Reis, S.; Lima, J.L.; Brezesinski, G. Binding of nonsteroidal anti-inflammatory drugs to DPPC: Structure and thermodynamic aspects. Langmuir 2008, 24, 4132–4139. [Google Scholar] [CrossRef]
- Hauet, N.; Artzner, F.; Boucher, F.; Grabielle-Madelmont, C.; Cloutier, I.; Keller, G.; Lesieur, P.; Durand, D.; Paternostre, M. Interaction between artificial membranes and enflurane, a general volatile anesthetic: DPPC-enflurane interaction. Biophys. J. 2003, 84, 3123–3137. [Google Scholar] [CrossRef]
- Arrondo, J.L.; Goñi, F.M.; Macarulla, J.M. Infrared spectroscopy of phosphatidylcholines in aqueous suspension. A study of the phosphate group vibrations. Biochim. Biophys. Acta 1984, 794, 165–168. [Google Scholar] [CrossRef]
- Schmid, M.; Wölk, C.; Giselbrecht, J.; Chan, K.L.A.; Harvey, R.D. A combined FTIR and DSC study on the bilayer-stabilising effect of electrostatic interactions in ion paired lipids. Colloids Surf. B Biointerfaces 2018, 169, 298–304. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Gamarnik, A.; Tripp, C.P. Identification of lipid aggregate structures on TiO2 surface using headgroup IR bands. J. Phys. Chem. B 2005, 109, 4539–4544. [Google Scholar] [CrossRef] [PubMed]
- Seu, K.J.; Cambrea, L.R.; Everly, R.M.; Hovis, J.S. Influence of lipid chemistry on membrane fluidity: Tail and headgroup interactions. Biophys. J. 2006, 91, 3727–3735. [Google Scholar] [CrossRef]
- Das Neves, J.; Bahia, M.F.; Amiji, M.M.; Sarmento, B. Mucoadhesive nanomedicines: Characterization and modulation of mucoadhesion at the nanoscale. Expert Opin. Drug Deliv. 2011, 8, 1085–1104. [Google Scholar] [CrossRef]
- Das Neves, J.; Amiji, M.; Sarmento, B. Mucoadhesive nanosystems for vaginal microbicide development: Friend or foe? Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2011, 3, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Das Neves, J.; Rocha, C.M.; Gonçalves, M.P.; Carrier, R.L.; Amiji, M.; Bahia, M.F.; Sarmento, B. Interactions of microbicide nanoparticles with a simulated vaginal fluid. Mol. Pharm. 2012, 9, 3347–3356. [Google Scholar] [CrossRef] [PubMed]
- Brandão, E.; Santos Silva, M.; García-Estévez, I.; Mateus, N.; de Freitas, V.; Soares, S. Molecular study of mucin-procyanidin interaction by fluorescence quenching and Saturation Transfer Difference (STD)-NMR. Food Chem. 2017, 228, 427–434. [Google Scholar] [CrossRef]
- Saltzman, W.M.; Radomsky, M.L.; Whaley, K.J.; Cone, R.A. Antibody diffusion in human cervical mucus. Biophys. J. 1994, 66, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Lai, S.K.; Wang, Y.Y.; Hanes, J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv. Drug Deliv. Rev. 2009, 61, 158–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, E.J.; Swartwout, J.R.; Boss, S. Effects of oral contraceptives on phospholipids of human cervical mucus. Am. J. Obs. Gynecol. 1972, 112, 285–291. [Google Scholar] [CrossRef]
- Ensign, L.M.; Tang, B.C.; Wang, Y.Y.; Tse, T.A.; Hoen, T.; Cone, R.; Hanes, J. Mucus-penetrating nanoparticles for vaginal drug delivery protect against herpes simplex virus. Sci. Transl. Med. 2012, 4, 138ra79. [Google Scholar] [CrossRef] [PubMed]
- Alves, A.C.; Ribeiro, D.; Horta, M.; Lima, J.; Nunes, C.; Reis, S. A biophysical approach to daunorubicin interaction with model membranes: Relevance for the drug’s biological activity. J. R. Soc. Interface 2017, 14, 20170408. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. SUPAC-SS: Nonsterile Semisolid Dosage Forms, Scale-up and Post-Approval Changes: Chemistry, Manufacturing and Controls; In Vitro Release Testing and In Vivo Bioequivalence Documentation; Food and Drug Administration: Silver Spring, MD, USA, 1997. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/supac-ss-nonsterile-semisolid-dosage-forms-scale-and-post-approval-changes-chemistry-manufacturing (accessed on 22 August 2019).
