In Vitro Dissolution and in Silico Modeling Shortcuts in Bioequivalence Testing


Abstract
:1. Introduction
2. Search Methodology
3. Literature Reviewed
4. Similarity and Difference Factors
5. GastroPlus™
6. SimCyp®
7. NONMEM®
8. PK-Sim®
9. Other in Silico Platforms
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAFE | Absolute average fold error |
| ANDA | Abbreviated new drug application |
| API | Active pharmaceutical ingredient |
| ASF | Absorption scale factor |
| AUC | Area under drug concentration-time curve |
| BA | Bioavailability |
| BE | Bioequivalence |
| BCS | Biopharmaceutics Classification System |
| Cmax | Maximum plasma drug concentration |
| CV | Coefficient of variation |
| DDIs | Drug-drug interactions |
| EMA | European Medicines Agency |
| ER | Extended-release |
| EU | European Union |
| ƒ1 | Difference factor |
| ƒ2 | Similarity factor |
| FDA | Food and Drug Administration |
| GI | Gastrointestinal |
| GIT | Gastrointestinal tract |
| IR | Immediate release |
| IV | Intravenous |
| IVIVC | In vitro-in vivo correlation |
| MR | Modified release |
| MSD | Multivariate statistical distance |
| NBE | Nonbioequivalence |
| NDA | New drug application |
| NONMEM | Nonlinear mixed effects modeling |
| OrBiTo | Oral biopharmaceutics tool |
| PBPK | Physiologically based pharmacokinetic |
| PD | Pharmacodynamic |
| Peff | Effective permeability |
| PK | Pharmacokinetic |
| PPI | Proton pump inhibitor |
| PRISMA | Preferred Reporting Items for Systematic Reviews and Meta-Analyses |
| SSD | Steady-state concentrations |
| USP | United States Pharmacopeia |
| WHO | World Health Organization |
References
- Gatarić, B.; Parojčić, J. Application of data mining approach to identify drug subclasses based on solubility and permeability. Biopharm. Drug Dispos. 2019, 40, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Cardot, J.-M.; Garcia Arieta, A.; Paixao, P.; Tasevska, I.; Davit, B. Implementing the Biopharmaceutics Classification System in Drug Development: Reconciling Similarities, Differences, and Shared Challenges in the EMA and US-FDA-Recommended Approaches. AAPS J. 2016, 18, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Tsume, Y.; Mudie, D.M.; Langguth, P.; Amidon, G.E.; Amidon, G.L. The Biopharmaceutics Classification System: Subclasses for in vivo predictive dissolution (IPD) methodology and IVIVC. Eur. J. Pharm. Sci. 2014, 57, 152–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Tabakha, M.; Fahelelbom, K.; Obaid, D.E.E.; Sayed, S. Quality Attributes and In Vitro Bioequivalence of Different Brands of Amoxicillin Trihydrate Tablets. Pharmaceutics 2017, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Leuratti, C.; Loprete, L.; Rossini, M.; Frangione, V.; Rovati, S.; Radicioni, M. Pharmacokinetics and Safety of a Diclofenac Sodium 75 mg/1 mL Solution (Akis®/Dicloin®) Administered as a Single Intravenous Bolus Injection in Healthy Men and Women. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 681–689. [Google Scholar] [CrossRef] [Green Version]
- Davit, B.M.; Kanfer, I.; Tsang, Y.C.; Cardot, J.-M. BCS Biowaivers: Similarities and Differences Among EMA, FDA, and WHO Requirements. AAPS J. 2016, 18, 612–618. [Google Scholar] [CrossRef] [Green Version]
- Center for Drug Evaluation and Research (CDER). Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System: Guidance for Industry. Available online: https://www.fda.gov/media/70963/download (accessed on 12 October 2019).
