Particle Detection and Characterization for Biopharmaceutical Applications: Current Principles of Established and Alternative Techniques


 ,
,  and
and
Abstract
:
1. Introduction
- (1)
- the nature and the properties of the particle sample (the particle and suspension medium) itself
- (2)
- the underlying measurement principle of the technique, with its sample-preparation requirements
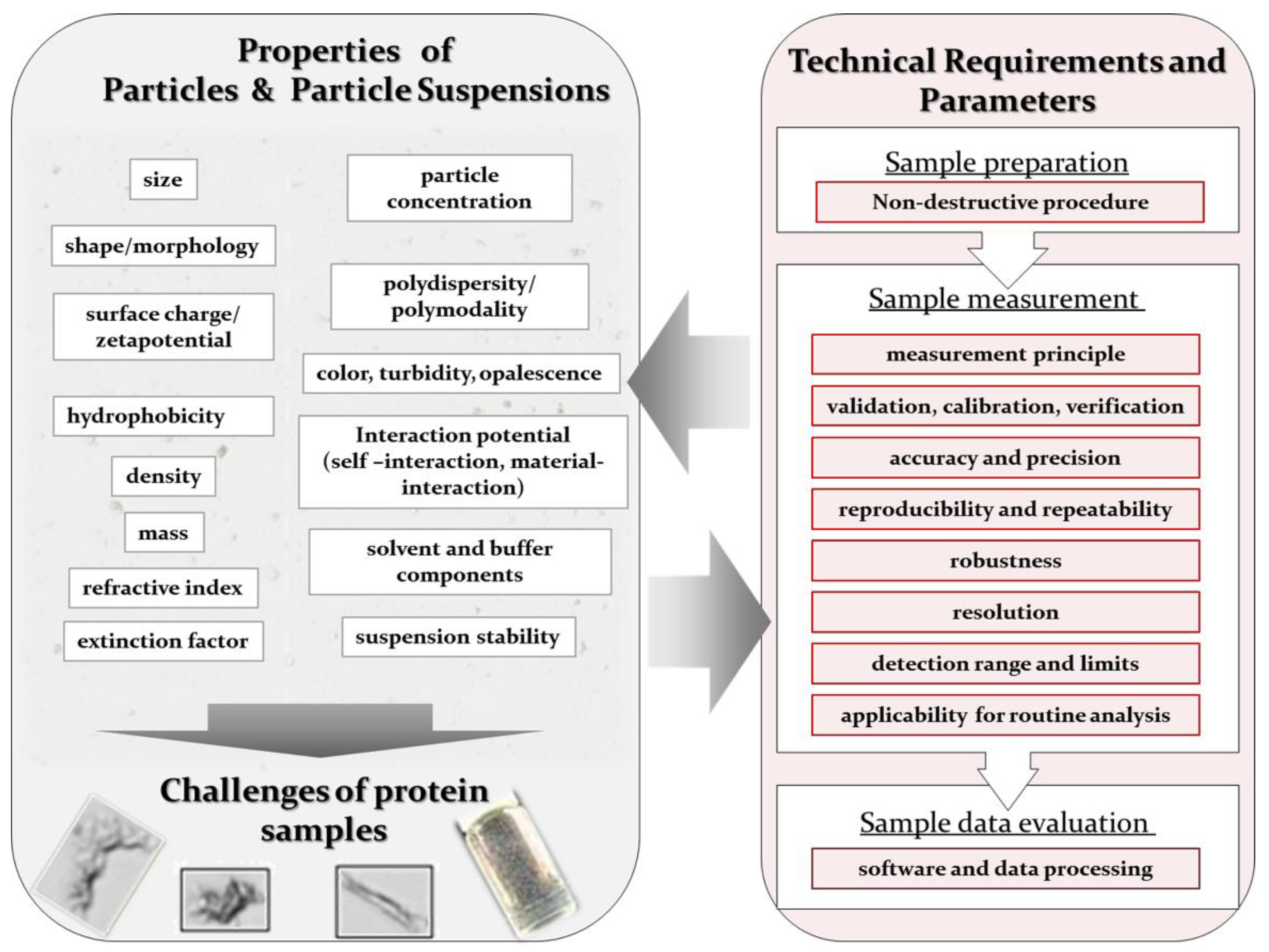
2. Particle Properties and Current Technical State
2.1. Common Properties of Particle Suspensions
2.2. Specific Properties and Challenges of Biopharmaceutical Protein Samples
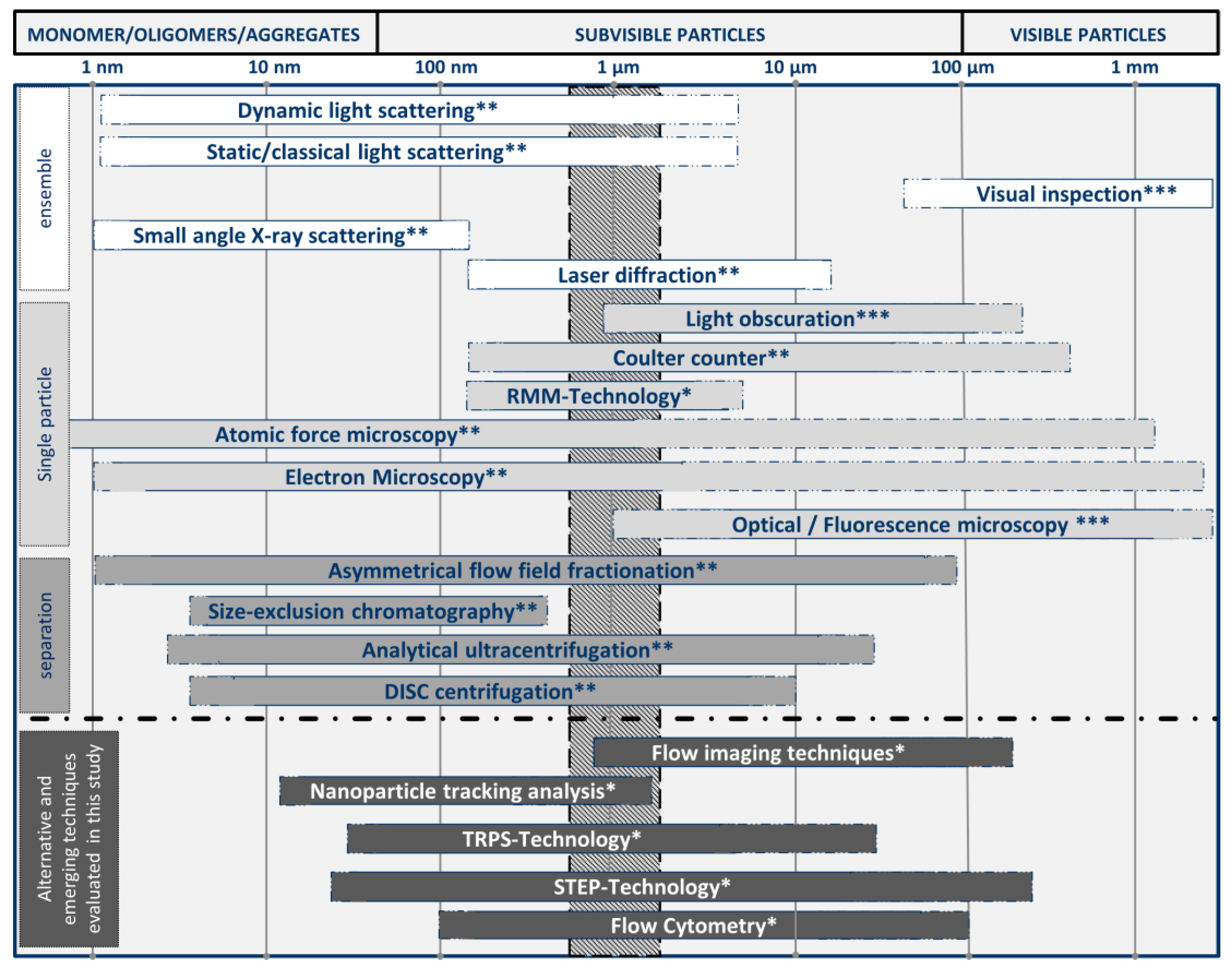
2.3. Particle Detection and Characterization in Biopharmaceutics
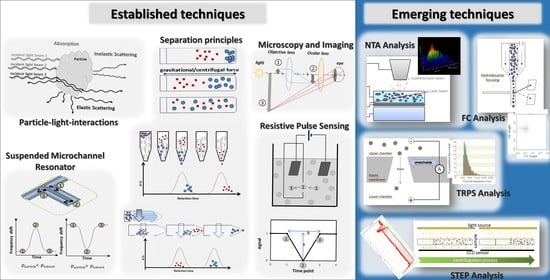
3. Principles of Common Particle Analysis, Detection, and Characterization Methods
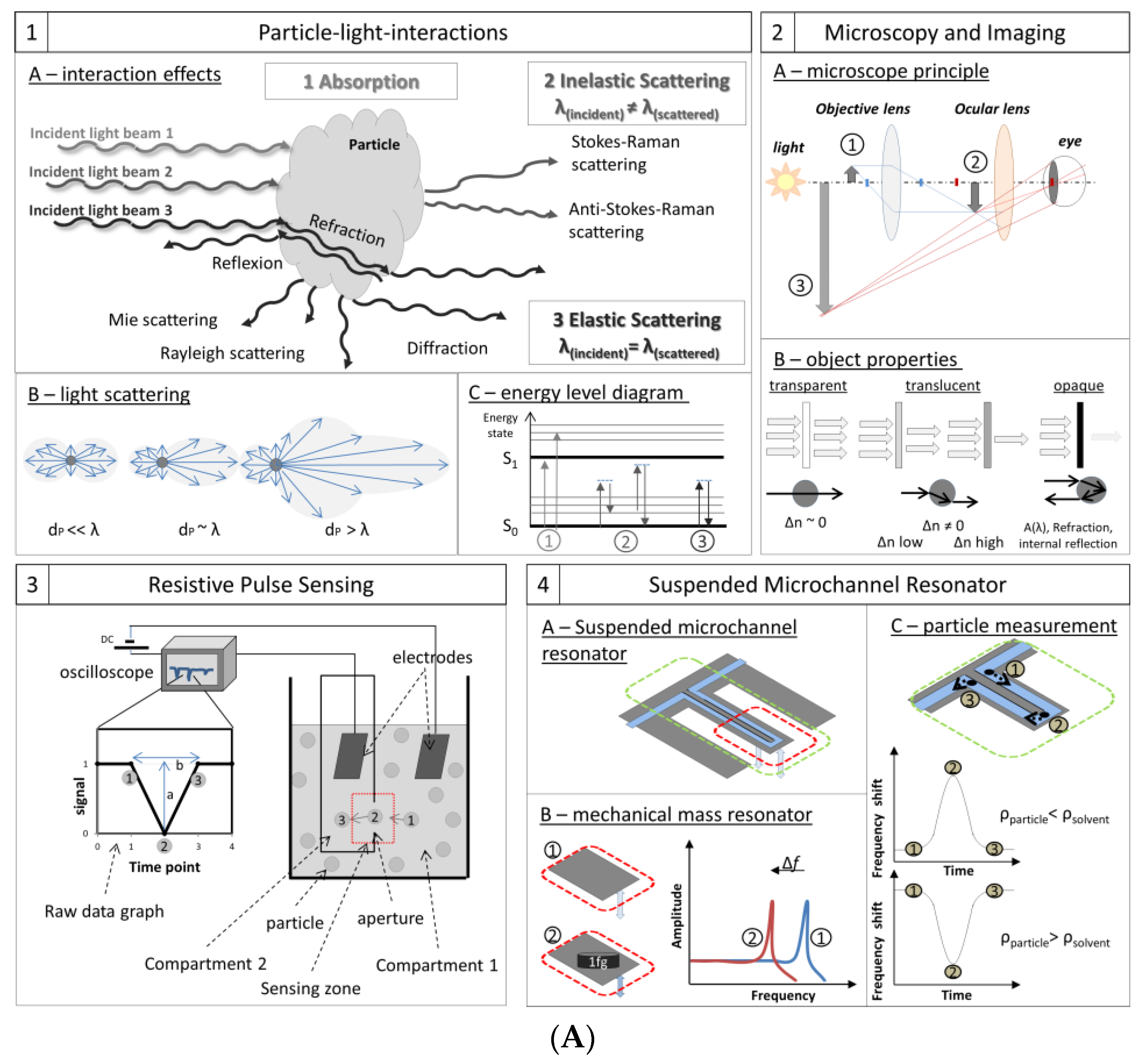
3.1. Particle–Light Interaction
3.1.1. Principles
3.1.2. General Advantages and Challenges
3.1.3. Applications and Techniques
3.2. Microscopy and Imaging
3.2.1. Principles
3.2.2. General Advantages and Challenges
3.2.3. Applications and Techniques
3.3. Resistive Pulse Sensing (RPS) or Coulter Principle
3.3.1. Principle
| R | resistance | D | pore diameter |
| ρ | solution resistivity/conductivity | L | pore length |
| A(z) | cross sectional area perpendicular to z | z | length coordinate |
| ΔR | resistance change caused by particle passage | D | pore diameter |
| ρ | solution resistivity/conductivity | d | particle diameter |
| J | event frequency | D | pore diameter |
| νS | particle velocity | C | particle concentration |
3.3.2. General Advantages and Challenges
3.3.3. Applications and Techniques
3.4. Resonant Mass Measurement (RMM) and Suspended Microchannel Resonators (SMR)
3.4.1. Principle
| f | resonance frequency | Δm | mass change upon sample addition |
| k | spring constant of resonator | m* | effective mass of the resonator |
| α | numerical constant depending on the geometric localization of Δm (as defined by [175,176]) | ||
| M | in vacuo or dry mass of the particle | ρF | fluid density |
| MB | buoyant mass | ρP | particle density |
| D | equivalent spherical particle diameter | ρF | fluid density |
| M | dry mass of the particle | ρP | particle density |
3.4.2. General Advantages and Challenges
3.4.3. Applications and Techniques
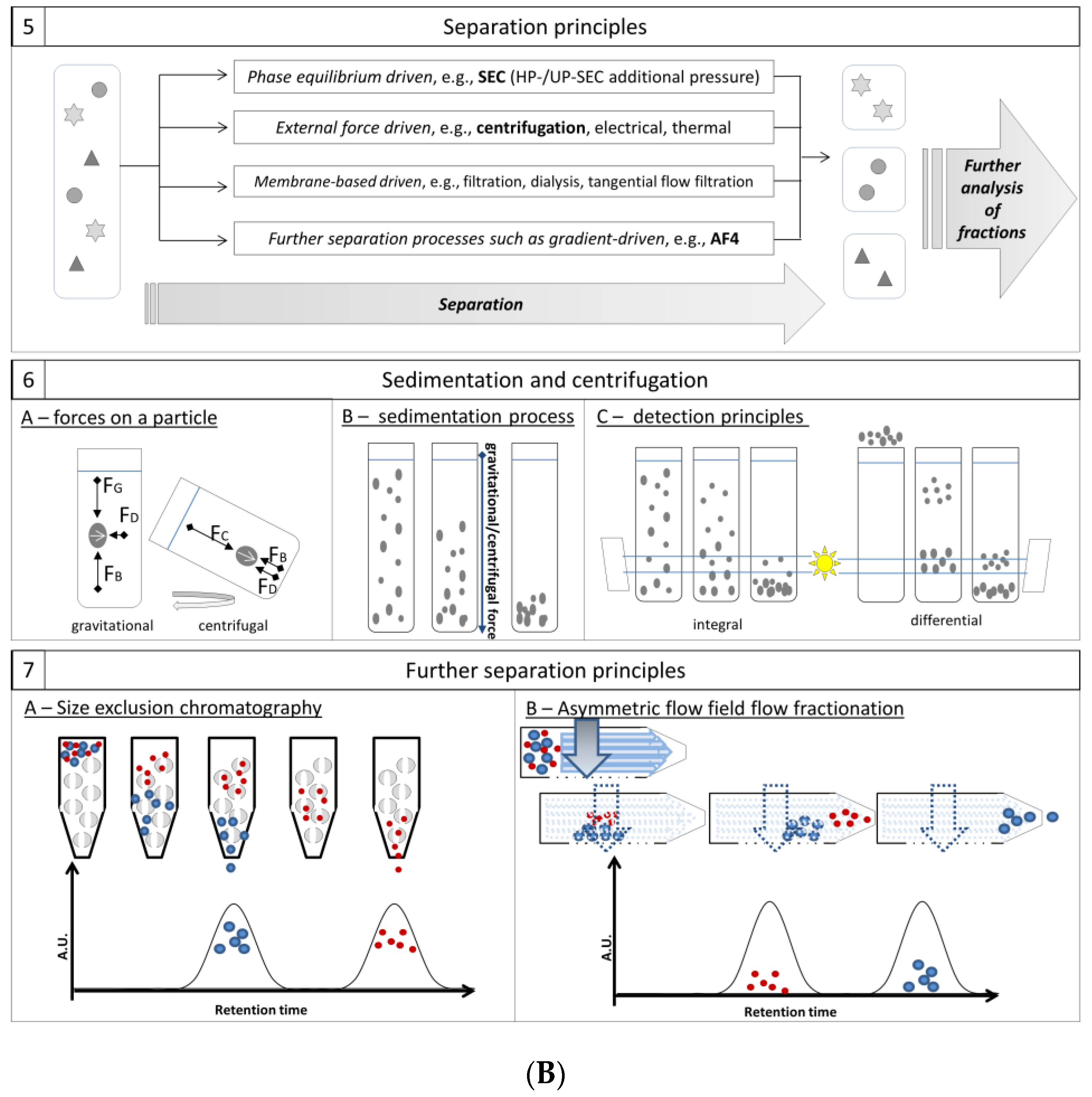
3.5. Separation Principles
3.5.1. Sedimentation and Centrifugation: Principle
| vS | sedimentation velocity | ρS | density of the solvent |
| g | acceleration due to gravity | DP | particle diameter |
| ρP | density of the particle | η | viscosity |
| DP | particle diameter | ρS | density of the solvent |
| η | viscosity | ρP | density of the particle |
| Rf | final radius of rotation | ω | rotational velocity |
| R0 | initial radius of rotation | t | time to move from R0 to Rf |
3.5.2. Centrifugation: Application and Techniques
| c | local concentration | s | sedimentation coefficient |
| t | time | ω | rotor angular velocity |
| r | radial position | D | diffusion coefficient |
3.5.3. Other Separation Principles
4. Examples of Emerging and Alternative Technical Approaches for Biopharmaceuticals
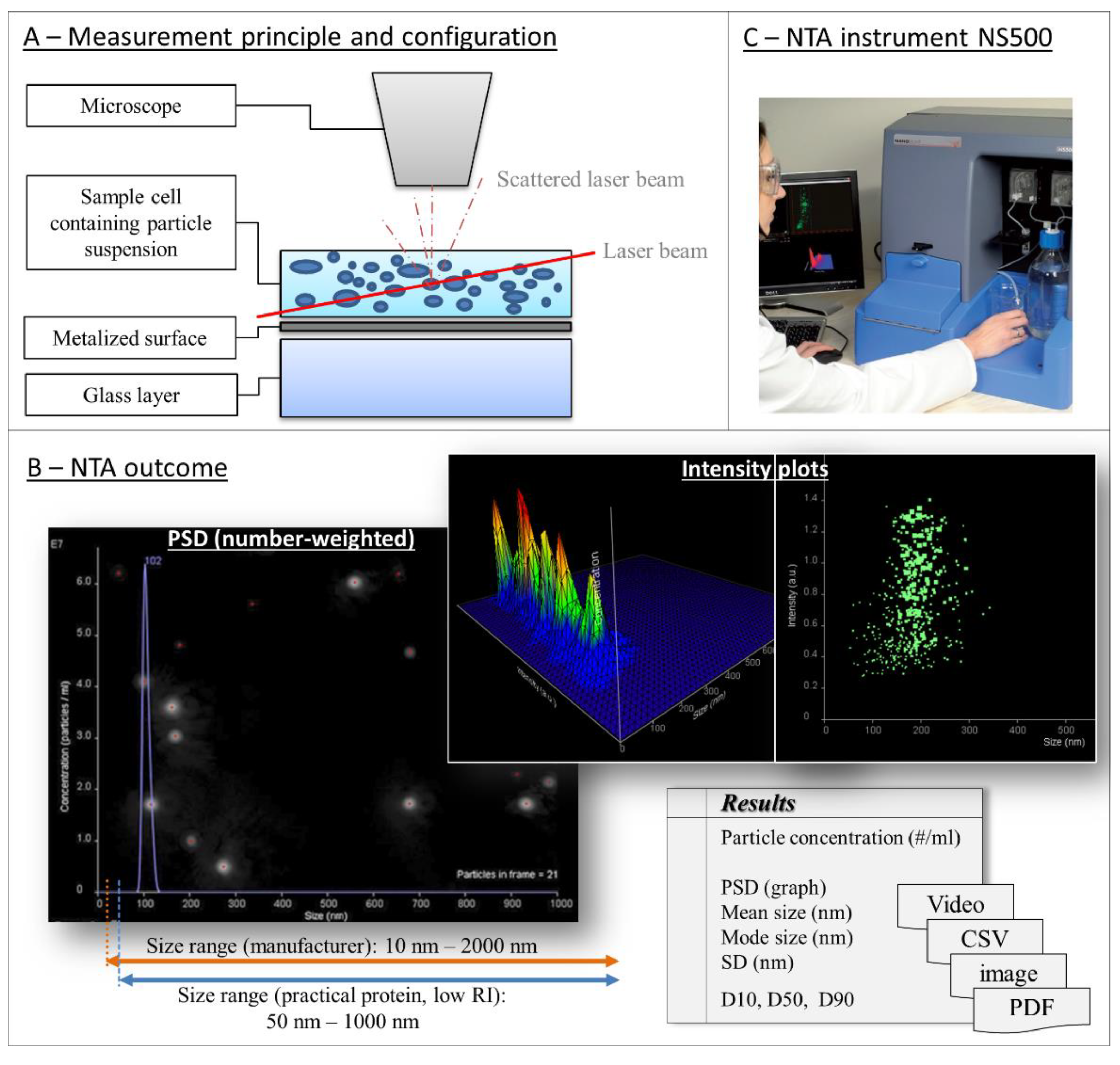
4.1. Nanoparticle Tracking Analysis
| D | diffusion coefficient | η | viscosity |
| T | absolute temperature | dH | spherical-equivalent hydrodynamic diameter |