- Levin, R.J. A journey through two lumens! Int. J. Impot. Res. 2003, 15, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesquita, L.; Galante, J.; Nunes, R.; Sarmento, B.; das Neves, J. Pharmaceutical vehicles for vaginal and rectal administration of anti-HIV microbicide nanosystems Pharmaceutics 2019, 11, 145. Pharmaceutics 2019, 11, 145. [Google Scholar] [CrossRef] [PubMed]
- Das Neves, J.; da Silva, M.V.; Gonçalves, M.P.; Amaral, M.H.; Bahia, M.F. Rheological properties of vaginal hydrophilic polymer gels. Curr. Drug Deliv. 2009, 6, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Mourtas, S.; Haikou, M.; Theodoropoulou, M.; Tsakiroglou, C.; Antimisiaris, S.G. The effect of added liposomes on the rheological properties of a hydrogel: A systematic study. J. Colloid Interface Sci. 2008, 317, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Pavelić, Ž.; Škalko-Basnet, N.; Schubert, R. Liposomal gels for vaginal drug delivery. Int. J. Pharm. 2001, 219, 139–149. [Google Scholar] [CrossRef]
- Pavelić, Ž.; Škalko-Basnet, N.; Filipovic-Grcic, J.; Martinac, A.; Jalsenjak, I. Development and in vitro evaluation of a liposomal vaginal delivery system for acyclovir. J. Control Release 2005, 106, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Vanić, Ž.; Hurler, J.; Ferderber, K.; Golja Gasparovic, P.; Škalko-Basnet, N.; Filipovic-Grcic, J. Novel vaginal