Insight into the Formation of Glimepiride Nanocrystals by Wet Media Milling



 ,
,  and
and 
Abstract
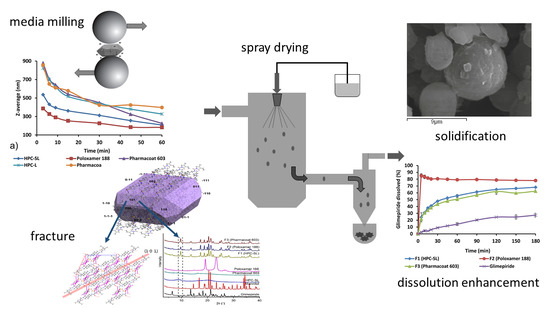
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of Glimepiride Nanosuspensions
2.2.2. Particle Size Measurements
2.2.3. Spray Drying of Nanosuspensions
2.2.4. Characterization of the Solidified Nanosuspensions
Redispersibility Testing
Differential Scanning Calorimetry (DSC)
Attenuated Total Reflectance Fourier Transform Infrared (ATR-FTIR) Spectroscopy
Powder X-ray Diffraction Analysis (PXRD)
Scanning Electron Microscopy (SEM)
In Vitro Dissolution Testing
2.2.5. Computational Study of Glimepiride’s Crystal Properties
Lattice Energy Frameworks
Crystal Morphology
3. Results
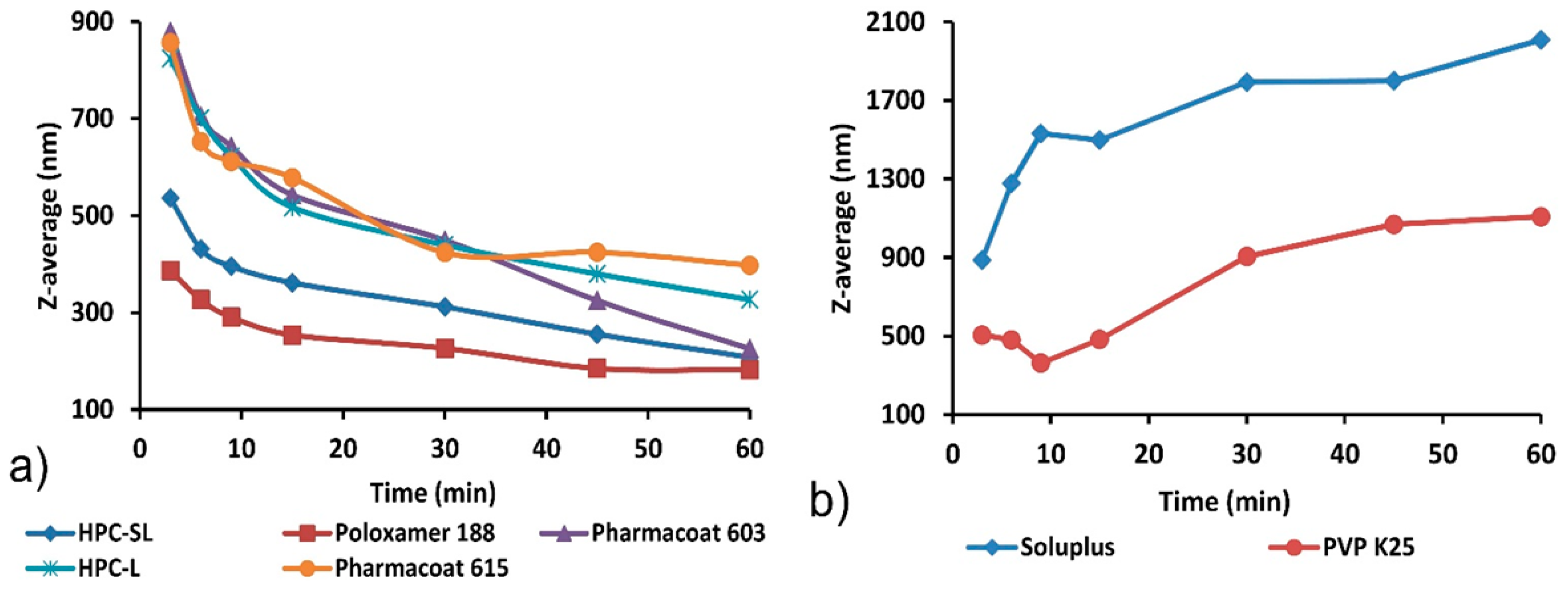
3.1. Wet Media Milling
3.2. Redispersibility Testing
3.3. Characterization of the Solidified Nanosuspensions
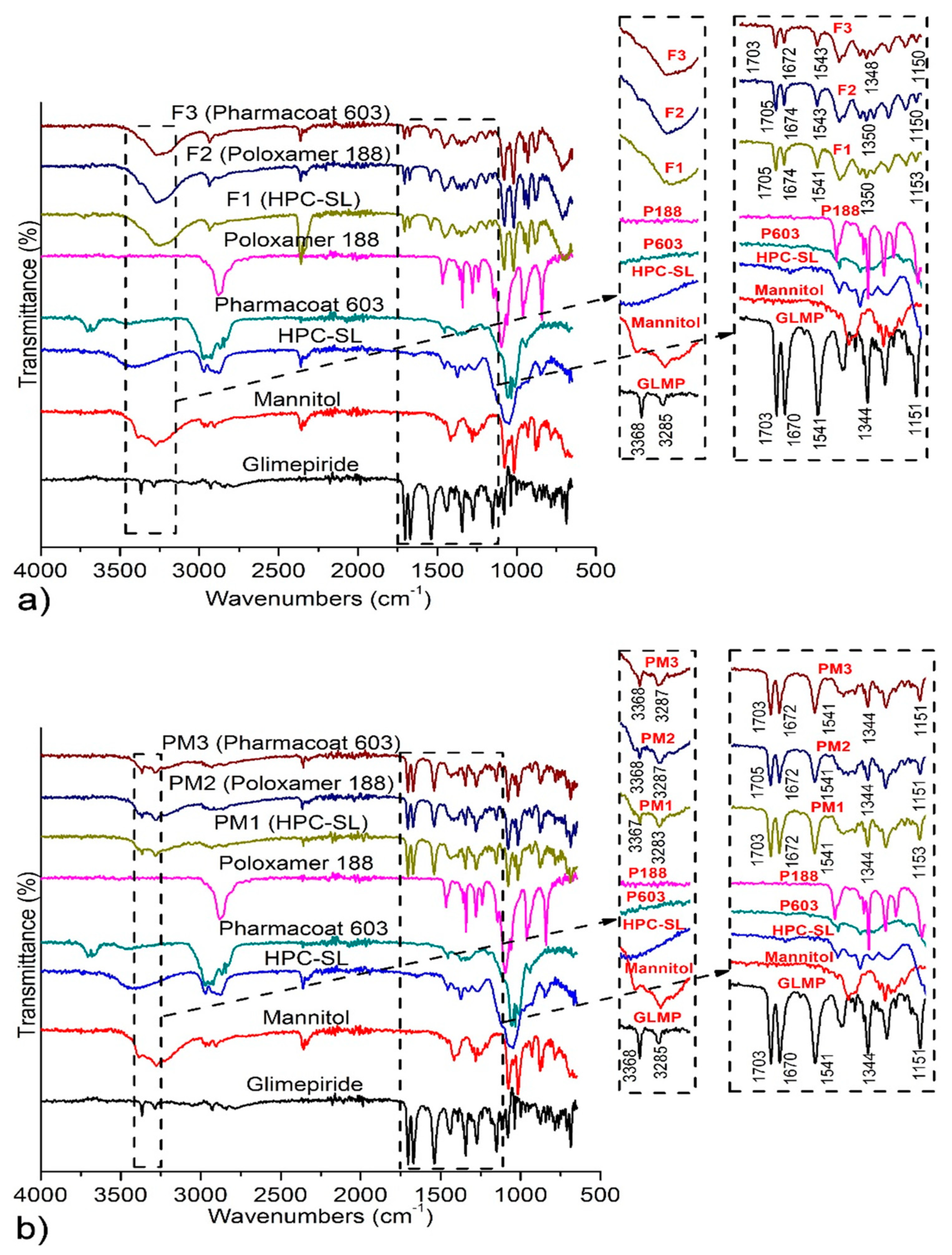
3.3.1. Attenuated Total Reflectance Fourier Transform Infrared (ATR-FTIR) Spectroscopy
3.3.2. Differential Scanning Calorimetry (DSC)
3.3.3. Powder X-ray Diffraction Analysis (PXRD)
3.3.4. Lattice Properties and Morphology Calculations
3.3.5. Scanning Electron Microscopy (SEM)
3.3.6. In Vitro Dissolution Testing
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ku, M.S.; Dulin, W. A biopharmaceutical classification-based Right-First-Time formulation approach to reduce human pharmacokinetic variability and project cycle time from First-In-Human to clinical Proof-Of-Concept. Pharm. Dev. Technol. 2012, 17, 285–302. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef]
- Vo, C.L.N.; Park, C.; Lee, B.J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef]
- Ahmed, T.A.; El-Say, K.M.; Aljaeid, B.M.; Fahmy, U.A.; Abd-Allah, F.I. Transdermal glimepiride delivery system based on optimized ethosomal nano-vesicles: Preparation, characterization, in vitro, ex vivo and clinical evaluation. Int. J. Pharm. 2016, 500, 245–254. [Google Scholar] [CrossRef]
- Kurkov, S.V.; Loftsson, T. Cyclodextrins. Int. J. Pharm. 2013, 453, 167–180. [Google Scholar] [CrossRef]
- Iwata, M.; Fukami, T.; Kawashima, D.; Sakai, M.; Furuishi, T.; Suzuki, T.; Tomono, K.; Ueda, H. Effectiveness of mechanochemical treatment with cyclodextrins on increasing solubility of glimepiride. Pharmazie 2009, 64, 390–394. [Google Scholar]
- Leleux, J.; Williams, R.O. Recent advancements in mechanical reduction methods: Particulate systems. Drug Dev. Ind. Pharm. 2014, 40, 289–300. [Google Scholar] [CrossRef]