| κB | Boltzmann´s constant |
4.2. Micro-Flow Imaging
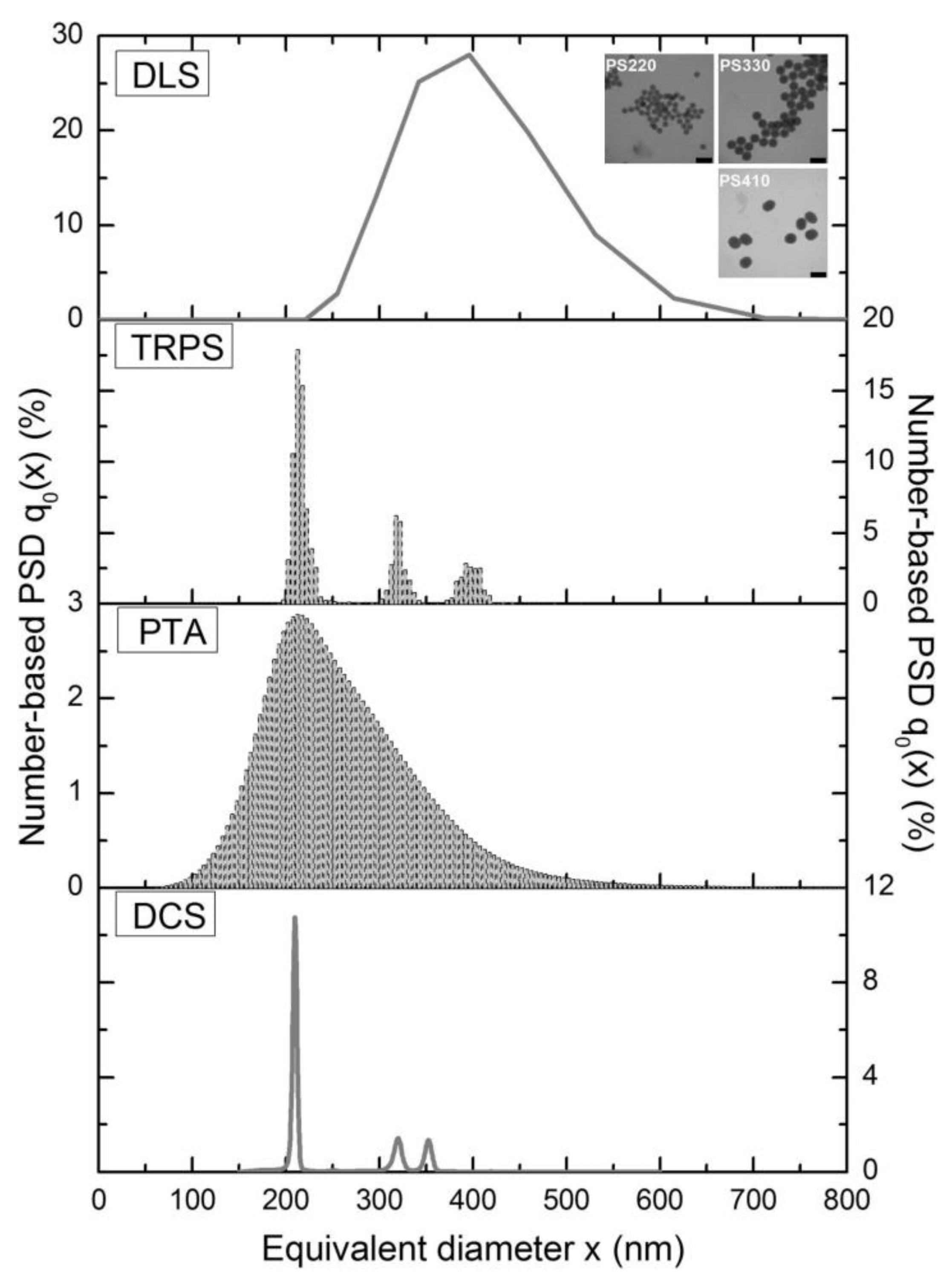
4.3. Tunable Resistive Pulse Sensing (TRPS)
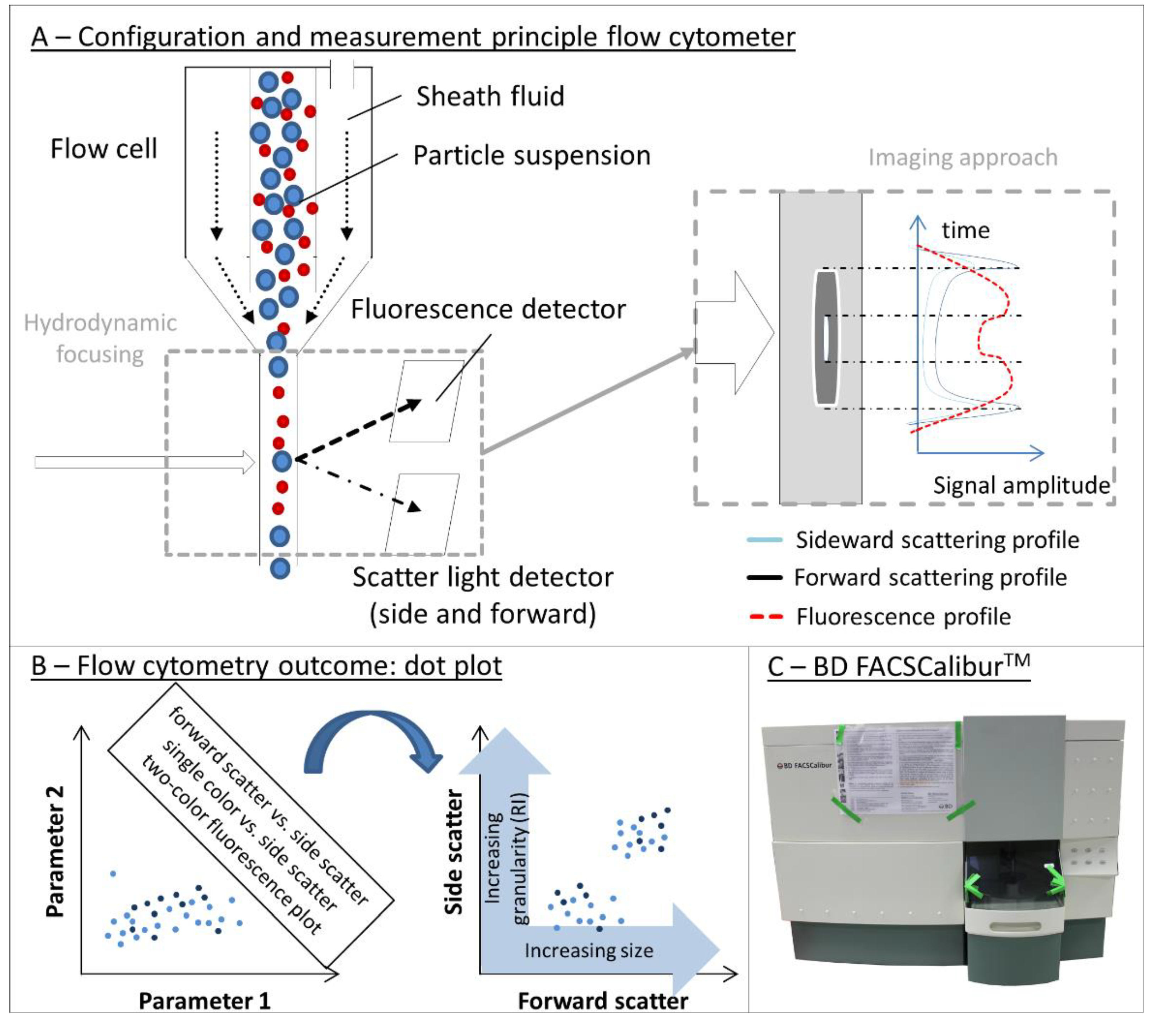
4.4. Flow Cytometry
4.5. Space- and Time-Resolved Extinction Profile (STEP®-Technology)
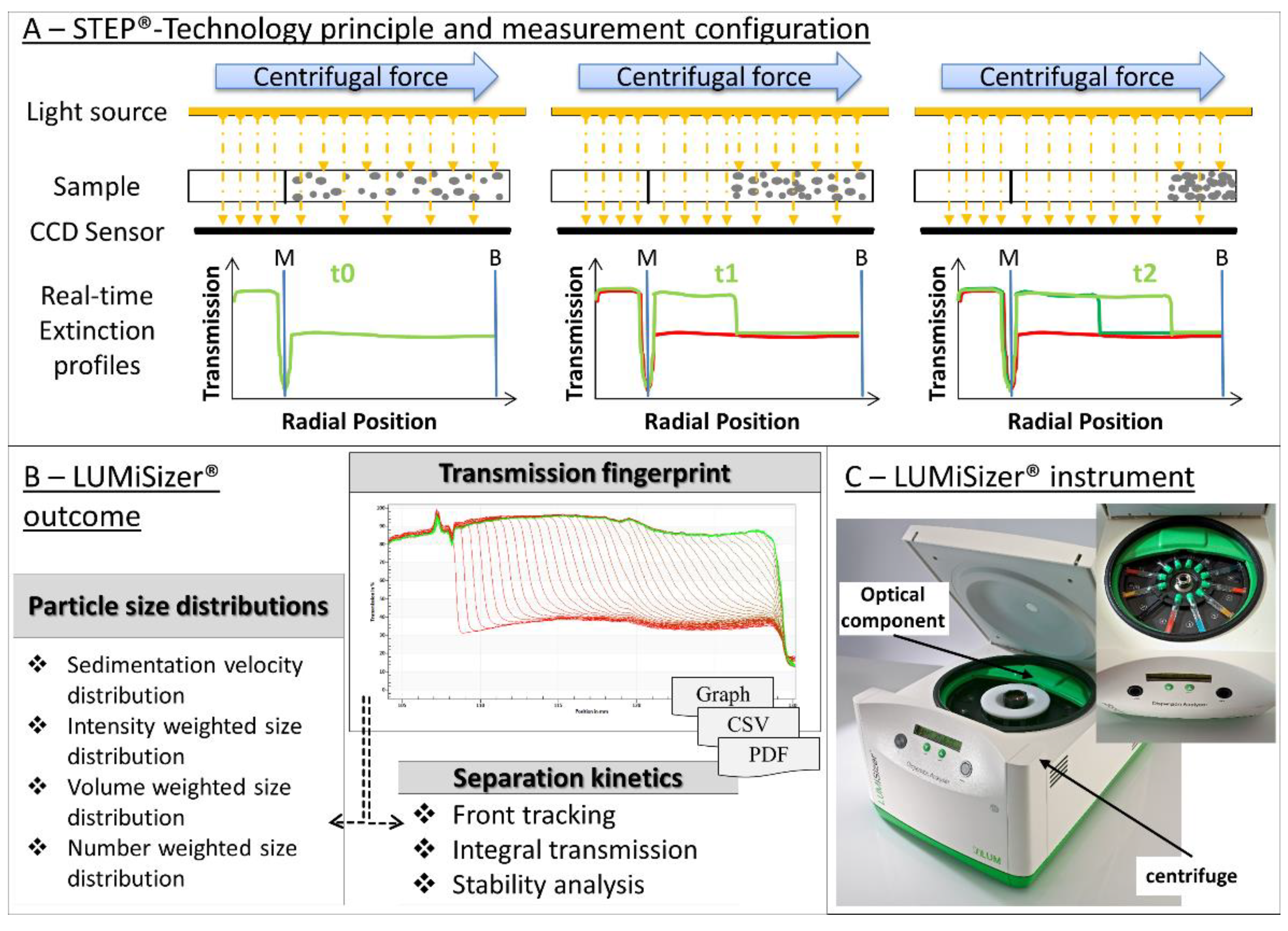
| u | sedimentation velocity | tm | measurement time |
| rm | measurement position | r0 | meniscus position |
5. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Singh, S.K.; Vaidya, Y.; Gulati, M.; Bhattacharya, S.; Garg, V.; Pandey, N.K. Nanosuspension: Principles, Perspectives and Practices. Curr. Drug Deliv. 2016, 13, 1222–1246. [Google Scholar] [CrossRef]
- Silva, A.L.; Soema, P.C.; Slütter, B.; Ossendorp, F.; Jiskoot, W. PLGA particulate delivery systems for subunit vaccines: Linking particle properties to immunogenicity. Hum. Vaccin. Immunother. 2016, 12, 1056–1069. [Google Scholar] [CrossRef]
- Cai, C.; Bakowsky, U.; Rytting, E.; Schaper, A.K.; Kissel, T. Charged nanoparticles as protein delivery systems: A feasibility study using lysozyme as model protein. Eur. J. Pharm. Biopharm. 2008, 69, 31–42. [Google Scholar] [CrossRef]
- Loureiro, J.A.; Gomes, B.; Fricker, G.; Coelho, M.A.N.; Rocha, S.; Pereira, M.C. Cellular uptake of PLGA nanoparticles targeted with anti-amyloid and anti-transferrin receptor antibodies for Alzheimer’s disease treatment. Colloids Surf. B Biointerfaces 2016, 145, 8–13. [Google Scholar] [CrossRef]
- Estep, P.; Caffry, I.; Yu, Y.; Sun, T.; Cao, Y.; Lynaugh, H.; Jain, T.; Vásquez, M.; Tessier, P.M.; Xu, Y. An alternative assay to hydrophobic interaction chromatography for high-throughput characterization of monoclonal antibodies. MAbs. 2015, 7, 553–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, P.; Yu, H.; Guntupalli, B.; Xiao, Y. Paper-Based Device for Rapid Visualization of NADH Based on Dissolution of Gold Nanoparticles. ACS Appl. Mater. Interfaces 2015, 7, 15023–15030. [Google Scholar] [CrossRef] [PubMed]
- Apetri, M.M.; Harkes, R.; Subramaniam, V.; Canters, G.W.; Schmidt, T.; Aartsma, T.J. Direct Observation of α-Synuclein Amyloid Aggregates in Endocytic Vesicles of Neuroblastoma Cells. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [Green Version]
- Cescatti, M.; Saverioni, D.; Capellari, S.; Tagliavini, F.; Kitamoto, T.; Ironside, J.; Giese, A.; Parchi, P. Analysis of Conformational Stability of Abnormal Prion Protein Aggregates across the Spectrum of Creutzfeldt-Jakob Disease Prions. J. Virol. 2016, 90, 6244–6254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eftekharzadeh, B.; Hyman, B.T.; Wegmann, S. Structural studies on the mechanism of protein aggregation in age related neurodegenerative diseases. Mech. Ageing Dev. 2016, 156, 1–13. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Bergink, S. Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 2016, 15, 748–759. [Google Scholar] [CrossRef]
- Polanco, J.C.; Scicluna, B.J.; Hill, A.F.; Götz, J. Extracellular Vesicles Isolated from the Brains of rTg4510 Mice Seed Tau Protein Aggregation in a Threshold-dependent Manner. J. Biol. Chem. 2016, 291, 12445–12466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogen-Shtern, N.; Ben David, T.; Lederkremer, G.Z. Protein aggregation and ER stress. Brain Res. 2016, 1648, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Langille, S.E. Particulate matter in injectable drug products. PDA J. Pharm. Sci. Technol. 2013, 67, 186–200. [Google Scholar] [CrossRef] [Green Version]