drug delivery system: Deformable propylene glycol liposomes-in-hydrogel. J. Liposome Res. 2014, 24, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Malana, M.A.; Zohra, R.; Khan, M.S. Rheological characterization of novel physically crosslinked terpolymeric hydrogels at different temperatures. Korea Aust. Rheol. J. 2012, 24, 155–162. [Google Scholar] [CrossRef]
- Ferreira, H.; Gonçalves, V.M.F.; Silva, R.; Tiritan, M.E.; Teixeira, M. Development of liposomes-in-hydrogel formulations containing betametha-sone for topical therapy. J. Pharm. Drug Deliv. Saf. 2017, 1, 3. [Google Scholar]
- Gali, Y.; Delezay, O.; Brouwers, J.; Addad, N.; Augustijns, P.; Bourlet, T.; Hamzeh-Cognasse, H.; Ariën, K.K.; Pozzetto, B.; Vanham, G. In vitro evaluation of viability, integrity and inflammation in genital epithelia upon exposure to pharmaceutical excipients and candidate microbicides. Antimicrob. Agents Chemother. 2010, 54, 5105–5114. [Google Scholar] [CrossRef] [PubMed]
- Schinazi, R.F.; McMillan, A.; Cannon, D.; Mathis, R.; Lloyd, R.M.; Peck, A.; Sommadossi, J.P.; St Clair, M.; Wilson, J.; Furman, P.A. Selective inhibition of human immunodeficiency viruses by racemates and enantiomers of cis-5-fluoro-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine. Antimicrob. Agents Chemother. 