- Gao, L.; Liu, G.; Ma, J.; Wang, X.; Zhou, L.; Li, X.; Wang, F. Application of drug nanocrystal technologies on oral drug delivery of poorly soluble drugs. Pharm. Res. 2013, 30, 307–324. [Google Scholar] [CrossRef]
- Müller, R.H.; Gohla, S.; Keck, C.M. State of the art of nanocrystals—Special features, production, nanotoxicology aspects and intracellular delivery. Eur. J. Pharm. Biopharm. 2011, 78, 1–9. [Google Scholar] [CrossRef]
- Gigliobianco, M.R.; Casadidio, C.; Censi, R.; Di Martino, P. Nanocrystals of poorly soluble drugs: Drug bioavailability and physicochemical stability. Pharmaceutics 2018, 10, 134. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Fang, Q.; Niu, B.; Wu, B.; Zhao, Y.; Quan, G.; Pan, X.; Wu, C. Comparative studies on amphotericin B nanosuspensions prepared by a high pressure homogenization method and an antisolvent precipitation method. Colloids Surf. B Biointerfaces 2018, 172, 372–379. [Google Scholar] [CrossRef]
- Möschwitzer, J.P. Drug nanocrystals in the commercial pharmaceutical development process. Int. J. Pharm. 2013, 453, 142–156. [Google Scholar] [CrossRef]
- Merisko-Liversidge, E.; Liversidge, G.G. Nanosizing for oral and parenteral drug delivery: A perspective on formulating poorly-water soluble compounds using wet media milling technology. Adv. Drug Deliv. Rev. 2011, 63, 427–440. [Google Scholar] [CrossRef]
- Roßkamp, R.; Wernicke-Panten, K.; Draeger, E. Clinical profile of the novel sulphonylurea glimepiride. Diabetes Res. Clin. Pract. 1996, 31, S33–S42. [Google Scholar] [CrossRef]
- Frick, A.; Möller, H.; Wirbitzki, E. Biopharmaceutical characterization of oral immediate release drug products. In vitro/in vivo comparison of phenoxymethylpenicillin potassium, glimepiride and levofloxacin. Eur. J. Pharm. Biopharm. 1998, 46, 305–311. [Google Scholar] [CrossRef]
- Matsuki, M.; Matsuda, M.; Kohara, K.; Shimoda, M.; Kanda, Y.; Tawaramoto, K.; Shigetoh, M.; Kawasaki, F.; Kotani, K.; Kaku, K. Pharmacokinetics and pharmacodynamics of glimepiride in type 2 diabetic patients: Compared effects of once- versus twice-daily dosing. Endocr. J. 2007, 54, 571–576. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Ma, L.; Li, X.; Cui, X.; Yang, W.; Shen, S.; Chen, M. A simple and effective method to improve bioavailability of glimepiride by utilizing hydrotropy technique. Eur. J. Pharm. Sci. 2015, 77, 154–160. [Google Scholar] [CrossRef]