- Islam, M.M.; Begum, M. Bootstrap confidence intervals for dissolution similarity factor f2. Biom. Biostat. Int. J. 2018, 7, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Shebley, M.; Sandhu, P.; Emami Riedmaier, A.; Jamei, M.; Narayanan, R.; Patel, A.; Peters, S.A.; Reddy, V.P.; Zheng, M.; de Zwart, L.; et al. Physiologically Based Pharmacokinetic Model Qualification and Reporting Procedures for Regulatory Submissions: A Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 88–110. [Google Scholar] [CrossRef]
- Peters, S.A.; Dolgos, H. Requirements to Establishing Confidence in Physiologically Based Pharmacokinetic (PBPK) Models and Overcoming Some of the Challenges to Meeting Them. Clin. Pharmacokinet. 2019, 58, 1355–1371. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Bartels, M.; Clark, A.; Erskine, T.; Auernhammer, T.; Bhhatarai, B.; Wilson, D.; Marty, S. Performance evaluation of the GastroPlusTM software tool for prediction of the toxicokinetic parameters of chemicals. SAR QSAR Environ. Res. 2018, 29, 875–893. [Google Scholar] [CrossRef]
- Wagner, C.; Pan, Y.; Hsu, V.; Hsu, V.; Sinha, V.; Zhao, P. Predicting the Effect of CYP3A Inducers on the Pharmacokinetics of Substrate Drugs Using Physiologically Based Pharmacokinetic (PBPK) Modeling: An Analysis of PBPK Submissions to the US FDA. Clin. Pharmacokinet. 2016, 55, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X. Bioequivalence: Modeling and Simulation. In FDA Bioequivalence Standards; Yu, L.X., Li, B.V., Eds.; AAPS Advances in the Pharmaceutical Sciences Series; Springer and American Association of Pharmaceutical Scientists: New York, NY, USA, 2014; pp. 395–417. [Google Scholar]
- Pidgeon, T.E.; Wellstead, G.; Sagoo, H.; Jafree, D.J.; Fowler, A.J.; Agha, R.A. An assessment of the compliance of systematic review articles published in craniofacial surgery with the PRISMA statement guidelines: A systematic review. J. Cranio-Maxillofac. Surg. 2016, 44, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Silva, N.E. Modeling and Simulation in Bioequivalence. Ph.D. Thesis, Universidade De Lisboa, Lisbon, Portugal, 2012. [Google Scholar]
- Xie, F.; Ji, S.; Cheng, Z. In vitro dissolution similarity factor (f2) and in vivo bioequivalence criteria, how and when do they match? Using a BCS class II drug as a simulation example. Eur. J. Pharm. Sci. 2015, 66, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Hasin, F. Comparative Bioequivalence Study of Different Brands of Valsartan Tablets Marketed in Bangladesh by Dissolution Modeling and Quality Control Tests. World J. Pharm. Pharm. Sci. 2017, 6, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.; Kesisoglou, F.; Dogterom, P. Application of absorption modeling to predict bioequivalence outcome of two batches of etoricoxib tablets. AAPS PharmSciTech 2015, 16, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Cuesta-Gragera, A.; Navarro-Fontestad, C.; Mangas-Sanjuan, V.; González-Álvarez, I.; García-Arieta, A.; Trocóniz, I.F.; Casabó, V.G.; Bermejo, M. Semi-physiologic model validation and bioequivalence trials simulation to select the best analyte for acetylsalicylic acid. Eur. J. Pharm. Sci. 2015, 74, 86–94. [Google Scholar] [CrossRef]
- Purohit, H.S.; Trasi, N.S.; Sun, D.D.; Chow, E.C.Y.; Wen, H.; Zhang, X.; Gao, Y.; Taylor, L.S. Investigating the Impact of Drug Crystallinity in Amorphous Tacrolimus Capsules on Pharmacokinetics and Bioequivalence Using Discriminatory In Vitro Dissolution Testing and Physiologically Based Pharmacokinetic Modeling and Simulation. J. Pharm. Sci. 2018, 107, 1330–1341. [Google Scholar] [CrossRef]