1992, 36, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, R.V.; Fridland, A. Antiviral activities of 9-R-2-phosphonomethoxypropyl adenine (PMPA) and bis(isopropyloxymethylcarbonyl)PMPA against various drug-resistant human immunodeficiency virus strains. Antimicrob. Agents Chemother. 1998, 42, 1484–1487. [Google Scholar] [CrossRef]
- Lee, W.A.; He, G.X.; Eisenberg, E.; Cihlar, T.; Swaminathan, S.; Mulato, A.; Cundy, K.C. Selective intracellular activation of a novel prodrug of the human immunodeficiency virus reverse transcriptase inhibitor tenofovir leads to preferential distribution and accumulation in lymphatic tissue. Antimicrob. Agents Chemother. 2005, 49, 1898–1906. [Google Scholar] [CrossRef]
- Tomitaka, A.; Arami, H.; Huang, Z.; Raymond, A.; Rodriguez, E.; Cai, Y.; Febo, M.; Takemura, Y.; Nair, M. Hybrid magneto-plasmonic liposomes for multimodal image-guided and brain-targeted HIV treatment. Nanoscale 2017, 10, 184–194. [Google Scholar] [CrossRef]
- Cautela, M.P.; Moshe, H.; Sosnik, A.; Sarmento, B.; das Neves, J. Composite films for vaginal delivery of tenofovir disoproxil fumarate and emtricitabine. Eur. J. Pharm. Biopharm. 2018, 138, 3–10. [Google Scholar] [CrossRef]
- Destache, C.J.; Mandal, S.; Yuan, Z.; Kang, G.; Date, A.A.; Lu, W.; Shibata, A.; Pham, R.; Bruck, P.; Rezich, M.; et al. Topical tenofovir disoproxil fumarate nanoparticles prevent HIV-1 vaginal transmission in a humanized mouse model. Antimicrob. Agents Chemother. 2016, 60, 3633–3639. [Google Scholar] [CrossRef]