- Shomali, M.; Freitag, A.; Engert, J.; Siedler, M.; Kaymakcalan, Z.; Winter, G.; Carpenter, J.F.; Randolph, T.W. Antibody responses in mice to particles formed from adsorption of a murine monoclonal antibody onto glass microparticles. J. Pharm. Sci. 2014, 103, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Shomali, M.; Tanriverdi, S.; Freitag, A.J.; Engert, J.; Winter, G.; Siedler, M.; Kaymakcalan, Z.; Carpenter, J.F.; Randolph, T.W. Dose levels in particulate-containing formulations impact anti-drug antibody responses to murine monoclonal antibody in mice. J. Pharm. Sci. 2015, 104, 1610–1621. [Google Scholar] [CrossRef]
- Shekunov, B.Y.; Chattopadhyay, P.; Tong, H.H.Y.; Chow, A.H.L. Particle size analysis in pharmaceutics: Principles, methods and applications. Pharm. Res. 2007, 24, 203–227. [Google Scholar] [CrossRef]
- Freitag, A.J.; Shomali, M.; Michalakis, S.; Biel, M.; Siedler, M.; Kaymakcalan, Z.; Carpenter, J.F.; Randolph, T.W.; Winter, G.; Engert, J. Investigation of the immunogenicity of different types of aggregates of a murine monoclonal antibody in mice. Pharm. Res. 2015, 32, 430–444. [Google Scholar] [CrossRef]
- Ahmadi, M.; Bryson, C.J.; Cloake, E.A.; Welch, K.; Filipe, V.; Romeijn, S.; Hawe, A.; Jiskoot, W.; Baker, M.P.; Fogg, M.H. Small amounts of sub-visible aggregates enhance the immunogenic potential of monoclonal antibody therapeutics. Pharm. Res. 2015, 32, 1383–1394. [Google Scholar] [CrossRef]
- Johnson, R.; Jiskoot, W. Models for evaluation of relative immunogenic potential of protein particles in biopharmaceutical protein formulations. J. Pharm. Sci. 2012, 101, 3586–3592. [Google Scholar] [CrossRef]
- Jiskoot, W.; Kijanka, G.; Randolph, T.W.; Carpenter, J.F.; Koulov, A.V.; Mahler, H.-C.; Joubert, M.K.; Jawa, V.; Narhi, L.O. Mouse Models for Assessing Protein Immunogenicity: Lessons and Challenges. J. Pharm. Sci. 2016, 105, 1567–1575. [Google Scholar] [CrossRef] [Green Version]
- Hermeling, S.; Crommelin, D.J.A.; Schellekens, H.; Jiskoot, W. Structure-immunogenicity relationships of therapeutic proteins. Pharm. Res. 2004, 21, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Filipe, V.; Jiskoot, W.; Basmeleh, A.H.; Halim, A.; Schellekens, H.; Brinks, V. Immunogenicity of different stressed IgG monoclonal antibody formulations in immune tolerant transgenic mice. MAbs 2012, 4, 740–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinks, V.; Weinbuch, D.; Baker, M.; Dean, Y.; Stas, P.; Kostense, S.; Rup, B.; Jiskoot, W. Preclinical models used for immunogenicity prediction of therapeutic proteins. Pharm. Res. 2013, 30, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Li, G.G.; Cao, S.; Jiao, N.; Wen, Z.-Q. Classification of glass particles in parenteral product vials by visual, microscopic, and spectroscopic methods. PDA J. Pharm. Sci. Technol. 2014, 68, 362–372. [Google Scholar] [CrossRef] [PubMed]
- United States Pharmacopeia Convention. USP<787>: Subvisible Particulate Matter in Therapeutic Protein Injections; United States Pharmacopeia Convention: Washington, DC, USA, 2014. [Google Scholar]
- United States Pharmacopeia Convention. USP<788>: Particulate Matter in Injections; United States Pharmacopeia Convention: Washington, DC, USA, 2014. [Google Scholar]