- Zhang, S.; Fang, M.; Zhang, Q.; Li, X.; Zhang, T. Evaluating the bioequivalence of metronidazole tablets and analyzing the effect of in vitro dissolution on in vivo absorption based on PBPK modeling. Drug Dev. Ind. Pharm. 2019, 45, 1646–1653. [Google Scholar] [CrossRef]
- Sperry, D.C.; Thomas, S.J.; Lobo, E. Dissolution modeling of bead formulations and predictions of bioequivalence for a highly soluble, highly permeable drug. Mol. Pharm. 2010, 7, 1450–1457. [Google Scholar] [CrossRef]
- Rebeka, J.; Jerneja, O.; Igor, L.; Boštjan, P.; Aleksander, B.; Simon, Ž.; Albin, K. PBPK Absorption Modeling of Food Effect and Bioequivalence in Fed State for Two Formulations with Crystalline and Amorphous Forms of BCS 2 Class Drug in Generic Drug Development. AAPS PharmSciTech 2019, 20, 59. [Google Scholar] [CrossRef]
- Duque, M.D.; Silva, D.A.; Issa, M.G.; Porta, V.; Löbenberg, R.; Ferraz, H.G. In silico prediction of plasma concentrations of fluconazole capsules with different dissolution profiles and bioequivalence study using population simulation. Pharmaceutics 2019, 11, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wen, H.; Fan, J.; Vince, B.; Li, T.; Gao, W.; Kinjo, M.; Brown, J.; Sun, W.; Jiang, W.; et al. Integrating In Vitro, Modeling, and In Vivo Approaches to Investigate Warfarin Bioequivalence. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 523–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.; Yang, H.; Fang, L.; Gonzalez-Sales, M.; Zhao, L.; Trame, M.N.; Lesko, L.; Schmidt, S. Physiologically Based Pharmacokinetic Modeling to Evaluate Formulation Factors Influencing Bioequivalence of Metoprolol Extended-Release Products. J. Clin. Pharmacol. 2019, 59, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Tsume, Y.; Amidon, G.L. The biowaiver extension for BCS class III drugs: The effect of dissolution rate on the bioequivalence of BCS class III immediate-release drugs predicted by computer simulation. Mol. Pharm. 2010, 7, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Pepin, X.J.H.; Flanagan, T.R.; Holt, D.J.; Eidelman, A.; Treacy, D.; Rowlings, C.E. Justification of Drug Product Dissolution Rate and Drug Substance Particle Size Specifications Based on Absorption PBPK Modeling for Lesinurad Immediate Release Tablets. Mol. Pharm. 2016, 13, 3256–3269. [Google Scholar] [CrossRef]
- Vaidhyanathan, S.; Wang, X.; Crison, J.; Varia, S.; Gao, J.Z.H.; Saxena, A.; Good, D. Bioequivalence Comparison of Pediatric Dasatinib Formulations and Elucidation of Absorption Mechanisms Through Integrated PBPK Modeling. J. Pharm. Sci. 2019, 108, 741–749. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Duan, J.; Kesisoglou, F.; Novakovic, J.; Amidon, G.L.; Jamei, M.; Lukacova, V.; Eissing, T.; Tsakalozou, E.; Zhao, L.; et al. Mechanistic oral absorption modeling and simulation for formulation development and bioequivalence evaluation: Report of an FDA public workshop. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 492–495. [Google Scholar] [CrossRef] [Green Version]
- Doki, K.; Darwich, A.S.; Patel, N.; Rostami-Hodjegan, A. Virtual bioequivalence for achlorhydric subjects: The use of PBPK modelling to assess the formulation-dependent effect of achlorhydria. Eur. J. Pharm. Sci. 2017, 109, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Zhang, X.; Zhao, L. Utility of Physiologically Based Pharmacokinetic Absorption Modeling to Predict the Impact of Salt-to-Base Conversion on Prasugrel HCl Product Bioequivalence in the Presence of Proton Pump Inhibitors. AAPS J. 2017, 19, 1479–1486. [Google Scholar] [CrossRef]