- Kamboj, S.; Saini, V.; Bala, S. Formulation and characterization of drug loaded nonionic surfactant vesicles (niosomes) for oral bioavailability enhancement. Sci. World J. 2014, 2014, 959741. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Romero, J.A.; Teleshova, N.; Zydowsky, T.M.; Robbiani, M. Preclinical assessments of vaginal microbicide candidate safety and efficacy. Adv. Drug Deliv. Rev. 2015, 92, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
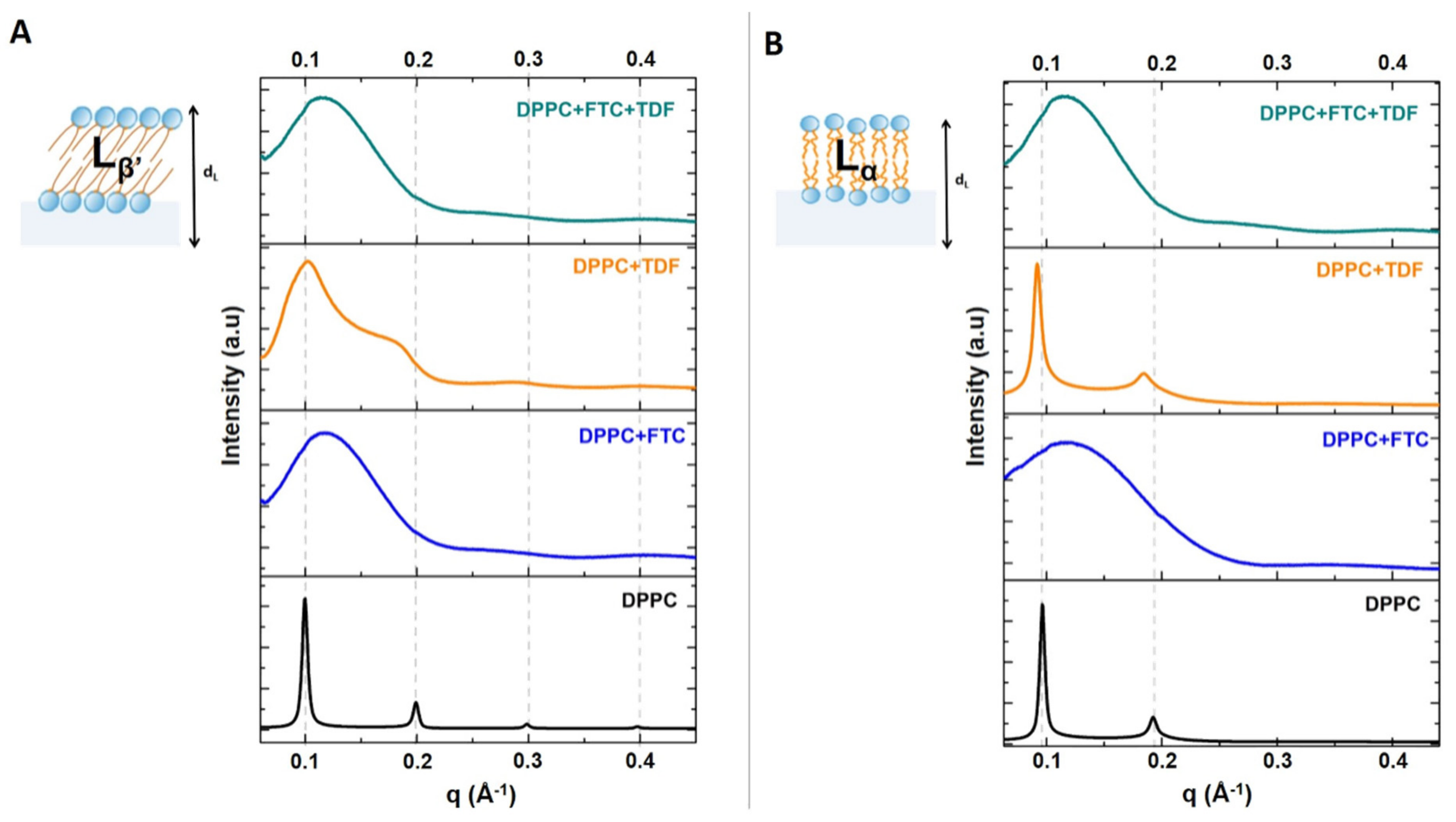{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Studies | Lipid Composition | Concentration (mM) | Temperature Used in Hydration and Extrusion Process (°C) |
|---|---|---|---|
| Mucoadhesion studies | Zwitterionic liposomes:DMPC | 0.4 | 40 |
| Anionic liposomes:DMPG | 40 | ||
| Cationic liposomes:DODAB | 60 | ||
| Encapsulation of TDF (40 μM) | DOPC | ||
| DOPC:CHOL (7:3) | 4 | Room temperature | |
| DOPC:CHOL (6:4) | |||
| DMPC | |||
| DMPC:CHOL (7:3) | 4 | 40 | |
| DMPC:CHOL (6:4) | |||
| DPPC | |||
| DPPC:CHOL (7:3) | 4 | 55 | |
| DPPC:CHOL (6:4) | |||
| DSPC | |||
| DSPC:CHOL (7:3) | 4 | 65 | |
| DSPC:CHOL (6:4) |
| Drug | MW (g·mol−1) | PSA (Å2) | VWSA (Å2) | logP | S (mg·mL−1) | pKa | H Donors | H Acceptors |
|---|---|---|---|---|---|---|---|---|
| TDF | 635.52 | 185.44 | 753.35 | 2.65 | 1.24 | 3.75 | 1 | 10 |