- Seedher, N.; Kanojia, M. Co-solvent solubilization of some poorly-soluble antidiabetic drugs Solubilization antidiabetic drugs. Pharm. Dev. Technol. 2009, 14, 185–192. [Google Scholar] [CrossRef]
- Mohd, A.B.; Sanka, K.; Bandi, S.; Diwan, P.V.; Shastri, N. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) for oral delivery of glimepiride: Development and antidiabetic activity in albino rabbits. Drug Deliv. 2015, 22, 499–508. [Google Scholar] [CrossRef]
- Ning, X.; Sun, J.; Han, X.; Wu, Y.; Yan, Z.; Han, J.; He, Z. Strategies to improve dissolution and oral absorption of glimepiride tablets: Solid dispersion versus micronization techniques. Drug Dev. Ind. Pharm. 2011, 37, 727–736. [Google Scholar] [CrossRef]
- Mohamed, E.A.; Meshali, M.M.; Foda, A.M.M.; Borg, T.M. Improvement of dissolution and hypoglycemic efficacy of glimepiride by different carriers. AAPS PharmSciTech 2012, 13, 1013–1023. [Google Scholar] [CrossRef] [Green Version]
- Reginald-Opara, J.N.; Attama, A.; Ofokansi, K.; Umeyor, C.; Kenechukwu, F. Molecular interaction between glimepiride and Soluplus®-PEG 4000 hybrid based solid dispersions: Characterisation and anti-diabetic studies. Int. J. Pharm. 2015, 496, 741–750. [Google Scholar] [CrossRef]
- Pahovnik, D.; Reven, S.; Grdadolnik, J.; Borstnar, R.; Mavri, J.; Zagar, E. Determination of the Interaction Between Glimepiride and Hyperbranched Polymers in Solid Dispersions. J. Pharm. Sci. 2013, 100, 4700–4709. [Google Scholar] [CrossRef]
- Ilić, I.; Dreu, R.; Burjak, M.; Homar, M.; Kerč, J.; Srčič, S. Microparticle size control and glimepiride microencapsulation using spray congealing technology. Int. J. Pharm. 2009, 381, 176–183. [Google Scholar] [CrossRef]
- Yadav, S.K.; Mishra, S.; Mishra, B. Eudragit-based nanosuspension of poorly water-soluble drug: Formulation and in vitro-in vivo evaluation. AAPS PharmSciTech 2012, 13, 1031–1044. [Google Scholar] [CrossRef] [Green Version]
- Du, B.; Shen, G.; Wang, D.; Pang, L.; Chen, Z.; Liu, Z. Development and characterization of glimepiride nanocrystal formulation and evaluation of its pharmacokinetic in rats. Drug Deliv. 2013, 20, 25–33. [Google Scholar] [CrossRef]
- Lee, J.; Choi, J.-Y.; Park, C.H. Characteristics of polymers enabling nano-comminution of water-insoluble drugs. Int. J. Pharm. 2008, 355, 328–336. [Google Scholar] [CrossRef]
- Yue, P.F.; Li, Y.; Wan, J.; Yang, M.; Zhu, W.F.; Wang, C.H. Study on formability of solid nanosuspensions during nanodispersion and solidification: I. Novel role of stabilizer/drug property. Int. J. Pharm. 2013, 454, 269–277. [Google Scholar] [CrossRef]