- Zhao, L.; Li, Q.; Li, X.; Yin, R.; Chen, X.; Geng, L.; Bi, K. Bioequivalence and population pharmacokinetic modeling of two forms of antibiotic, cefuroxime lysine and cefuroxime sodium, after intravenous infusion in beagle dogs. J. Biomed. Biotechnol. 2012, 2012, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chitnis, S.D.; Han, Y.; Yamaguchi, M.; Mita, S.; Zhao, R.; Sunkara, G.; Kulmatycki, K. Population pharmacokinetic modeling and noncompartmental analysis demonstrated bioequivalence between metformin component of metformin/vildagliptin fixed-dose combination products and metformin immediate-release tablet sourced from various countries. Clin. Pharmacol. Drug Dev. 2016, 5, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Fang, L.; Yu, J.; Meng, Z.; Trame, M.N.; Schmidt, S.; Lesko, L.J.; Zhao, L. Is Bioequivalence Established Based on the Reference-Scaled Average Bioequivalence Approach Relevant to Chronic Administration of Phenytoin? Perspectives Based on Population Pharmacokinetic Modeling and Simulations. J. Clin. Pharmacol. 2019, 59, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Simionato, L.D.; Petrone, L.; Baldut, M.; Bonafede, S.L.; Segall, A.I. Comparison between the dissolution profiles of nine meloxicam tablet brands commercially available in Buenos Aires, Argentina. Saudi Pharm. J. 2018, 26, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Dennison, T.J.; Smith, J.C.; Badhan, R.K.; Mohammed, A.R. Fixed-dose combination orally disintegrating tablets to treat cardiovascular disease: Formulation, in vitro characterization and physiologically based pharmacokinetic modeling to assess bioavailability. Drug Des. Dev. Ther. 2017, 11, 811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristofoletti, R.; Chiann, C.; Dressman, J.B.; Storpirtis, S. A comparative analysis of biopharmaceutics classification system and biopharmaceutics drug disposition classification system: A cross-sectional survey with 500 bioequivalence studies. J. Pharm. Sci. 2013, 102, 3136–3144. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, E.; Laosa, O.; Guerra, P.; Duque, B.; Mosquera, B.; Borobia, A.M.; Lei, S.H.; Carcas, A.J.; Frias, J. Acceptability and characteristics of 124 human bioequivalence studies with active substances classified according to the Biopharmaceutic Classification System. Br. J. Clin. Pharmacol. 2010, 70, 694–702. [Google Scholar] [CrossRef] [Green Version]
- Paixão, P.; Gouveia, L.F.; Silva, N.; Morais, J.A.G. Evaluation of dissolution profile similarity—Comparison between the f2, the multivariate statistical distance and the f2 bootstrapping methods. Eur. J. Pharm. Biopharm. 2017, 112, 67–74. [Google Scholar] [CrossRef]
- Cardot, J.M.; Roudier, B.; Schütz, H. Dissolution comparisons using a Multivariate Statistical Distance (MSD) test and a comparison of various approaches for calculating the measurements of dissolution profile comparison. AAPS J. 2017, 19, 1091–1101. [Google Scholar] [CrossRef]
- Kuemmel, C.; Yang, Y.; Zhang, X.; Florian, J.; Zhu, H.; Tegenge, M.; Huang, S.M.; Wang, Y.; Morrison, T.; Zineh, I. Consideration of a Credibility Assessment Framework in Model-Informed Drug Development: Potential Application to Physiologically-Based Pharmacokinetic Modeling and Simulation. CPT Pharmacomet. Syst. Pharmacol. 2019. Available online: https://ascpt.onlinelibrary.wiley.com/doi/abs/10.1002/psp4.12479 (accessed on 25 October 2019). [CrossRef] [Green Version]
- Li, M.; Zhao, P.; Pan, Y.; Wagner, C. Predictive Performance of Physiologically Based Pharmacokinetic Models for the Effect of Food on Oral Drug Absorption: Current Status. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 82–89. [Google Scholar] [CrossRef]
