1. Introduction
Imaging biological events and states is important to resolve biological questions. Many researchers have developed various in vitro and in vivo imaging techniques [
1,
2,
3,
4]. Multiscale imaging, from whole body to sub-organelle levels, is required for clarification. Results from cell cultures are often misrepresentative of in vivo phenomena [
5]. However, live animal imaging has difficulties, especially in visualizing the submicron level. Ex vivo observation of tissues is suitable for three-dimensional imaging at a targeted moment in time course experiments and has various merits, such as reflecting in vivo phenomena, submicron imaging, and visualizing spatial relationships among biological events and states. Tissue optical clearing methods facilitate three-dimensional multicolor deep imaging using fluorescence, and have been widely applied to neurology and pharmaceutics [
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21].
Methodologies of tissue optical clearing can be categorized into electrophoretic methods and sequential immersion in tissue clearing solutions. A representative of the former is the CLARITY protocol, which has an extreme tissue clearing efficiency [
6]. In CLARITY, once a tissue is infused with formaldehyde and hydrogel monomers, it polymerizes into a hydrogel. Then, electrophoresis with a high sodium dodecyl sulfate concentration is performed to extract lipids, which are the main reason for opacity. Overall, this protocol requires almost 10 days. Therefore, CLARITY is a cumbersome and time-consuming task. However, immersion in tissue clearing solutions can be performed without specialized devices. Among them, aqueous solutions are useful to avoid inactivation of fluorescent proteins. In 2011, Hama et al. developed a urea-based tissue clearing formulation, Sca
leA2, which optically cleared the brain in 2 weeks [
7]. However, Sca
leA2 had some problems, including an insufficient tissue clearing efficiency, slow rate of tissue clearing, fragility, and swelling. Susaki et al. developed CUBIC (clear, unobstructed brain imaging cocktails and computational analysis), which greatly improved tissue clearing efficiency using high concentrations of a detergent, urea, and amino alcohols [
8]. However, CUBIC affects the ultrastructure of specimens, mainly because of the high concentration of the detergent [
11]. Hama et al. developed Sca
leS with urea and sorbitol, a mild tissue-permeant sugar alcohol [
11]. Sca
leS contains minimal detergent and preserves ultrastructures. However, Sca
leS requires sequential immersion into a series of solutions (Sca
leS0, S1, S2, S3, and S4) and is slightly cumbersome and time consuming (more than 3 days). Chen et al. developed UBasM (Urea-Based Amino-Sugar Mixture), which contains urea, sugar amine meglumine, and a low concentration of detergent [
12]. Here, even a low concentration of detergent can theoretically affect lipid structures. Detergent-free methods have also been developed. The
ClearT2 protocol includes 50% formamide and 10–20% polyethylene glycol [
13]. SeeDB (See Deep Brain) uses an extremely high concentration of fructose (gradually increased from 20 to 115
w/
v%) [
14]. Unfortunately, the tissue clearing efficiencies of
ClearT2 and SeeDB for adult tissues are insufficient for deep imaging with confocal microscopy [
16,
19]. However, the combination of fructose and urea, such as in FRUIT [
20] and FUnGI (fructose, urea, and glycerol for imaging) [
21], appears to be a useful strategy. Researchers must select a tissue optical clearing method by compromising between transparency and ultrastructure preservation.
Not only amides such as urea, but also amines are often used in tissue clearing. CUBIC uses two amines,
N,N,N′,N′-tetrakis(2-hydroxypropyl)ethylenediamine and 2,2′,2′′-nitrilotriethanol, and UBasM uses meglumine. Although these amines have a buffering capacity, a tissue clearing method applicable to various pHs is not available, even though many fluorescent dyes and proteins are dependent on pH. In this study, we focused on polyethylenimine (PEI), which has a unique buffering capacity over an appreciable range of pH [
22]. Here, using PEI, we developed a novel tissue optical clearing solution, Seebest (SEE Biological Events and States in Tissues), which is a pH-adjustable method. To observe biological events and states using fluorescent dyes, it is preferable to preserve lipid structures to maintain barrier functions against leakage of fluorescent dyes. Therefore, in this study, Seebest without detergents was developed. Seebest provided sufficient transparency while preserving lipid ultrastructures.
2. Materials and Methods
2.1. Materials
PEI with average molecular weights of 600, 1800, and 10,000 was obtained from Wako Pure Chemical Industries, Osaka, Japan (Product number: 163-17835, 169-17815, and 166-17825, respectively). Propylene oxide-denatured PEI (PP-061) was kindly provided by Nippon Shokubai Co., Ltd., Tokyo, Japan).
2.2. Plasmid
pZsGreen1-N1 expressing a variant of the Zoanthus sp. green fluorescent protein, ZsGreen1, was purchased from Clontech Laboratories (Mountain View, CA, USA). pZsGreen1-N1 was amplified in Escherichia coli strain DH5α, isolated, and purified using an EndoFree Plasmid Giga Kit (Qiagen GmbH, Hilden, Germany). Plasmid DNA dissolved in water was stored at −20 °C prior to experiments.
2.3. Mice
Male ddY mice (5-week-old, 24.0–29.5 g) were obtained from Japan SLC, Inc. (Shizuoka, Japan). Anesthesia was induced by intraperitoneal injection of a mixture of 0.75 mg/kg medetomidine, 5 mg/kg butorphanol, and 4 mg/kg midazolam. Animal experiments were performed in accordance with the guidelines of Nagasaki University and approved by the university committee (Protocol Approval No. 1603101289). This work complied with all relevant ethical regulations regarding animal research.
2.4. Preparation of Seebest Solutions
Once PEI was dissolved in distilled water, the pH of the solution was adjusted with 8 M hydrochloric acid (Nacalai Tesque Inc., Kyoto, Japan, 18321-05) on ice to prevent colorization. Then, urea (Wako Pure Chemical Industries, 210-01185) was dissolved in the pH-adjusted PEI solution at approximately 60 °C using a hot stirrer. Finally, pH of the solution was finely adjusted at 30 °C. Typical final concentrations of PEI and urea were 20 w/v% and 8 M, respectively. Seebest-PP (Propylene oxide-denatured PEI) was prepared in a similar fashion except for the use of propylene oxide-denatured PEI instead of PEI. 4′,6-diamidino-2-phenylindole (DAPI) (Sigma Aldrich Co. LLC., Saint Louis, MO, USA) was optionally added to the Seebest solution at a concentration of 5 µg/mL. It was necessary to use freshly prepared Seebest due to the stability and urea crystallization.
2.5. Tissue Clearing in Seebest
After pre-warming the Seebest solution, a tissue specimen was set in the solution and incubated at 37 °C. Incubation time was 1 day unless otherwise indicated. During incubation, a tube rotator was used to stir the solution.