- United States Pharmacopeia Convention. USP<1787>: Subvisible Particulate Matter in Therapeutic Protein Injections; United States Pharmacopeia Convention: Washington, DC, USA, 2015. [Google Scholar]
- Syvitski, J.P.M. (Ed.) Principles, Methods, and Application of Particle Size Analysis; Reprinted; Cambridge Univ. Press: Cambridge, UK, 1997; ISBN 978-0-521-36472-0. [Google Scholar]
- Bauer, K.C.; Schermeyer, M.-T.; Seidel, J.; Hubbuch, J. Impact of polymer surface characteristics on the microrheological measurement quality of protein solutions A tracer particle screening. Int. J. Pharm. 2016, 505, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Hirn, S.; Semmler-Behnke, M.; Schleh, C.; Wenk, A.; Lipka, J.; Schäffler, M.; Takenaka, S.; Möller, W.; Schmid, G.; Simon, U.; et al. Particle size-dependent and surface charge-dependent biodistribution of gold nanoparticles after intravenous administration. Eur. J. Pharm. Biopharm. 2011, 77, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Narhi, L.O.; Schmit, J.; Bechtold-Peters, K.; Sharma, D. Classification of protein aggregates. J. Pharm. Sci. 2012, 101, 493–498. [Google Scholar] [CrossRef]
- Cavicchi, R.E.; Carrier, M.J.; Cohen, J.B.; Boger, S.; Montgomery, C.B.; Hu, Z.; Ripple, D.C. Particle shape effects on subvisible particle sizing measurements. J. Pharm. Sci. 2015, 104, 971–987. [Google Scholar] [CrossRef]
- Olsen, S.N.; Andersen, K.B.; Randolph, T.W.; Carpenter, J.F.; Westh, P. Role of electrostatic repulsion on colloidal stability of Bacillus halmapalus alpha-amylase. Biochim. Biophys. Acta 2009, 1794, 1058–1065. [Google Scholar] [CrossRef]
- Anderson, W.; Kozak, D.; Coleman, V.A.; Jämting, Å.K.; Trau, M. A comparative study of submicron particle sizing platforms: Accuracy, precision and resolution analysis of polydisperse particle size distributions. J. Colloid Interface Sci. 2013, 405, 322–330. [Google Scholar] [CrossRef]
- Zölls, S.; Gregoritza, M.; Tantipolphan, R.; Wiggenhorn, M.; Winter, G.; Friess, W.; Hawe, A. How subvisible particles become invisible-relevance of the refractive index for protein particle analysis. J. Pharm. Sci. 2013, 102, 1434–1446. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, C.; Shaw, M.; Hole, P.; Smith, J.; Tannetta, D.; Redman, C.W.; Sargent, I.L. Measurement of refractive index by nanoparticle tracking analysis reveals heterogeneity in extracellular vesicles. J. Extracell. Vesicles 2014, 3, 25361. [Google Scholar] [CrossRef] [PubMed]
- Planken, K.L.; Cölfen, H. Analytical ultracentrifugation of colloids. Nanoscale 2010, 2, 1849–1869. [Google Scholar] [CrossRef] [PubMed]
- Folzer, E.; Khan, T.A.; Schmidt, R.; Finkler, C.; Huwyler, J.; Mahler, H.-C.; Koulov, A.V. Determination of the Density of Protein Particles Using a Suspended Microchannel Resonator. J. Pharm. Sci. 2015, 104, 4034–4040. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, C.F.; Nguyen, B.H.; Soucie, K.R.; Torres, R.M.; Carpenter, J.F.; Randolph, T.W. In Vivo Analysis of the Potency of Silicone Oil Microdroplets as Immunological Adjuvants in Protein Formulations. J. Pharm. Sci. 2015, 104, 3681–3690. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Ammar, D.A.; Ross, L.A.; Mandava, N.; Kahook, M.Y.; Carpenter, J.F. Silicone oil microdroplets and protein aggregates in repackaged bevacizumab and ranibizumab: Effects of long-term storage and product mishandling. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1023–1034. [Google Scholar] [CrossRef]