- Mitra, A.; Petek, B.; Bajc, A.; Velagapudi, R.; Legen, I. Physiologically based absorption modeling to predict bioequivalence of controlled release and immediate release oral products. Eur. J. Pharm. Biopharm. 2019, 134, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Honório, T.; Pinto, E.C.; Rocha, H.V.A.; Esteves, V.S.D.; dos Santos, T.C.; Castro, H.C.R.; Rodrigues, C.R.; de Sousa, V.P.; Cabral, L.M. In Vitro–In Vivo Correlation of Efavirenz Tablets Using GastroPlus®. AAPS PharmSciTech 2013, 14, 1244–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, A.; Shakeel, F.; Singh, S.K.; Alsarra, I.A.; Faruk, A.; Alanazi, F.K.; Peter Christoper, G.V. Solidified SNEDDS for the oral delivery of rifampicin: Evaluation, proof of concept, in vivo kinetics, and in silico GastroPlusTM simulation. Int. J. Pharm. 2019, 566, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Kovačević, I.; Parojčić, J.; Homšek, I.; Tubić-Grozdanis, M.; Langguth, P. Justification of Biowaiver for Carbamazepine, a Low Soluble High Permeable Compound, in Solid Dosage Forms Based on IVIVC and Gastrointestinal Simulation. Mol. Pharm. 2009, 6, 40–47. [Google Scholar] [CrossRef]
- Pepin, X.J.H.; Moir, A.J.; Mann, J.C.; Sanderson, N.J.; Barker, R.; Meehan, E.; Plumb, A.P.; Bailey, G.R.; Murphy, D.S.; Krejsa, C.M.; et al. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part II. A mechanistic PBPK model for IR formulation comparison, proton pump inhibitor drug interactions, and administration with acidic juices. Eur. J. Pharm. Biopharm. 2019, 142, 435–448. [Google Scholar] [CrossRef]
- Kesisoglou, F.; Xia, B.; Agrawal, N.G.B. Comparison of Deconvolution-Based and Absorption Modeling IVIVC for Extended Release Formulations of a BCS III Drug Development Candidate. AAPS J. 2015, 17, 1492–1500. [Google Scholar] [CrossRef] [Green Version]
- Marsousi, N.; Desmeules, J.A.; Rudaz, S.; Daali, Y. Prediction of drug-drug interactions using physiologically-based pharmacokinetic models of CYP450 modulators included in Simcyp software. Biopharm. Drug Dispos. 2018, 39, 3–17. [Google Scholar] [CrossRef]
- Gidal, B.E.; Maganti, R.; Laurenza, A.; Yang, H.; Verbel, D.A.; Schuck, E.; Ferry, J. Effect of enzyme inhibition on perampanel pharmacokinetics: Why study design matters. Epilepsy Res. 2017, 134, 41–48. [Google Scholar] [CrossRef]
- Costello, C.; Rossenu, S.; Vermeulen, A.; Cleton, A.; Dunne, A. A time scaling approach to develop an in vitro-in vivo correlation (IVIVC) model using a convolution-based technique. J. Pharmacokinet. Pharmacodyn. 2011, 38, 519–539. [Google Scholar] [CrossRef]
- Idkaidek, N.; Arafat, T. Saliva versus plasma bioequivalence of rusovastatin in humans: Validation of class III drugs of the salivary excretion classification system. Drugs R & D 2015, 15, 79–83. [Google Scholar]
- Chetty, M.; Johnson, T.N.; Polak, S.; Salem, F.; Doki, K.; Rostami-Hodjegan, A. Physiologically based pharmacokinetic modelling to guide drug delivery in older people. Adv. Drug Deliv. Rev. 2018, 135, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Sjögren, E.; Thörn, H.; Tannergren, C. In Silico Modeling of Gastrointestinal Drug Absorption: Predictive Performance of Three Physiologically Based Absorption Models. Mol. Pharm. 2016, 13, 1763–1778. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.B.; Liu, B.; Patel, N.; Pathak, S.M.; Polak, S.; Jamei, M.; Dressman, J.; Rostami-Hodjegan, A. Comment on in Silico Modeling of Gastrointestinal Drug Absorption: Predictive Performance of Three Physiologically-Based Absorption Models. Mol. Pharm. 2017, 14, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Välitalo, P.; Ranta, V.-P.; Hooker, A.C.; Kokki, M.; Kokki, H. Population pharmacometrics in support of analgesics studies. Acta Anaesthesiol. Scand. 2014, 58, 143–156. [Google Scholar] [CrossRef]
- Fairman, K.; Chaturvedula, A.; Srinivasan, M. Evaluation of the Sensitivity of Concluding Bioequivalence Using Stochastic Simulation and Estimation. Available online: https://hdl.handle.net/20.500.12503/27885 (accessed on 3 September 2019).