- Shishkin, O.V.; Dyakonenko, V.V.; Maleev, A.V. Supramolecular architecture of crystals of fused hydrocarbons based on topology of intermolecular interactions. CrystEngComm 2012, 14, 1795–1804. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Shishkin, O.V.; Medvediev, V.V.; Zubatyuk, R.I. Supramolecular architecture of molecular crystals possessing shearing mechanical properties: Columns versus layers. CrystEngComm 2013, 15, 160–167. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Van De Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- McArdle, P.; Hu, Y.; Lyons, A.; Dark, R. Predicting and understanding crystal morphology: The morphology of benzoic acid and the polymorphs of sulfathiazole. CrystEngComm 2010, 12, 3119–3125. [Google Scholar] [CrossRef]
- Lifson, S.; Hagler, A.T.; Dauber, P. Consistent Force Field Studies of Intermolecular Forces in Hydrogen-Bonded Crystals. 1. Carboxylic Acids, Amides and the C=O…H-Hydrogen Bonds. J. Am. Chem. Soc. 1979, 101, 5111–5121. [Google Scholar] [CrossRef]
- Rappé, A.K.; Goddard, W.A. Charge equilibration for molecular dynamics simulations. J. Phys. Chem. 1991, 95, 3358–3363. [Google Scholar] [CrossRef]
- Gale, J.D.; Rohl, A.L. The General Utility Lattice Program (GULP). Mol. Simul. 2003, 29, 291–341. [Google Scholar] [CrossRef]
- Patel, G.V.; Patel, V.B.; Pathak, A.; Rajput, S.J. Nanosuspension of efavirenz for improved oral bioavailability: Formulation optimization, in vitro, in situ and in vivo evaluation. Drug Dev. Ind. Pharm. 2014, 40, 80–91. [Google Scholar] [CrossRef]
- Knieke, C.; Azad, M.A.; Davé, R.N.; Bilgili, E. A study of the physical stability of wet media-milled fenofibrate suspensions using dynamic equilibrium curves. Chem. Eng. Res. Des. 2013, 91, 1245–1258. [Google Scholar] [CrossRef]
- Medarević, D.; Djuriš, J.; Ibrić, S.; Mitrić, M.; Kachrimanis, K. Optimization of formulation and process parameters for the production of carvedilol nanosuspension by wet media milling. Int. J. Pharm. 2018, 540, 150–161. [Google Scholar] [CrossRef]
- Verma, S.; Kumar, S.; Gokhale, R.; Burgess, D.J. Physical stability of nanosuspensions: Investigation of the role of stabilizers on Ostwald ripening. Int. J. Pharm. 2011, 406, 145–152. [Google Scholar] [CrossRef]
- Leone, F.; Cavalli, R. Drug nanosuspensions: A ZIP tool between traditional and innovative pharmaceutical formulations. Expert Opin. Drug Deliv. 2015, 12, 1607–1625. [Google Scholar] [CrossRef] [PubMed]