- Liu, L.; Randolph, T.W.; Carpenter, J.F. Particles shed from syringe filters and their effects on agitation-induced protein aggregation. J. Pharm. Sci. 2012, 101, 2952–2959. [Google Scholar] [CrossRef]
- Nicoud, L.; Owczarz, M.; Arosio, P.; Morbidelli, M. A multiscale view of therapeutic protein aggregation: A colloid science perspective. Biotechnol. J. 2015, 10, 367–378. [Google Scholar] [CrossRef]
- Salinas, B.A.; Sathish, H.A.; Bishop, S.M.; Harn, N.; Carpenter, J.F.; Randolph, T.W. Understanding and modulating opalescence and viscosity in a monoclonal antibody formulation. J. Pharm. Sci. 2010, 99, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Asherie, N.; Lomakin, A.; Benedek, G.B. Phase Diagram of Colloidal Solutions. Phys. Rev. Lett. 1996, 77, 4832–4835. [Google Scholar] [CrossRef] [Green Version]
- Basheva, E.S.; Kralchevsky, P.A.; Danov, K.D.; Ananthapadmanabhan, K.P.; Lips, A. The colloid structural forces as a tool for particle characterization and control of dispersion stability. Phys. Chem. Chem. Phys. 2007, 9, 5183–5198. [Google Scholar] [CrossRef] [PubMed]
- Garidel, P.; Blume, A.; Wagner, M. Prediction of colloidal stability of high concentration protein formulations. Pharm. Dev. Technol. 2015, 20, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Le Brun, V.; Friess, W.; Bassarab, S.; Garidel, P. Correlation of protein-protein interactions as assessed by affinity chromatography with colloidal protein stability: A case study with lysozyme. Pharm. Dev. Technol. 2010, 15, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Connolly, B.D.; Petry, C.; Yadav, S.; Demeule, B.; Ciaccio, N.; Moore, J.M.R.; Shire, S.J.; Gokarn, Y.R. Weak interactions govern the viscosity of concentrated antibody solutions: High-throughput analysis using the diffusion interaction parameter. Biophys. J. 2012, 103, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, T.; Kita, Y.; Carpenter, J.F. Protein--solvent interactions in pharmaceutical formulations. Pharm. Res. 1991, 8, 285–291. [Google Scholar] [CrossRef]
- Gebauer, J.S.; Malissek, M.; Simon, S.; Knauer, S.K.; Maskos, M.; Stauber, R.H.; Peukert, W.; Treuel, L. Impact of the nanoparticle-protein corona on colloidal stability and protein structure. Langmuir 2012, 28, 9673–9679. [Google Scholar] [CrossRef]
- Donoso, M.D.; Haskell, R.J.; Schartman, R.R. Surfactant choice and the physical stability of nanosuspensions as a function of pH. Int. J. Pharm. 2012, 439, 1–7. [Google Scholar] [CrossRef]
- Mathonet, S.; Mahler, H.-C.; Esswein, S.T.; Mazaheri, M.; Cash, P.W.; Wuchner, K.; Kallmeyer, G.; Das, T.K.; Finkler, C.; Lennard, A. A Biopharmaceutical Industry Perspective on the Control of Visible Particles in Biotechnology-Derived Injectable Drug Products. PDA J. Pharm. Sci. Technol. 2016, 70, 392–408. [Google Scholar] [CrossRef]
- Chi, E.Y.; Krishnan, S.; Randolph, T.W.; Carpenter, J.F. Physical stability of proteins in aqueous solution: Mechanism and driving forces in nonnative protein aggregation. Pharm. Res. 2003, 20, 1325–1336. [Google Scholar] [CrossRef]
- Gokarn, Y.R.; Fesinmeyer, R.M.; Saluja, A.; Cao, S.; Dankberg, J.; Goetze, A.; Remmele, R.L.; Narhi, L.O.; Brems, D.N. Ion-specific modulation of protein interactions: Anion-induced, reversible oligomerization of a fusion protein. Protein Sci. 2009, 18, 169–179. [Google Scholar] [CrossRef] [Green Version]