- Choi, S.; Jeon, S.; Han, S. Population pharmacokinetic analysis of metformin administered as fixed-dose combination in Korean healthy adults. Transl. Clin. Pharmacol. 2018, 26, 25. [Google Scholar] [CrossRef] [Green Version]
- Hsu, L. Investigation of the Discriminatory Ability of Pharmacokinetic Metrics for the Bioequivalence Assessment of PEGylated Liposomal Doxorubicin. Pharm. Res. 2018, 35, 106. [Google Scholar] [CrossRef]
- Mangas-Sanjuan, V.; Navarro-Fontestad, C.; García-Arieta, A.; Trocóniz, I.F.; Bermejo, M. Computer simulations for bioequivalence trials: Selection of analyte in BCS class II and IV drugs with first-pass metabolism, two metabolic pathways and intestinal efflux transporter. Eur. J. Pharm. Sci. 2018, 117, 193–203. [Google Scholar] [CrossRef]
- Kim, S.; Sharma, V.D.; Lingineni, K.; Farhan, N.; Fang, L.; Zhao, L.; Brown, J.D.; Cristofoletti, R.; Vozmediano, V.; Ait-Oudhia, S.; et al. Evaluating the Clinical Impact of Formulation Variability: A Metoprolol Extended-Release Case Study. J. Clin. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Gaynor, C.; Dunne, A.; Costello, C.; Davis, J. A population approach to in vitro-in vivo correlation modelling for compounds with nonlinear kinetics. J. Pharmacokinet. Pharmacodyn. 2011, 38, 317–332. [Google Scholar] [CrossRef]
- Ibarra, M.; Valiante, C.; Sopeña, P.; Schiavo, A.; Lorier, M.; Vázquez, M.; Fagiolino, P. Integration of in vitro biorelevant dissolution and in silico PBPK model of carvedilol to predict bioequivalence of oral drug products. Eur. J. Pharm. Sci. 2018, 118, 176–182. [Google Scholar] [CrossRef]
- Idkaidek, N.; Arafat, T.; Hamadi, H.; Hamadi, S.; Al-Adham, I. Saliva Versus Plasma Bioequivalence of Azithromycin in Humans: Validation of Class I Drugs of the Salivary Excretion Classification System. Drugs R & D 2017, 17, 219–224. [Google Scholar]