2.6. Comparison with Other Clearing Methods
Each clearing method was performed in accordance with the literature (Sca
leA2 [
7], Sca
leS, and Sca
leSQ(0) [
11], CUBIC [
8],
ClearT2 [
12], and FUnGI [
21]). The composition of each solution was as follows. Sca
leA2, urea (4 M), glycerol (10.0
w/
v%), and Triton
x-100 (0.1
v/
v%). Sca
leS0,
d-(−)-sorbitol (20
w/
v%), glycerol (5
w/
v%), methyl-β-cyclodextrin (1 mM), γ-cyclodextrin (1 mM),
N-acetyl-L-hydroxyproline (1
w/
v%), and dimethyl sulfoxide (DMSO, Nacalai Tesque, 13445-45) (3
v/
v%) in PBS(−)). Sca
leS1,
d-(−)-sorbitol (20
w/
v%), glycerol (10
w/
v%), urea (4 M), and Triton
x-100 (0.2
w/
v%). Sca
leS2,
d-(−)-sorbitol (27
w/
v%), urea (2.7 M), Triton
x-100 (0.1
w/
v%), and DMSO (8.3
v/
v %). Sca
leS3,
d-(−)-sorbitol (36.4
w/
v%), urea (2.7 M), and DMSO (9.1
v/
v%). Sca
leS4,
d-(−)-sorbitol (40
w/
v%), glycerol (10
w/
v%), urea (4 M), Triton
x-100 (0.2
w/
v%), and DMSO (15
v/v%). Sca
leSQ(0), D-(−)-sorbitol (22.5
w/
v%) and urea (9.1 M). CUBIC-1, urea (25
w/
v%),
N,
N,
N′,
N′-tetrakis(2-hydroxypropyl)ethylenediamine (25
w/
v%), and Triton
x-100 (15
w/
v%). CUBIC-2, sucrose (50
w/
v%), urea (25
w/
v%), 2,2′,2″-nitrilotriethanol (10
w/
v%) and Triton
x-100 (0.1
v/v%).
ClearT2, formamide (50
v/v%) polyethylene glycol (average molecular weight 8000, 20
w/
v%). To prepare FUnGI, 10 g fructose was dissolved in a mixture of 2.33 mL Tris-EDTA (100 mM Tris and 10 mM EDTA disodium salt, pH 8.0) and 11 mL glycerol for 1 day at room temperature. Then, 3.31 g urea was added to the mixture and dissolved for further 1 day, producing approximately 20 mL FUnGI.
2.7. Confocal Microscopy
Cleared specimens were imaged under an inverted confocal laser scanning microscope (LSM710, Carl Zeiss Microscopy GmbH, Cologne, Germany). Laser wavelengths were 405, 488, 543, and 633 nm. Lenses were a ×10 EC Plan-Neofluar (numerical aperture (NA): 0.30; working distance (WD): 5.2 mm), ×20 EC Plan-Neofluar ×20 (NA: 0.50; WD: 2.0 mm), ×25 LD LCI Plan-Apochromat (NA: 0.8; WD: 0.57 mm), and ×40 LD C-Apochromat (NA: 1.1; WD: 0.62 mm). Images were acquired by Zen Black software (version 2012, Carl Zeiss Microscopy).
2.8. Transmission Electron Microscopy
Mice underwent hepatic perfusion fixation under anesthesia. Perfusates were 10 mL PBS (−) containing 10 U/mL heparin, 30 mL PBS (−) and 40 mL fixative in this order. For transmission electron microscopy, the fixative was 4% paraformaldehyde (PFA) with 0.1% glutaraldehyde (GA) in PBS (−). Then, the liver specimens were immersed in the same fixative overnight at 4 °C. The specimens were washed twice in PBS (−) and immersed in Seebest (pH 7.3 or 11) or PBS (−) for 1 day. After that, the specimens were washed twice in PBS (−) and incubated in PBS (−) for 1 day. The specimens were sectioned at 0.5 mm thicknesses, post-fixed with 1% osmium tetroxide in 0.1 M phosphate buffer (pH 7.4) for 2 h, dehydrated in acetone, and embedded in Epon812 resin (TAAB, Berkshire, UK). Ultrathin sections (70 nm thick) were prepared with a diamond knife, stained with uranyl acetate and lead citrate, and observed under a transmission electron microscope (H7650, Hitachi High-Technologies Corporation, Tokyo, Japan).