- DePaz, R.A.; Barnett, C.C.; Dale, D.A.; Carpenter, J.F.; Gaertner, A.L.; Randolph, T.W. The excluding effects of sucrose on a protein chemical degradation pathway: Methionine oxidation in subtilisin. Arch. Biochem. Biophys. 2000, 384, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Pikal-Cleland, K.A.; Cleland, J.L.; Anchordoquy, T.J.; Carpenter, J.F. Effect of glycine on pH changes and protein stability during freeze-thawing in phosphate buffer systems. J. Pharm. Sci. 2002, 91, 1969–1979. [Google Scholar] [CrossRef] [PubMed]
- Tessier, P.M.; Jinkoji, J.; Cheng, Y.-C.; Prentice, J.L.; Lenhoff, A.M. Self-interaction nanoparticle spectroscopy: A nanoparticle-based protein interaction assay. J. Am. Chem. Soc. 2008, 130, 3106–3112. [Google Scholar] [CrossRef] [PubMed]
- Godavarti, M.; Rodriguez, J.J.; Yopp, T.A.; Lambert, G.M.; Galbraith, D.W. Automated particle classification based on digital acquisition and analysis of flow cytometric pulse waveforms. Cytometry 1996, 24, 330–339. [Google Scholar] [CrossRef]
- Joubert, M.K.; Luo, Q.; Nashed-Samuel, Y.; Wypych, J.; Narhi, L.O. Classification and characterization of therapeutic antibody aggregates. J. Biol. Chem. 2011, 286, 25118–25133. [Google Scholar] [CrossRef] [Green Version]
- Noel, V.; Chepfer, H.; Ledanois, G.; Delaval, A.; Flamant, P.H. Classification of particle effective shape ratios in cirrus clouds based on the lidar depolarization ratio. Appl. Opt. 2002, 41, 4245–4257. [Google Scholar] [CrossRef]
- Sanchez-Ruiz, J.M. Protein kinetic stability. Biophys. Chem. 2010, 148, 1–15. [Google Scholar] [CrossRef]
- Philo, J.S.; Arakawa, T. Mechanisms of protein aggregation. Curr. Pharm. Biotechnol. 2009, 10, 348–351. [Google Scholar] [CrossRef]
- Nicoud, L.; Arosio, P.; Sozo, M.; Yates, A.; Norrant, E.; Morbidelli, M. Kinetic analysis of the multistep aggregation mechanism of monoclonal antibodies. J. Phys. Chem. B 2014, 118, 10595–10606. [Google Scholar] [CrossRef]
- Morris, A.M.; Watzky, M.A.; Finke, R.G. Protein aggregation kinetics, mechanism, and curve-fitting: A review of the literature. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2009, 1794, 375–397. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Randolph, T.W.; Stevens, F.J.; Carpenter, J.F. Kinetics and energetics of assembly, nucleation, and growth of aggregates and fibrils for an amyloidogenic protein. Insights into transition states from pressure, temperature, and co-solute studies. J. Biol. Chem. 2002, 277, 27240–27246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frieden, C. Protein aggregation processes: In search of the mechanism. Protein Sci. 2007, 16, 2334–2344. [Google Scholar] [CrossRef] [PubMed]
- De Jong, W.H.; Hagens, W.I.; Krystek, P.; Burger, M.C.; Sips, A.J.A.M.; Geertsma, R.E. Particle size-dependent organ distribution of gold nanoparticles after intravenous administration. Biomaterials 2008, 29, 1912–1919. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Heller, D.A.; Sharma, R.; Strano, M.S. Size-dependent cellular uptake and expulsion of single-walled carbon nanotubes: Single particle tracking and a generic uptake model for nanoparticles. ACS Nano 2009, 3, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Koide, H.; Asai, T.; Hatanaka, K.; Urakami, T.; Ishii, T.; Kenjo, E.; Nishihara, M.; Yokoyama, M.; Ishida, T.; Kiwada, H.; et al. Particle size-dependent triggering of accelerated blood clearance phenomenon. Int. J. Pharm. 2008, 362, 197–200. [Google Scholar] [CrossRef]