- Galgatte, U.C.; Jamdade, V.R.; Aute, P.P.; Chaudhari, P.D. Study on requirements of bioequivalence for registration of pharmaceutical products in USA, Europe and Canada. Saudi Pharm. J. 2014, 22, 391–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolskee, A.; Darwich, A.S.; Galetin, A.; Rostami-Hodjegan, A.; Aarons, L. Deconvolution and IVIVC: Exploring the Role of Rate-Limiting Conditions. AAPS J. 2016, 18, 321–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsamandouras, N.; Dickinson, G.; Guo, Y.; Hall, S.; Rostami-Hodjegan, A.; Galetin, A.; Aarons, L. Development and application of a mechanistic pharmacokinetic model for simvastatin and its active metabolite simvastatin acid using an integrated population PBPK approach. Pharm. Res. 2015, 32, 1864–1883. [Google Scholar] [CrossRef] [PubMed]
- Sjögren, E.; Westergren, J.; Grant, I.; Hanisch, G.; Lindfors, L.; Lennernäs, H.; Abrahamsson, B.; Tannergren, C. In silico predictions of gastrointestinal drug absorption in pharmaceutical product development: Application of the mechanistic absorption model GI-Sim. Eur. J. Pharm. Sci. 2013, 49, 679–698. [Google Scholar] [CrossRef] [PubMed]
- Bouzom, F.; Ball, K.; Perdaems, N.; Walther, B. Physiologically based pharmacokinetic (PBPK) modelling tools: How to fit with our needs? Biopharm. Drug Dispos. 2012, 33, 55–71. [Google Scholar] [CrossRef]
- MacPherson, M.; Burgess, J.; McMillan, B.; Daviau, T.; Tipparaju, S.M. Artificial Neural Networks in Drug Transport Modeling and Simulation-I. In Artificial Neural Network for Drug Design, Delivery and Disposition; Elsevier Inc.: Amsterdam, The Netherlands, 2016; pp. 221–241. ISBN 9780128015599. [Google Scholar]
- Jones, H.M.; Dickins, M.; Youdim, K.; Gosset, J.R.; Attkins, N.J.; Hay, T.L.; Gurrell, I.K.; Logan, Y.R.; Bungay, P.J.; Jones, B.C.; et al. Application of PBPK modelling in drug discovery and development at Pfizer. Xenobiotica 2012, 42, 94–106. [Google Scholar] [CrossRef]