2.9. Dox Visualization
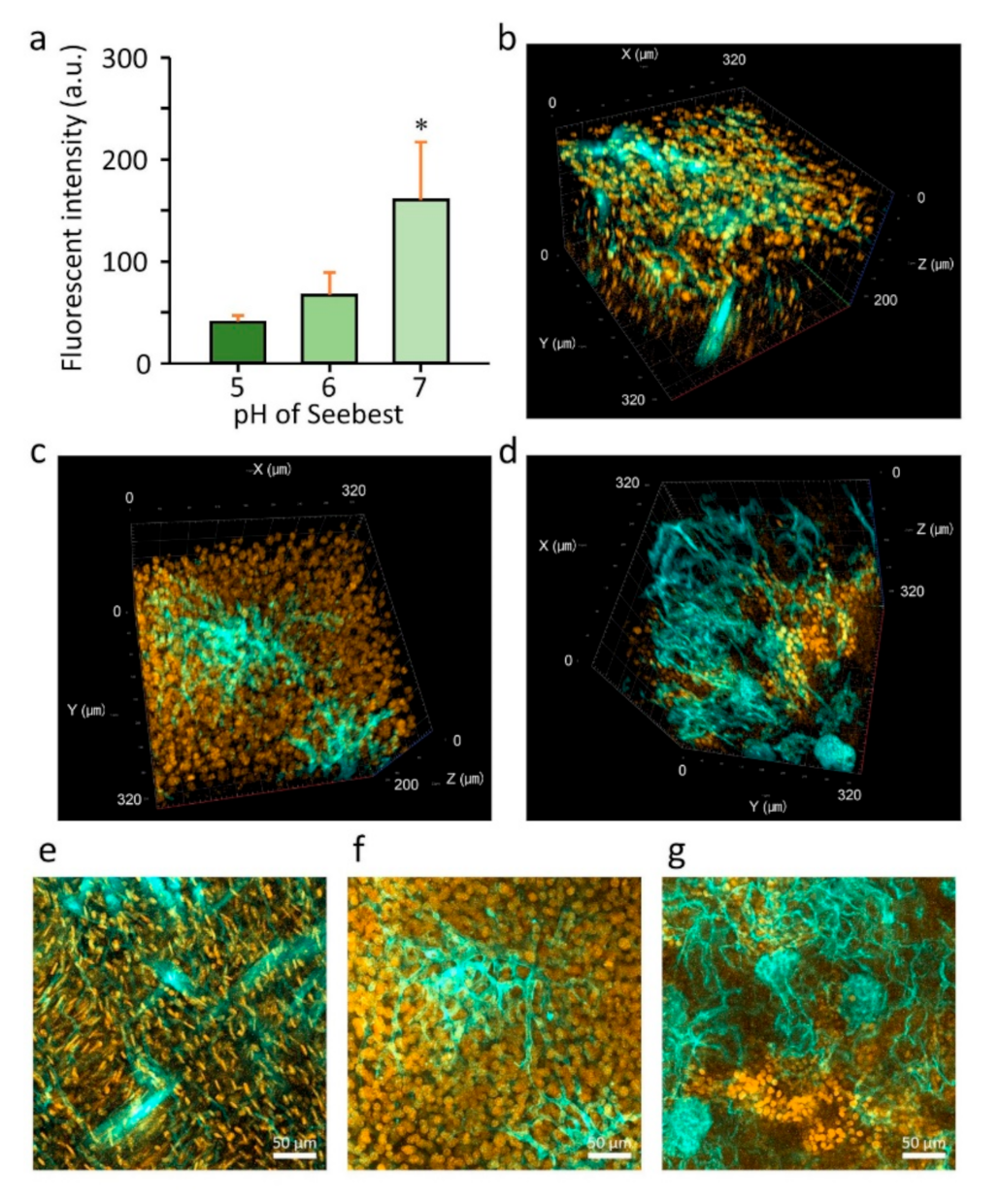
Doxorubicin (Dox) hydrochloride (Tokyo Chemical Industry Co., Ltd., Japan, D4193) was dissolved in DMSO as a stock solution (10 mg/mL). To measure Dox release during tissue clearing, the stock solution of Dox was diluted in saline at a concentration of 1 mg/mL and then intravenously injected into mice (10 mg/kg). Mice were subjected to cardiac perfusion fixation under anesthesia. Perfusates were 10 mL PBS (−) containing 10 U/mL heparin, 50 mL PBS (−), and 160 mL 4% PFA in this order. Then, the liver was cut into small blocks. The blocks of liver were weighed, and 20 µL/mg Seebest solutions (pH 5, 6, and 7) were added to the blocks. One day after incubation at 37 °C on the tube rotator, fluorescence in the external fluid was measured at excitation and emission wavelengths of 488 and 570 nm, respectively. For three-dimensional observation, the stock solution of Dox was diluted in saline at a concentration of 2 mg/mL, and then intravenously injected into mice (20 mg/kg). Cardiac perfusion was performed. Perfusates (filtrated with a 0.45 µm cellulose acetate filter) were 10 mL PBS (−) containing 10 U/mL heparin, 50 mL PBS (−), 5 mL 5% glucose, 10 mL 0.12 mg/mL 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine (DiD) (perchlorate salt, Thermo Fisher Scientific Inc., Bedford, MA, USA, D307) in 5% glucose, 5 mL 5% glucose and 160 mL 4% PFA in this order. Then the heart, liver, and kidney were subjected to post-fixation at 4 °C overnight. Tissues were washed twice in PBS (−) and then immersed in Seebest solution at 37 °C. Confocal microscopy was performed within 4 h of immersion with Seebest.
2.10. Liver Ischemia/Reperfusion Injury
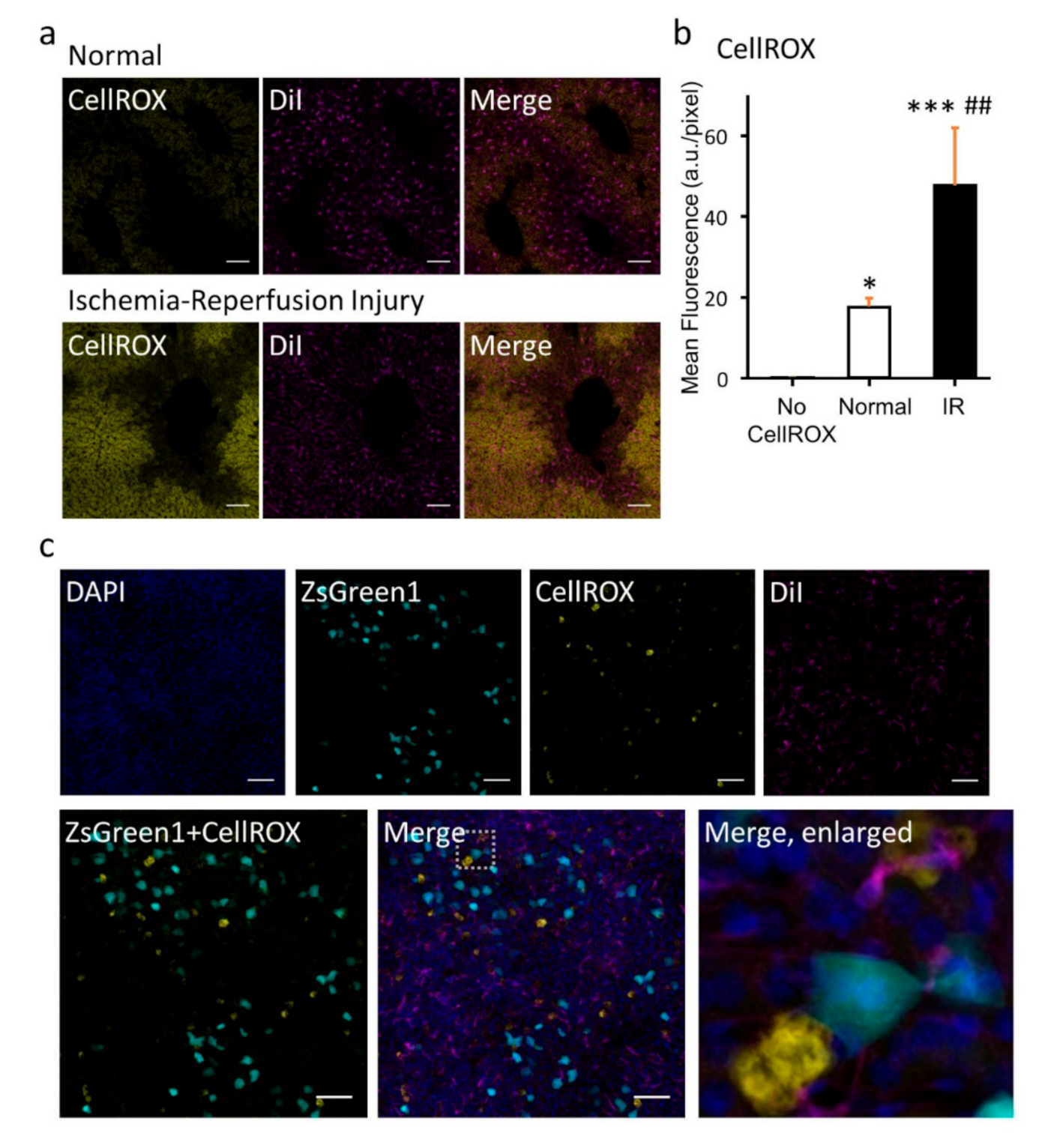
Reactive oxygen species (ROS) probe CellROX Deep Red (125 µmole, Thermo Fisher Scientific Inc., Bedford, MA, USA, C10422) was injected intraperitoneally. One hour after injection, mice were anesthetized, and the portal vein of mice was clamped for 30 min and then reperfused for 1 h. Intravenous injection of 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine (DiI) (perchlorate salt, Sigma Aldrich Co. LLC., 42364) in 5% glucose (0.5 mg/mL, 4.8 mg/kg) and perfusion fixation were performed. A liver specimen was subjected to post-fixation at 4 °C overnight. Tissues were washed twice in PBS (−) and then immersed in Seebest solution at 37 °C. Confocal microscopy was performed at 1 day after immersion in Seebest.