| FTC | 247.24 | 88.15 | 279.83 | -0.90 | 7.67 | 1.75 | 2 | 5 |
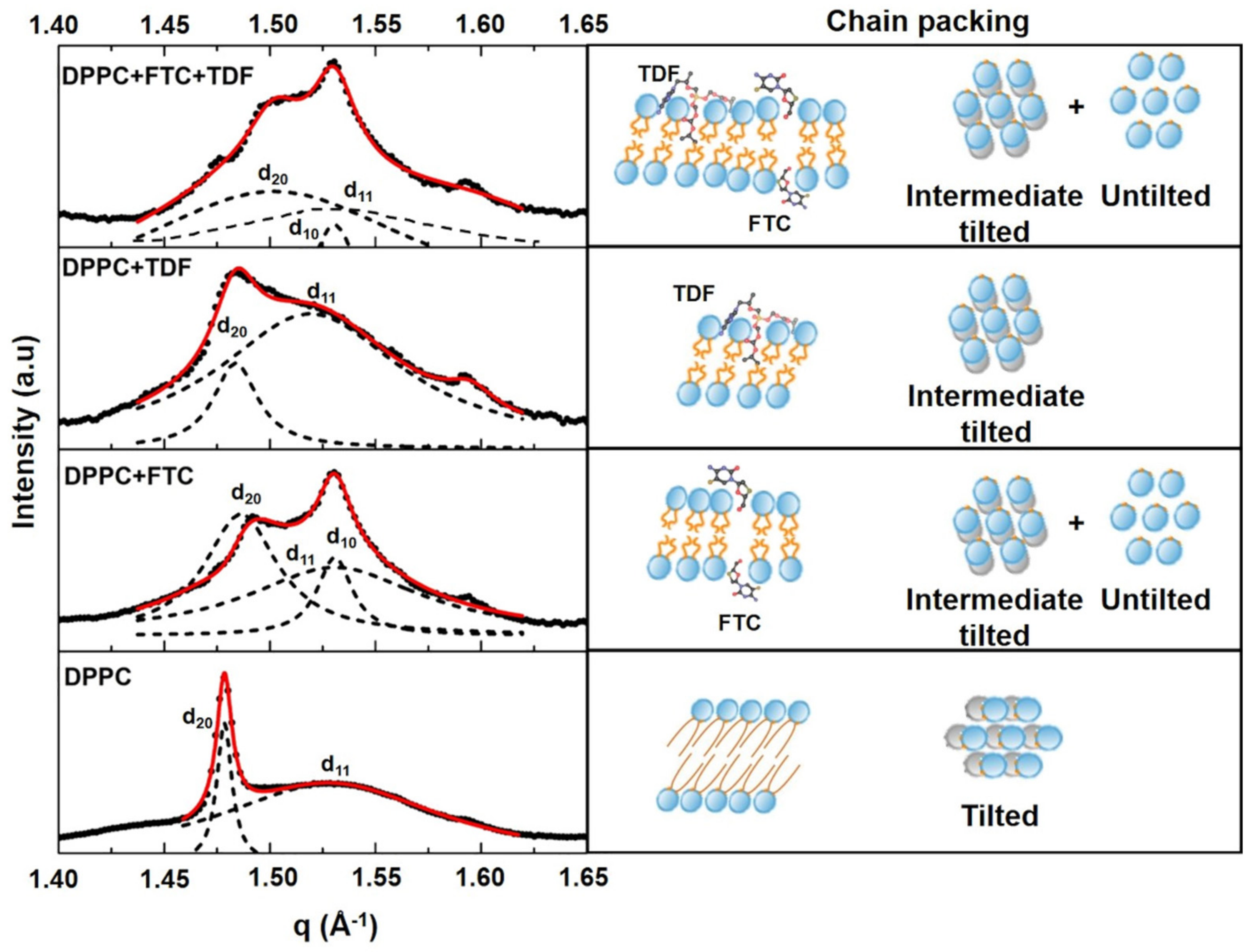
| Lipid Bilayers in the Absence or Presence of Drugs | Long-Spacing Values (SAXS) 1 | Short-Spacing Values (WAXS) | ||||
|---|---|---|---|---|---|---|
| dL 1 (Å) | ξ | dS (Å) | dS (Å) | |||
| Lβ′ | Lα | Lβ′ | Lα | Lβ′ | Lβ′ | |
| DPPC | 62.91 | 65.15 | 1084 | 1018 | 4.25 | 4.11 |
| DPPC + FTC | 51.80 | 54.21 | 41 | 24 | 4.21 | 4.11 |
| DPPC + TDF | 62.70 | 68.14 | 96 | 688 | 4.24 | 4.14 |
| DPPC + FTC + TDF | 53.36 | 53.55 | 41 | 42 | 4.19 | 4.11 |
| Type of Characterization | Parameters Evaluated | DPPC | DPPC+TDF | ||
|---|---|---|---|---|---|
| Incubation | Hydration | Direct Mixing | |||
| Physicochemical | EE (%) | - | 84.63 ± 5.09 ns | 87.31 ± 4.68 ns | 84.79 ± 6.51 ns |
| Z-average (nm) | 123.97 ± 3.62 | 113.13 ± 0.38 ns | 113.47 ± 1.07 ** | 129.83 ± 6.69 ** | |
| PDI | 0.11 ± 0.01 | 0.06 ± 0.01 ** | 0.10 ± 0.01 *** | 0.24 ± 0.01 *** | |
| Zeta potential (mV) | −3.55 ± 0.29 | +6.00 ± 0.28 *** | +0.80 ± 0.33 *** | −5.29 ± 0.280 ns | |
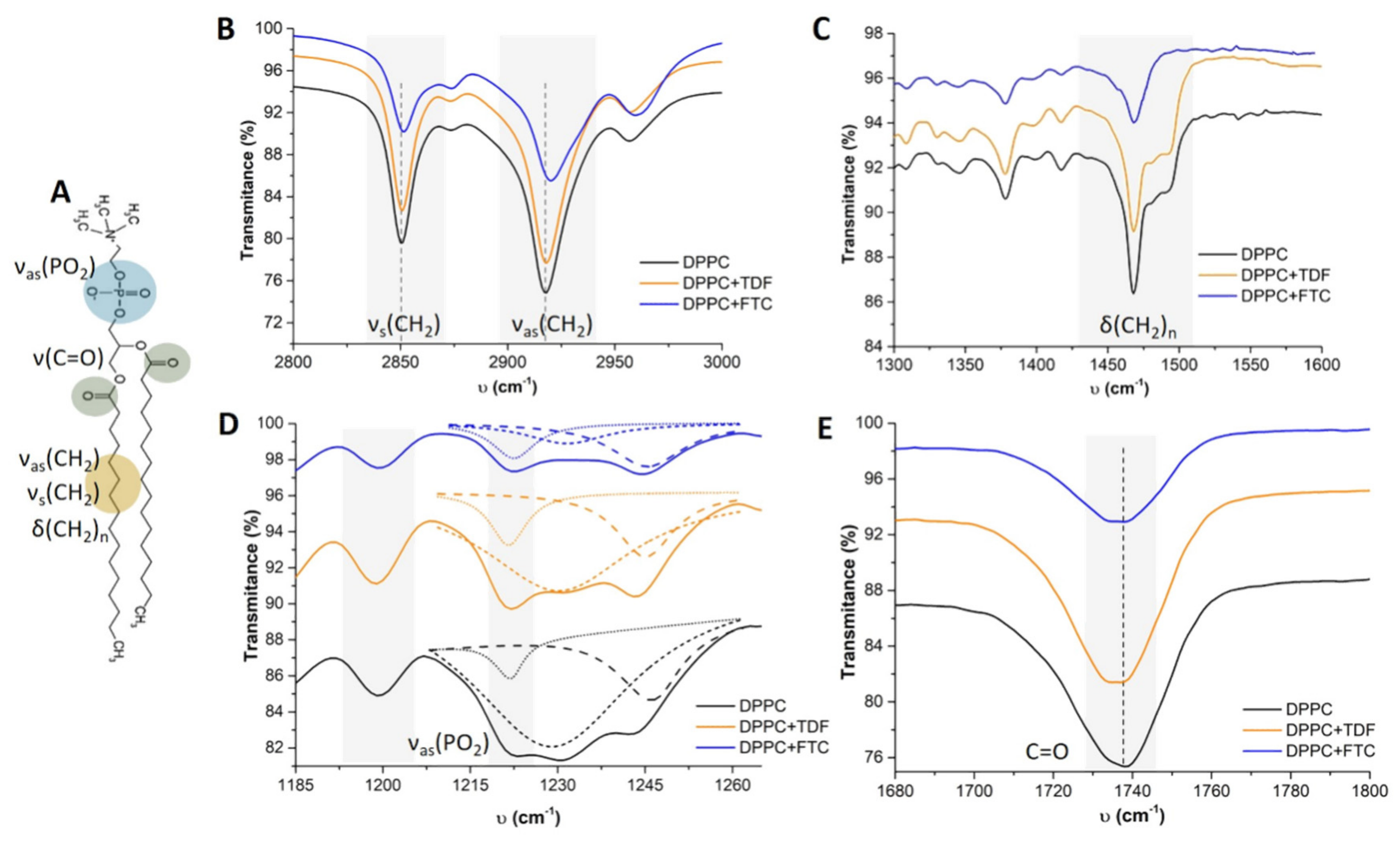
| Biophysical | Tm | 41.05 ± 0.03 | 41.19 ± 0.05 ns | 41.27 ± 0.02 ns | 41.49 ± 0.03 ns |
| B | 2781 ± 234 | 4117 ± 724 ns | 3922 ± 238 ns | 3135 ± 298 ns | |