- Rong, Y.; Zhou, T.; Cheng, W.; Guo, J.; Cui, X.; Liu, Y.; Chen, W. Particle-size-dependent cytokine responses and cell damage induced by silica particles and macrophages-derived mediators in endothelial cell. Environ. Toxicol. Pharmacol. 2013, 36, 921–928. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Ito, A.; Sogo, Y.; Ohno, T. Particle-size-dependent toxicity and immunogenic activity of mesoporous silica-based adjuvants for tumor immunotherapy. Acta Biomater. 2013, 9, 7480–7489. [Google Scholar] [CrossRef]
- Slütter, B.; Jiskoot, W. Sizing the optimal dimensions of a vaccine delivery system: A particulate matter. Expert Opin. Drug Deliv. 2016, 13, 167–170. [Google Scholar] [CrossRef]
- Haskell, R.J. Characterization of submicron systems via optical methods. J. Pharm. Sci. 1998, 87, 125–129. [Google Scholar] [CrossRef]
- Albrechtsson, U.; Hansson, G.A.; Olin, T. Vascular occlusion with a ferromagnetic particle suspension. An experimental investigation in rabbits. Acta Radiol. Diagn. 1977, 18, 279–291. [Google Scholar] [CrossRef]
- Bernard, S.L.; Obermiller, T.; Polissar, N.L.; Mendenhall, J.M.; Butler, J.; Lakshminarayan, S. Fifteen micrometer microspheres reflux up the pulmonary veins during pulmonary artery occlusion. Microvasc. Res. 1993, 45, 262–268. [Google Scholar] [CrossRef]
- Zheng, S.; Bai, Y.-Y.; Changyi, Y.; Gao, X.; Zhang, W.; Wang, Y.; Zhou, L.; Ju, S.; Li, C. Multimodal nanoprobes evaluating physiological pore size of brain vasculatures in ischemic stroke models. Adv. Healthc. Mater. 2014, 3, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Garidel, P.; Hegyi, M.; Bassarab, S.; Weichel, M. A rapid, sensitive and economical assessment of monoclonal antibody conformational stability by intrinsic tryptophan fluorescence spectroscopy. Biotechnol. J. 2008, 3, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Verwey, E.J.W. Theory of the stability of lyophobic colloids. J. Phys. Colloid Chem. 1947, 51, 631–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derjaguin, B. On the repulsive forces between charged colloid particles and on the theory of slow coagulation and stability of lyophobe sols. Trans. Faraday Soc. 1940, 35, 203–215. [Google Scholar] [CrossRef]
- Le Brun, V.; Friess, W.; Bassarab, S.; Mühlau, S.; Garidel, P. A critical evaluation of self-interaction chromatography as a predictive tool for the assessment of protein-protein interactions in protein formulation development: A case study of a therapeutic monoclonal antibody. Eur. J. Pharm. Biopharm. 2010, 75, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Jiao, N.; Jiang, Y.; Mire-Sluis, A.; Narhi, L.O. Sub-visible particle quantitation in protein therapeutics. Pharmeur. Biol. Sci. Notes 2009, 2009, 73–79. [Google Scholar]
- Wagner, M.; Reiche, K.; Blume, A.; Garidel, P. Viscosity measurements of antibody solutions by photon correlation spectroscopy: An indirect approach limitations and applicability for high-concentration liquid protein solutions. Pharm. Dev. Technol. 2013, 18, 963–970. [Google Scholar] [CrossRef]
- Karow, A.R.; Götzl, J.; Garidel, P. Resolving power of dynamic light scattering for protein and polystyrene nanoparticles. Pharm. Dev. Technol. 2015, 20, 84–89. [Google Scholar] [CrossRef]