2.11. Hydrodynamics-Based In Vivo Transfection to the Liver
Mice were injected with a plasmid DNA (pZsGreen1-N1) solution in saline (10 µg/2.2 mL/mouse) within 5 sec via the tail vein. One hour prior to plasmid DNA injection, CellROX Deep Red (125 µmole) was injected intraperitoneally. Twelve hours after plasmid DNA injection, mice were intravenously injected with a DiI solution in 5% glucose (0.5 mg/mL, 4.8 mg/kg). Thirty minutes after DiI injection, mice underwent hepatic perfusion fixation. A liver specimen was cleared in Seebest (pH 8.5) for 1 day and then confocal microscopy was performed.
2.12. Preparation and Characterization of Liposomes for Spatial Distribution
Traditional Bangham and limit size liposomes were prepared as follows. Traditional Bangham liposomes were prepared using thin lipid film hydration (Bangham’s method). Egg lecithin (Wako Pure Chemical Industries, Osaka City, Osaka Prefecture, Japan 124-05031), cholesterol (Nacalai Tesque Inc., Kyoto, Japan 08721-62), and 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) (Sigma Aldrich Co. LLC., D4292) were dissolved in methanol at a weight ratio of 75:25:1.42. Then, a thin lipid film was formed using a rotary evaporator, which was subsequently desiccated overnight. To obtain liposomes, the thin lipid film was rehydrated in a 5% glucose solution at a final lipid concentration of 2 mg/mL. Liposomes were extruded through polycarbonate membrane filters (200-nm pore sizes, 11 times) using a commercially available instrument (Mini-Extruder, Avanti Polar Lipids, Inc., Alabaster, AL, USA). Limit size liposomes were prepared based on microfluidics using a NanoAssemblr instrument (Precision Nanosystems, Vancouver, BC, Canada). Briefly, anhydrous ethanol was prepared by dehydration of ethanol (Kanto Chemical Co., Inc., Tokyo, Japan, 14033-70) using a molecular sieve (3A, powder, Nacalai Tesque, 04176-55). Egg lecithin (7.5 mg), cholesterol (2.5 mg), and DiO (0.142 mg) were dissolved in anhydrous ethanol and evaporated using the rotary evaporator. Then, the lipids were again dissolved in 1 mL anhydrous ethanol, and this solution served as the ethanol phase. Glucose (Nacalai Tesque, 16806-25) was dissolved in distilled water at a concentration of 5 w/v%, and this solution served as the water phase. Ethanol and water phases (volume ratio 1:5) were mixed together using the NanoAssemblr instrument at a flow rate of 12 mL/min and dialyzed overnight using a dialyzing tube (Spectra/Por Float-A-Lyzer G2, 1 mL size, 3.5–5 KDa, Spectrum Laboratories Inc., Piscataway, NJ, USA) against a 5% glucose solution to produce limit size liposomes. The particle size of liposomes was measured using a Zetasizer Nano ZS instrument (Malvern Instruments Ltd., Worcestershire, UK).
2.13. Lipophilic Carbocyanine Dye Staining
To stain blood vessels, DiI perfusion was performed as reported previously [
23] with slight modifications. Originally, DiI was dissolved in ethanol. However, dissolution rate of DiI in ethanol appeared to be slow. Before perfusion, the DiI stock solution (8 mg/mL in DMSO) was diluted in 5% glucose solution to produce a 0.12 mg/mL solution. DiD perfusion was performed in the same fashion.
2.14. Fixation
Fixation was performed by cardiac or hepatic perfusion under anesthesia. For cardiac perfusion, a bent needle (26G, Nipro Corporation, Osaka, Japan) equipped with a tube and a peristatic pump was inserted to the left ventricular. Then, right atrial was cut with surgical scissors. Immediately, perfusion was started. For hepatic perfusion, the portal vein was catheterized with a Surflo® I.V. Catheter, 22 G × 1 1/4″ (Terumo Co., Tokyo, Japan). Immediately, the liver was perfused with perfusates. The fixative was 4% PFA in PBS (−), except for electron microscopy (4% PFA with 0.1% GA). Tissue specimens (indicated in each figure) were subjected to post-fixation at 4 °C overnight. Before tissue clearing in Seebest, tissue specimens were washed twice in PBS (−).