| R2 | 0.999 | 0.999 | 0.999 | 0.999 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faria, M.J.; Machado, R.; Ribeiro, A.; Gonçalves, H.; Real Oliveira, M.E.C.D.; Viseu, T.; das Neves, J.; Lúcio, M. Rational Development of Liposomal Hydrogels: A Strategy for Topical Vaginal Antiretroviral Drug Delivery in the Context of HIV Prevention. Pharmaceutics 2019, 11, 485. https://doi.org/10.3390/pharmaceutics11090485
Faria MJ, Machado R, Ribeiro A, Gonçalves H, Real Oliveira MECD, Viseu T, das Neves J, Lúcio M. Rational Development of Liposomal Hydrogels: A Strategy for Topical Vaginal Antiretroviral Drug Delivery in the Context of HIV Prevention. Pharmaceutics. 2019; 11(9):485. https://doi.org/10.3390/pharmaceutics11090485
Chicago/Turabian StyleFaria, Maria J., Raul Machado, Artur Ribeiro, Hugo Gonçalves, Maria Elisabete C. D. Real Oliveira, Teresa Viseu, José das Neves, and Marlene Lúcio. 2019. "Rational Development of Liposomal Hydrogels: A Strategy for Topical Vaginal Antiretroviral Drug Delivery in the Context of HIV Prevention" Pharmaceutics 11, no. 9: 485. https://doi.org/10.3390/pharmaceutics11090485
APA StyleFaria, M. J., Machado, R., Ribeiro, A., Gonçalves, H., Real Oliveira, M. E. C. D., Viseu, T., das Neves, J., & Lúcio, M. (2019). Rational Development of Liposomal Hydrogels: A Strategy for Topical Vaginal Antiretroviral Drug Delivery in the Context of HIV Prevention. Pharmaceutics, 11(9), 485. https://doi.org/10.3390/pharmaceutics11090485