- Malamatari, M.; Somavarapu, S.; Taylor, K.M.G.; Buckton, G. Solidification of nanosuspensions for the production of solid oral dosage forms and inhalable dry powders. Expert Opin. Drug Deliv. 2016, 5247, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Bhakay, A.; Azad, M.; Bilgili, E.; Dave, R. Redispersible fast dissolving nanocomposite microparticles of poorly water-soluble drugs. Int. J. Pharm. 2014, 461, 367–379. [Google Scholar] [CrossRef]
- Reven, S.; Grdadolnik, J.; Kristl, J.; Žagar, E. Hyperbranched poly(esteramides) as solubility enhancers for poorly water-soluble drug glimepiride. Int. J. Pharm. 2010, 396, 119–126. [Google Scholar] [CrossRef]
- Bonfilio, R.; Pires, S.A.; Ferreira, L.M.B.; De Almeida, A.E.; Doriguetto, A.C.; De Araújo, M.B.; Salgado, H.R.N. A Discriminating Dissolution Method for Glimepiride Polymorphs. J. Pharm. Sci. 2012, 101, 794–804. [Google Scholar] [CrossRef]
- Remko, M. Theoretical study of molecular structure, pKa, lipophilicity, solubility, absorption, and polar surface area of some hypoglycemic agents. J. Mol. Struct. THEOCHEM 2009, 897, 73–82. [Google Scholar] [CrossRef]
- Jackson, C.L.; McKenna, G.B. The melting behavior of organic materials confined in porous solids. J. Chem. Phys. 1990, 93, 9002–9011. [Google Scholar] [CrossRef]
- Kadam, S.M.; Tarur, R.; Naik, S.J.; Gavhane, S.B. Process for Preparation of Substantially Pure Glimepiride. U.S. Patent Application No. 11/156,343, 12 April 2007. [Google Scholar]
- York, P.; Ticehurst, M.D.; Osborn, J.C.; Roberts, R.J.; Rowe, R.C. Characterisation of the surface energetics of milled dl-propranolol hydrochloride using inverse gas chromatography and molecular modelling. Int. J. Pharm. 1998, 174, 179–186. [Google Scholar] [CrossRef]
- Heng, J.Y.Y.; Thielmann, F.; Williams, D.R. The effects of milling on the surface properties of form I paracetamol crystals. Pharm. Res. 2006, 23, 1918–1927. [Google Scholar] [CrossRef]
- Peltonen, L.; Hirvonen, J. Pharmaceutical nanocrystals by nanomilling: Critical process parameters, particle fracturing and stabilization methods. J. Pharm. Pharmacol. 2010, 62, 1569–1579. [Google Scholar] [CrossRef]
- Wang, C.; Sun, C.C. Identifying Slip Planes in Organic Polymorphs by Combined Energy Framework Calculations and Topology Analysis. Cryst. Growth Des. 2018, 18, 1909–1916. [Google Scholar] [CrossRef]