2.15. FRET Analysis for Integrity of Lipid Membranes
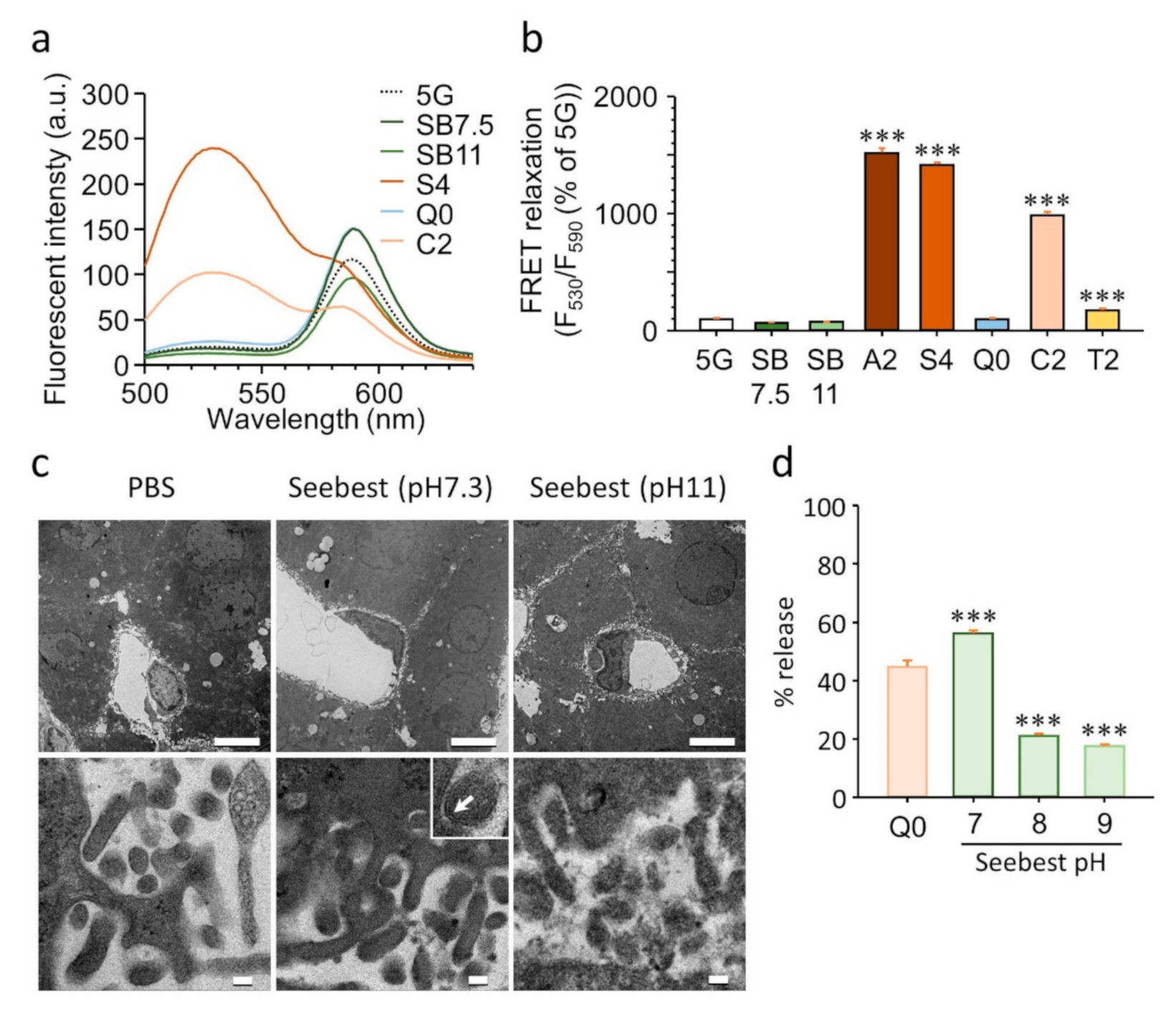
Förster resonance energy transfer (FRET) liposomes were prepared using thin lipid film hydration (Bangham’s method). Egg lecithin, cholesterol, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt, Avanti Polar Lipids, 810145), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt, Avanti Polar Lipids, 810150) were dissolved in methanol at a weight ratio of 100:50:2.4:3.4. Then, a thin lipid film was formed using a rotary evaporator and subsequently desiccated overnight. To obtain FRET liposomes, the thin lipid film was rehydrated in a 5% glucose solution at a final lipid concentration of 0.4 mg/mL. Liposomes were extruded through polycarbonate membrane filters (100- and 50-nm pore sizes, 11 times for each pore size) using a commercially available instrument (Mini-Extruder, Avanti Polar Lipids, Inc.). FRET relaxation of liposomes after mixing with tissue clearing solution was monitored at an excitation wavelength of 460 nm using a spectrofluorometer (RF-6000, Shimadzu Corporation, Kyoto, Japan).
2.16. Dye Leakage from Liposomes
To evaluate membrane integrity and the importance of pH adjustment, dye leakage from phenolsulfonphthalein (PSP)-containing liposomes was measured. PSP-containing liposomes were prepared using Bangham’s method. The 1,2-Dioleoyl-sn-glycero-3-phosphocholine and cholesterol were dissolved in methanol at a molar ratio of 70:30. Then, a thin lipid film was formed using a rotary evaporator and subsequently desiccated overnight. To obtain PSP-containing liposomes, the thin lipid film was rehydrated in 5 mg/mL PSP (Nacalai Tesque, 26807-92) in carbonate buffer solution (10 mM sodium carbonate, 150 mM sodium chloride, pH 10) at a final lipid concentration of 20 mg/mL. Liposomes were extruded through polycarbonate membrane filters (200-nm pore sizes, 11 times for each pore size) using a commercially available instrument (Mini-Extruder). To remove PSP in the outer phase, a desalting column (PD-10, GE Healthcare Lifesciences, Cheshire, UK) was used. Then, 200 µL of liposome dispersion was mixed with 1 mL tissue clearing solution, and the mixture was incubated for 1 h at 37 °C. Subsequently, 1 mL of the mixture was diluted with 1.5 mL of the carbonate buffer solution and desalted with a PD-10 column. The eluate (500 µL) was diluted with 1 mL of 1 M NaOH, and PSP absorbance at 550 nm was determined using a UV-vis spectrometer (UV-1850, Shimadzu Corporation). For the intact liposome group, liposomes were diluted with the carbonate buffer solution. For the 100% release group, 1% Triton X-100 in the carbonate buffer solution was used as the diluent. The percentage of released PSP was calculated using the following equation.
2.17. Supplementary Experiments
2.18. Data Analysis
Exact p-values for each experiment were calculated using R (version 3.5.1). Two-tailed, unpaired t-tests or one-way ANOVA followed by Dunnett’s or Tukey’s test were performed to compare experimental groups as indicated in the figure legends. Numerical conversion of fluorescence in images were performed using Fiji (Image J, NIH). Fiji, Zen Black, and Zen Blue (Carl Zeiss Microscopy) software were used for image visualization including scale bar drawing. Zen Blue software was used for maximum intensity projections, average intensity projections, and three-dimensional (3D) renderings. Adobe Photoshop (Adobe Systems Inc., San Jose, CA, USA) was used for inputs of scale information to 3D-rendered images. Microsoft PowerPoint (Microsoft Corporation, Redmond, WA, USA) was used for figure arrangements. Adobe Photoshop was used for resolution adjustments without image resampling.