- Zhao, L.; Seo, P.; Lionberger, R. Current Scientific Considerations to Verify Physiologically-Based Pharmacokinetic Models and Their Implications for Locally Acting Products. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 347–351. [Google Scholar] [CrossRef] [Green Version]
- Andreas, C.J.; Rosenberger, J.; Butler, J.; Augustijns, P.; McAllister, M.; Abrahamsson, B.; Dressman, J. Introduction to the OrBiTo decision tree to select the most appropriate in vitro methodology for release testing of solid oral dosage forms during development. Eur. J. Pharm. Biopharm. 2018, 130, 207–213. [Google Scholar] [CrossRef]
- Zhang, L. FY2018 Generic Drug Regulatory Science Initiatives Public Workshop: FDA Research Update on the FY18 Initiatives. Available online: https://www.fda.gov/media/113597/download+&cd=6&hl=en&ct=clnk&gl=ae (accessed on 6 October 2019).





{kind=link}
{kind=link}
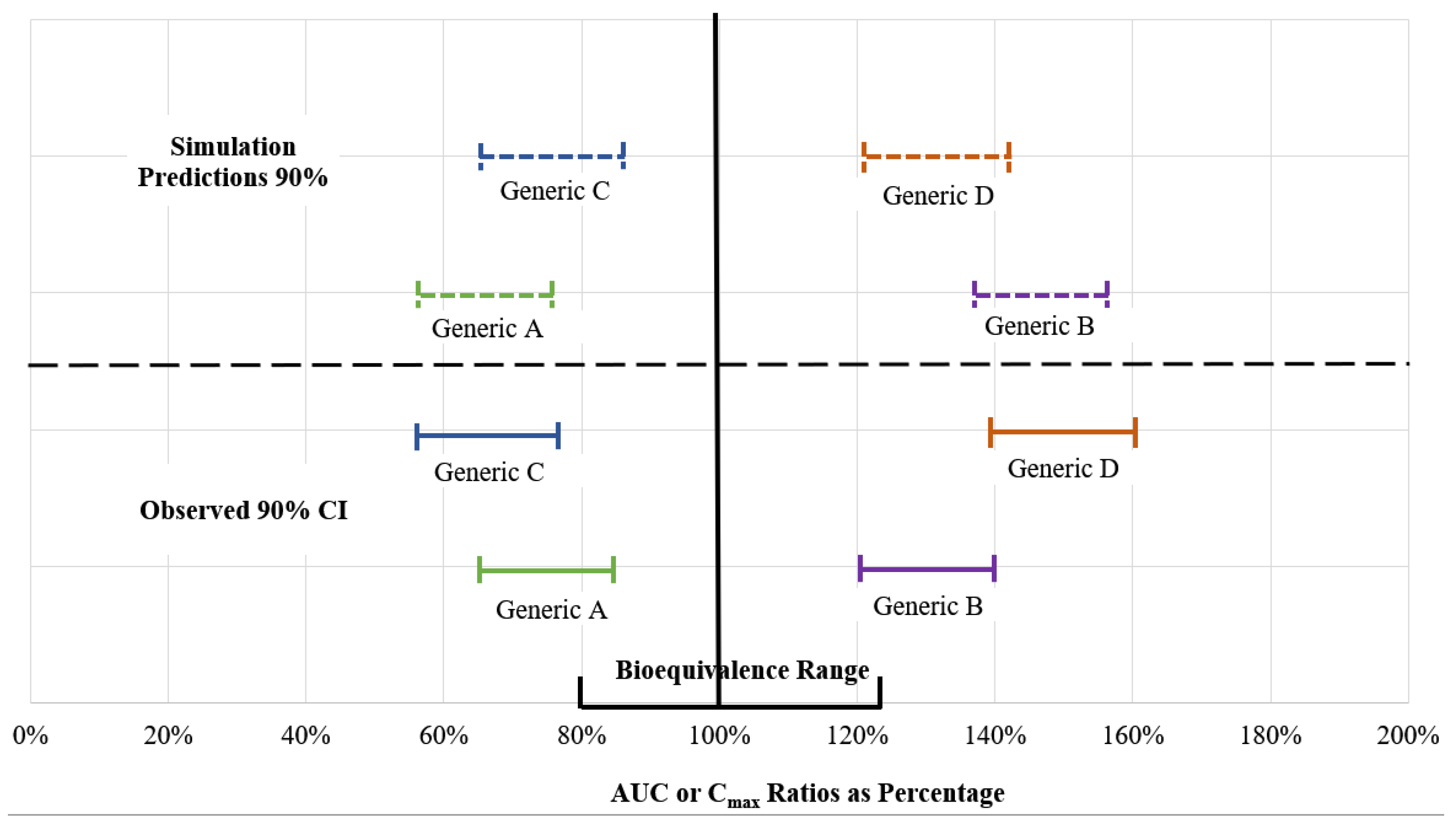{kind=link}
{kind=link}
| In Vitro/In Silico | Number of Times Used in the Reviewed Literature |
|---|---|
| Similarity/Difference factors (ƒ1/ƒ2) | 5 [15,16,17,18,19] |
| GastroPlus™ | 12 [13,18,20,21,22,23,24,25,26,27,28,29] |
| SimCyp® | 6 [13,15,23,30,31,32] |
| NONMEM® | 4 [19,33,34,35] |
| PK-Sim® | 2 [13,30] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Tabakha, M.M.; Alomar, M.J. In Vitro Dissolution and in Silico Modeling Shortcuts in Bioequivalence Testing. Pharmaceutics 2020, 12, 45. https://doi.org/10.3390/pharmaceutics12010045
Al-Tabakha MM, Alomar MJ. In Vitro Dissolution and in Silico Modeling Shortcuts in Bioequivalence Testing. Pharmaceutics. 2020; 12(1):45. https://doi.org/10.3390/pharmaceutics12010045
Chicago/Turabian StyleAl-Tabakha, Moawia M., and Muaed J. Alomar. 2020. "In Vitro Dissolution and in Silico Modeling Shortcuts in Bioequivalence Testing" Pharmaceutics 12, no. 1: 45. https://doi.org/10.3390/pharmaceutics12010045
APA StyleAl-Tabakha, M. M., & Alomar, M. J. (2020). In Vitro Dissolution and in Silico Modeling Shortcuts in Bioequivalence Testing. Pharmaceutics, 12(1), 45. https://doi.org/10.3390/pharmaceutics12010045