- Vehring, R. Pharmaceutical particle engineering via spray drying. Pharm. Res. 2008, 25, 999–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stabilizer | Particle Size (nm) | PDI | Particle Size (nm) after 7 Days | PDI after 7 Days |
|---|---|---|---|---|
| HPC SL® | 207.7 ± 1.8 | 0.213 ± 0.007 | 183.3 ± 6.6 | 0.254 ± 0.025 |
| Poloxamer 188 | 182.3 ± 11.8 | 0.181 ± 0.055 | 181.7 ± 3.5 | 0.196 ± 0.007 |
| Pharmacoat® 603 | 225.7 ± 11.7 | 0.209 ± 0.035 | 238.7 ± 5.7 | 0.248 ± 0.007 |
| HPC L | 326.7 ± 8.8 | 0.169 ± 0.020 | 311.3 ± 30.9 | 0.266 ± 0.039 |
| Pharmacoat® 615 | 397.7 ± 8.3 | 0.206 ± 0.011 | 379.5 ± 9.1 | 0.165 ± 0.015 |
| PVP K25 | 1108 ± 156.4 | 0.597 ± 0.096 | 1715 ± 82.6 | 0.262 ± 0.069 |
| Soluplus® | 2008.3 ± 110 | 0.324 ± 0.090 | 1340 ± 23.4 | 0.265 ± 0.074 |
| Dimer | Symmetry Operator | Interaction Energy (kcal/mol) |
|---|---|---|
| 1 | 1 + x, y, z 0 0 | −8.7 |
| 2 | 1 + x, −1 + y, z 0 0 | −3.1 |
| 3 | −1 + x, y, z 0 0 | −8.7 |
| 4 | −1 + x, 1 + y, z 0 0 | −3.1 |
| 5 | 1/2 − x, 1/2 + y, 1/2 − z 0 0 | −12.0 |
| 6 | 1/2 − x, −1/2 + y, 1/2 − z 0 0 | −12.0 |
| 7 | 3/2 − x, 1/2 + y, 1/2 − z 0 0 | −2.2 |
| 8 | 3/2 − x, −1/2 + y, 1/2 − z 0 0 | −2.2 |
| 9 | −1/2 − x, 1/2 + y, 1/2 − z 0 0 | −1.4 |
| 10 | −1/2 − x, −1/2 + y, 1/2 − z 0 0 | −1.4 |
| 11 | −x, 1 − y, −z 0 0 | −17.5 |
| 12 | 1 − x, −y, −z 0 0 | −21.5 |
| 13 | 1 − x, 1 − y, −z 0 0 | −11.1 |
| 14 | 2−x, −y, −z 0 0 | −2.7 |
| 15 | 1/2 + x, 1/2 − y, −1/2 + z 0 0 | −6.2 |
| 16 | −1/2 + x, 1/2 − y, 1/2 + z 0 0 | −6.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medarević, D.; Ibrić, S.; Vardaka, E.; Mitrić, M.; Nikolakakis, I.; Kachrimanis, K. Insight into the Formation of Glimepiride Nanocrystals by Wet Media Milling. Pharmaceutics 2020, 12, 53. https://doi.org/10.3390/pharmaceutics12010053
Medarević D, Ibrić S, Vardaka E, Mitrić M, Nikolakakis I, Kachrimanis K. Insight into the Formation of Glimepiride Nanocrystals by Wet Media Milling. Pharmaceutics. 2020; 12(1):53. https://doi.org/10.3390/pharmaceutics12010053
Chicago/Turabian StyleMedarević, Djordje, Svetlana Ibrić, Elisavet Vardaka, Miodrag Mitrić, Ioannis Nikolakakis, and Kyriakos Kachrimanis. 2020. "Insight into the Formation of Glimepiride Nanocrystals by Wet Media Milling" Pharmaceutics 12, no. 1: 53. https://doi.org/10.3390/pharmaceutics12010053
APA StyleMedarević, D., Ibrić, S., Vardaka, E., Mitrić, M., Nikolakakis, I., & Kachrimanis, K. (2020). Insight into the Formation of Glimepiride Nanocrystals by Wet Media Milling. Pharmaceutics, 12(1), 53. https://doi.org/10.3390/pharmaceutics12010053