4. Discussion
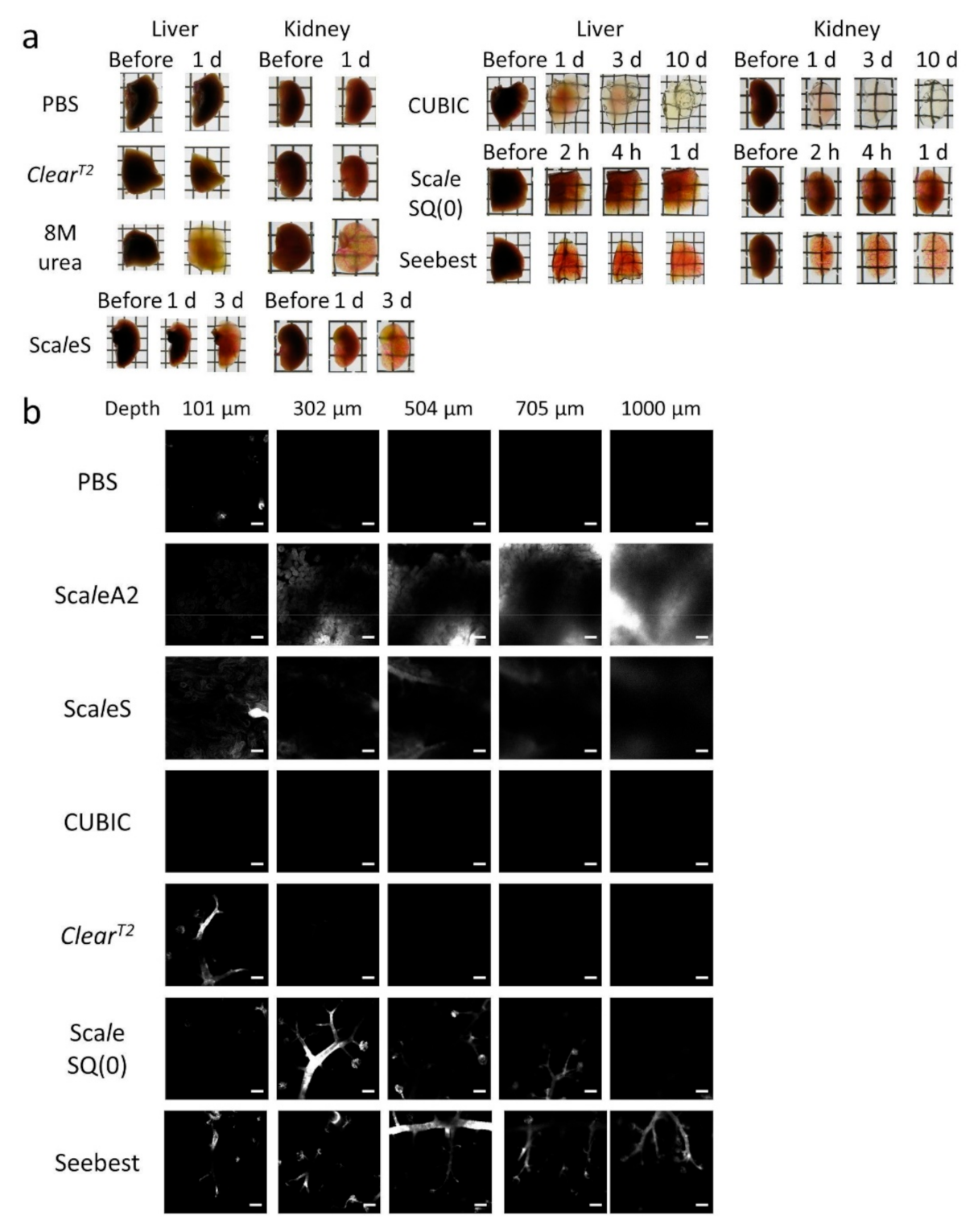
We succeeded in development of the novel tissue optical solution Seebest. Blood vessels stained with a lipophilic carbocyanine dye were deeply observable with Seebest (
Figure 2 and
Figure S6, Supplementary Materials). Seebest was applicable to multiple tissues including the brain, liver, kidney, spleen, heart, lung, stomach, and small and large intestines (
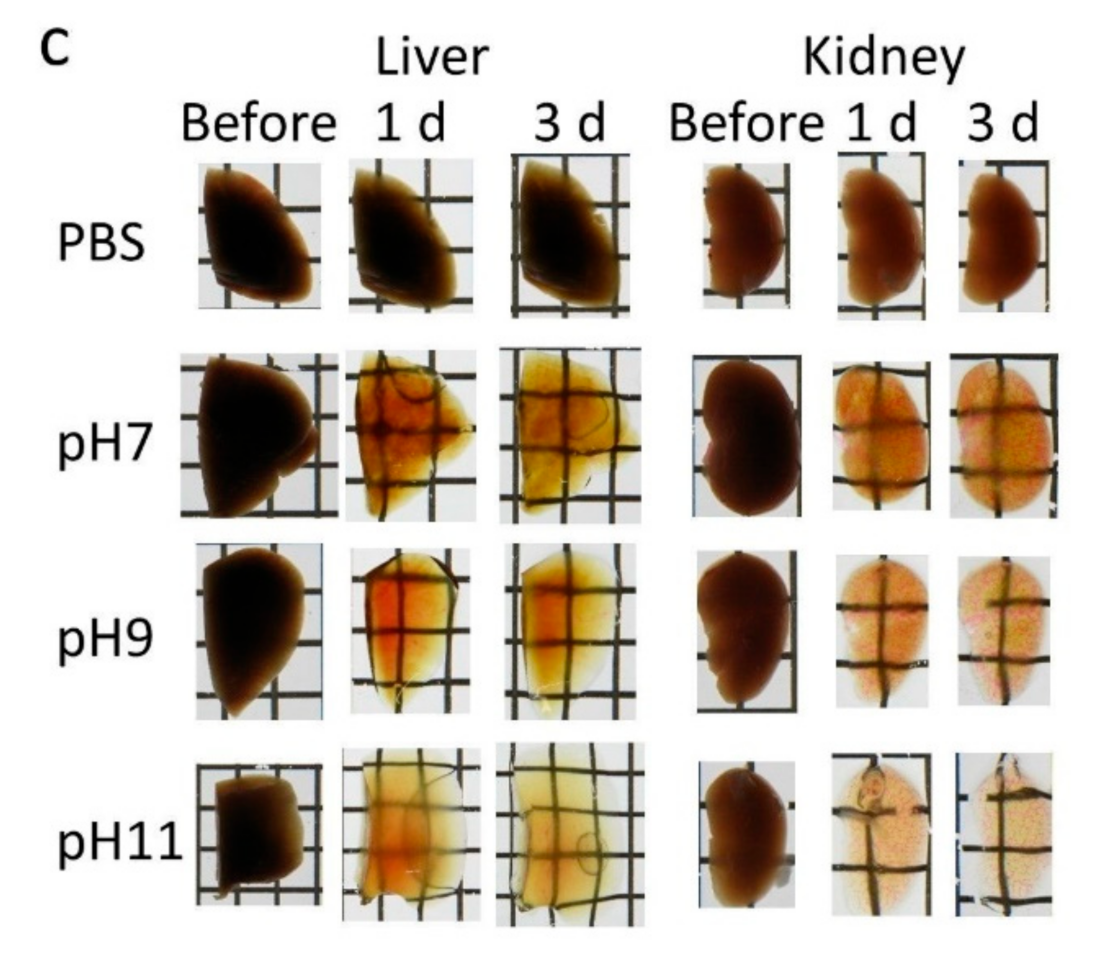
Figure S6, Supplementary Materials). Tissue clearing efficiency of Seebest was dependent on pH, but Seebest substantially cleared tissues even at neutral pH (
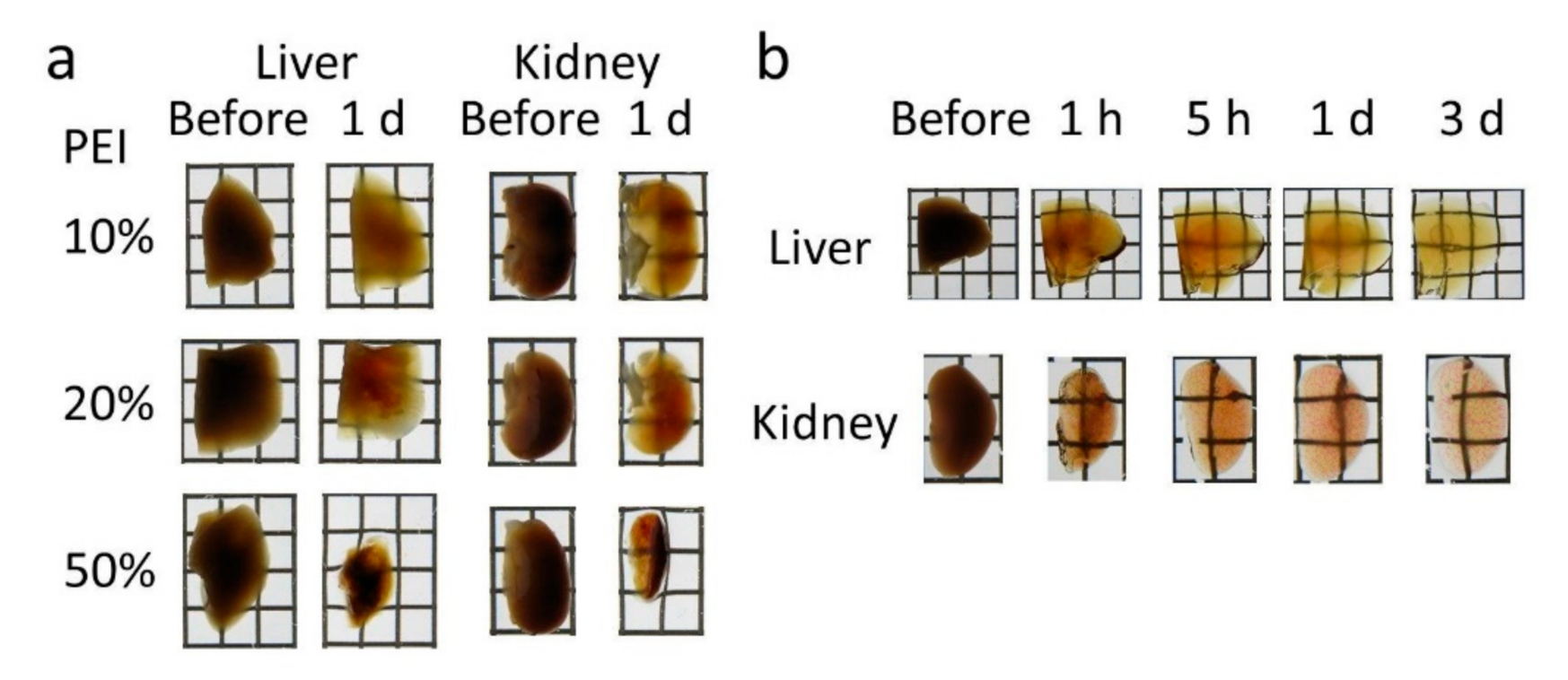
Figure 1c). The clearing procedure of Seebest was simple, that is, immersion in only one solution. The clearing speed of Seebest was very high (
Figure 1b,
Figure S9 and Table S1, Supplementary Materials). Importantly, Seebest around neutral pH only slightly changed the sample size during clearing (
Figure 1 and
Figure S5, Supplementary Materials) and preserved lipid ultrastructures at the electron microscopy level (
Figure 3). We applied Seebest to visualization of the relative spatial distribution of gene expression and oxidative stress with nuclear and sinusoidal fluorescence staining (
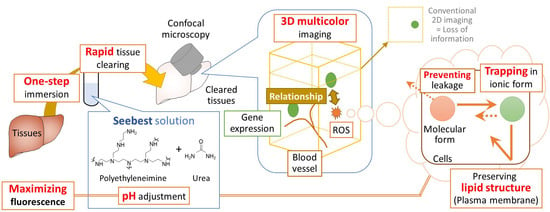
Figure 5). Thus, Seebest provides pH adjustable, simple, rapid and effective tissue optical clearing while preserving lipid ultrastructures for multicolor deep bioimaging.
The most important advancement in tissue clearing using Seebest was the pH adjustment. Numerous fluorescent dyes and proteins have been developed. Although most fluorescent dyes and proteins are sensitive to pH in terms of fluorescence spectrum, intensity, and stability, little attention has previously focused on pH during tissue clearing. Simultaneously, Seebest preserved plasma membranes, which enabled us to stain lipidic structures with fluorescent dyes. Considering the pH adjustment and preserved plasma membranes, we expected that Seebest could retain small ionic fluorescent molecules in cells during tissue clearing (
Figure S16, Supplementary Materials). Conventional tissue optical clearing methods with detergents, including CLARITY, CUBIC, Sca
leS and UBasM, affect the plasma membrane. Consequently, it is impossible to retain free fluorescent dyes in cells. Although there are several detergent-free tissue clearing methods, including
ClearT2, SeeDB, and Sca
leSQ(0), pH during tissue clearing is not adjusted in these protocols. Therefore, the equilibrium should shift from ionic to molecular forms because of the loss of the molecular form of probes. Sample size changes during immersion in a clearing solution might be another important factor to prevent dye leakage. The pH of Sca
leSQ(0) is above the pKa of PSP, but Sca
leSQ(0) did not prevent PSP leakage from liposomes (
Figure 3d). This might be attributed to the increase in sample size during immersion in Sca
leSQ(0) (
Figure S5, Supplementary Materials). To date, tissue clearing requires fixation of fluorescent molecules. Seebest could minimize the loss of small fluorescent molecules during tissue clearing by adjusting the pH to maximize the proportion of the ionic form. Tissue clearing using Seebest was extremely quick. Thus, we succeeded in observing the small molecule Dox before its loss (
Figure 4). However, the suppression of dye leakage was incomplete even in Seebest (
Figure 3d and
Figure S12, Supplementary Materials). This limitation might be attributed to the slight changes in sample size during immersion in Seebest (
Figure S5, Supplementary Materials).
Seebest’s tissue clearing mechanism is unclear. Essentially, tissues are opaque mainly because of light scattering by various components with different refractive indices. Tissue clearing methods generally homogenize these refractive indices [
29]. Several strategies can be used to adjust the refractive index, such as dehydration, lipid removal, and use of materials with high refractive indices such as sugars and urea. PEI itself had a partial clearing effect (
Figure 1a). This effect might be due to dehydration because the molecular weight of PEI is too high to penetrate cells. The refractive index of the PEI solution was not as high as that of the urea solution (
Table 1). Obviously, the combination of PEI and urea synergistically enhanced the tissue clearing capacity (
Figure 1 and
Figure 2). Urea penetration might be an important factor for tissue clearing using Seebest. Except for
ClearT2, absolute clearing capacities for tissue homogenates did not vary significantly among tissue clearing solutions (
Figure S8, Supplementary Materials). Additionally, the refractive indices of these solutions were comparable (
Table 1). Thus, not only the refractive index of the solution, but also other factors, such as penetration speed should be involved in effective tissue clearing. Observable depths for DiI-stained blood vessels depended on tissues. Although the clearing capacity for the brain homogenate was comparable to those for liver and kidney homogenates (
Figure S2, Supplementary Materials), the observable depth of the brain was much deeper. This result might be due to the contents of heme iron and elastic fibers. In the case of the lung, air in the trachea and alveoli might interfere with tissue clearing.
Usability of 400–700 nm wavelengths is preferable for multicolor deep imaging using confocal microscopy. Although Seebest employing PEI was compatible to four-color imaging (
Figure 5c), it had slight difficulty in observing DAPI with the 405-nm excitation laser. This should be attributed to the light absorbance at the low wavelength (
Figure S15a, Supplementary Materials). The cause of light absorbance at a low wavelength might be the production of impurities that were probably azo compounds. To reduce the reactivity of PEI, we used propylene oxide-denatured PEI. Use of propylene oxide-denatured PEI to prepare the Seebest solution (Seebest-PP) successfully reduced light absorbance at low wavelengths (
Figure S15b, Supplementary Materials). This improved the observable depth of DAPI (
Figure S15c, Supplementary Materials) and indicates an increase in the multicolor deep imaging potential. Using Seebest-PP, we observed spatial distributions of liposomes in relation to blood vessels (
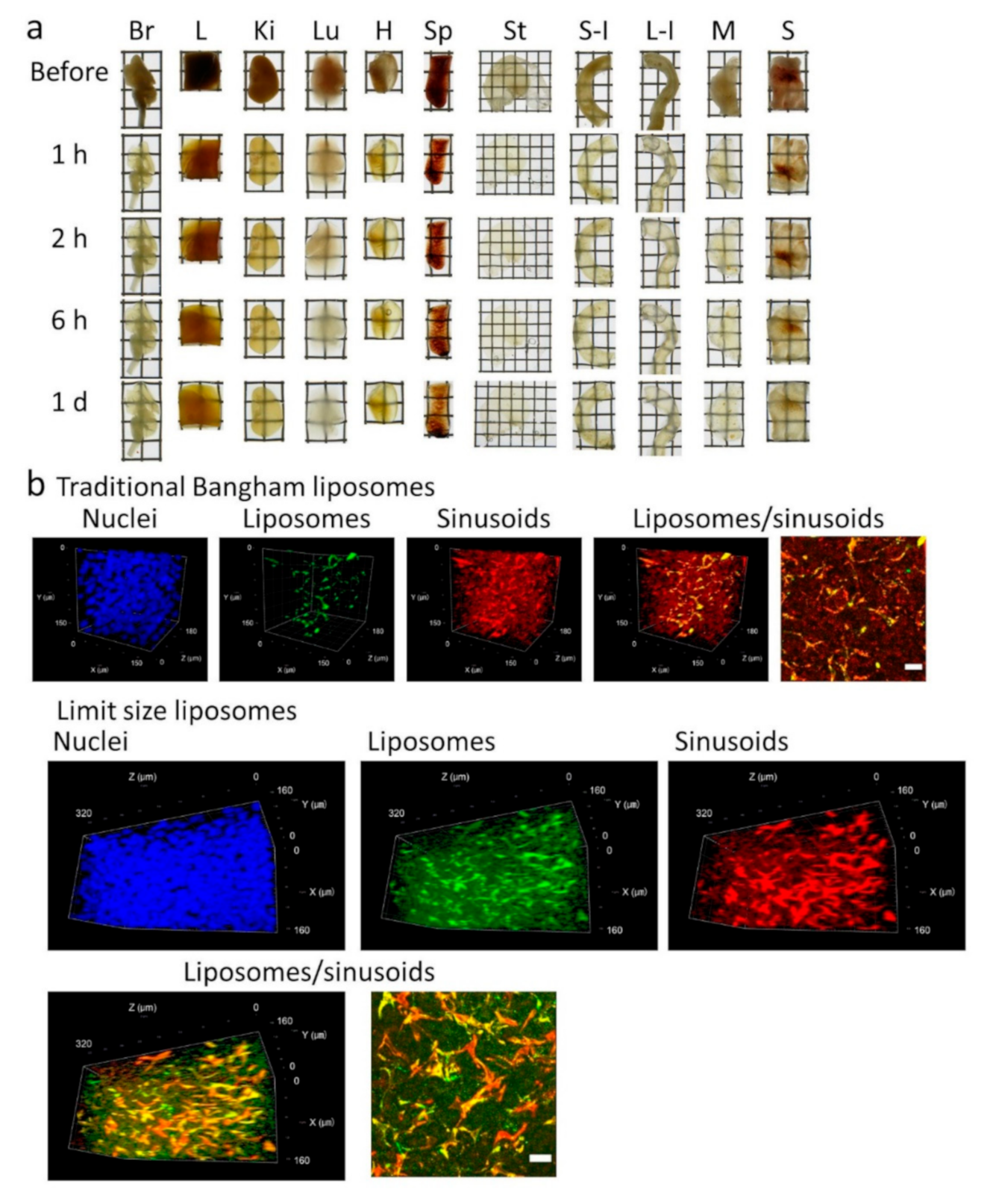
Figure 6b). We also successfully elucidated different spatial distribution characteristics between traditional Bangham and limit size liposomes. Thus, Seebest-PP may be useful to visualize the spatial distribution of lipidic drug delivery system (DDS) carriers. About evaluation of spatial distribution in DDS fields, we have reported several applications using lipid-preserving tissue clearing methods
ClearT2 and Sca
leSQ(0) [
30,
31,
32,
33,
34,
35]. In these reports, we have evaluated positional relationship of spatial distribution of gene expression with blood vessels or tissue surface stained with lipidic carbocyanine dyes DiI. Seebest and Seebest-PP are thought to be applicable to such evaluation. Moreover, Seebest-PP would maximize multicolor deep imaging potency to simultaneously evaluate positional relationship of gene expression and/or lipidic carriers (such as liposomes and lipid nanoparticles) with biological structures (such as blood vessels, tissue surface and elastic fibers), oxidative stress and so on. Moreover, we have developed the quantification method of DDS carrier concentration in tissue homogenates using tissue clearing solutions [
36]. We are currently optimizing the composition of Seebest-PP to determine the concentration of lipidic DDS carriers in tissues. Thus, Seebest solution would realize not only multicolor deep imaging of spatial distribution, but also easy quantification of various DDS carriers [
37].
Clearing using Seebest did not reduce antigenicity. Of course, Seebest was incompatible with three-dimensional immunohistochemistry because of the preserved plasma membrane. The permeabilization procedure during immunohistochemistry destroys lipid structures. Therefore, preservation of lipid structures and immunohistochemistry contradict each other. However, sequential observations before and after immunohistochemistry would enable us to perform ultra-multicolor deep imaging, namely three-dimensional correlative twice light microscopy. At least one color should serve as a reference color for the first observation. After the first observation, the other colors are removed by solubilization and/or photobleaching and staining with antibodies is performed. In the second observation, the corresponding region with the first observation can be observed according to the pattern of the reference color. We also plan to apply Seebest to clinical biopsies such as cancers and non-alcoholic steatohepatitis.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}