- Den Engelsman, J.; Kebbel, F.; Garidel, P. Laser Light Scattering-Based Techniques Used for the Characterization of Protein Therapeutics. In Analysis of Aggregates and Particles in Protein Pharmaceuticals; Mahler, H.-C., Jiskoot, W., Eds.; John Wiley & Sons: Chicester, UK, 2012; pp. 37–60. ISBN 9781118150573. [Google Scholar]
- Van der Pol, E.; Hoekstra, A.G.; Sturk, A.; Otto, C.; van Leeuwen, T.G.; Nieuwland, R. Optical and non-optical methods for detection and characterization of microparticles and exosomes. J. Thromb. Haemost. 2010, 8, 2596–2607. [Google Scholar] [CrossRef]
- Sung, J.J.; Pardeshi, N.N.; Mulder, A.M.; Mulligan, S.K.; Quispe, J.; On, K.; Carragher, B.; Potter, C.S.; Carpenter, J.F.; Schneemann, A. Transmission electron microscopy as an orthogonal method to characterize protein aggregates. J. Pharm. Sci. 2015, 104, 750–759. [Google Scholar] [CrossRef] [Green Version]
- Demeule, B.; Messick, S.; Shire, S.J.; Liu, J. Characterization of particles in protein solutions: Reaching the limits of current technologies. AAPS J. 2010, 12, 708–715. [Google Scholar] [CrossRef] [Green Version]
- Mehta, S.B.; Oldenbourg, R. Image simulation for biological microscopy: Microlith. Biomed. Opt. Express 2014, 5, 1822–1838. [Google Scholar] [CrossRef] [Green Version]
- Wilson, G.A.; Manning, M.C. Flow imaging: Moving toward best practices for subvisible particle quantitation in protein products. J. Pharm. Sci. 2013, 102, 1133–1134. [Google Scholar] [CrossRef]
- Sawyer, N.B.E.; Morgan, S.P.; Somekh, M.G.; See, C.W.; Astrakharchik-Farrimond, E.; Shekunov, B.Y. Amplitude and phase microscopy for sizing of spherical particles. Appl. Opt. 2003, 42, 4488–4498. [Google Scholar] [CrossRef]
- Kumru, O.S.; Liu, J.; Ji, J.A.; Cheng, W.; Wang, Y.J.; Wang, T.; Joshi, S.B.; Middaugh, C.R.; Volkin, D.B. Compatibility, physical stability, and characterization of an IgG4 monoclonal antibody after dilution into different intravenous administration bags. J. Pharm. Sci. 2012, 101, 3636–3650. [Google Scholar] [CrossRef]
- Telikepalli, S.N.; Kumru, O.S.; Kalonia, C.; Esfandiary, R.; Joshi, S.B.; Middaugh, C.R.; Volkin, D.B. Structural characterization of IgG1 mAb aggregates and particles generated under various stress conditions. J. Pharm. Sci. 2014, 103, 796–809. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Kumru, O.S.; Yi, L.; Wang, Y.J.; Zhang, J.; Kim, J.H.; Joshi, S.B.; Middaugh, C.R.; Volkin, D.B. Effect of ionic strength and pH on the physical and chemical stability of a monoclonal antibody antigen-binding fragment. J. Pharm. Sci. 2013, 102, 2520–2537. [Google Scholar] [CrossRef]
- Wang, W.; Singh, S.; Zeng, D.L.; King, K.; Nema, S. Antibody structure, instability, and formulation. J. Pharm. Sci. 2007, 96, 1–26. [Google Scholar] [CrossRef]
- Corvari, V.; Narhi, L.O.; Spitznagel, T.M.; Afonina, N.; Cao, S.; Cash, P.; Cecchini, I.; DeFelippis, M.R.; Garidel, P.; Herre, A.; et al. Subvisible (2–100 μm) particle analysis during biotherapeutic drug product development: Part 2, experience with the application of subvisible particle analysis. Biologicals 2015, 43, 457–473. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.R.; Lau, D.; Liu, J. Quantification and characterization of micrometer and submicrometer subvisible particles in protein therapeutics by use of a suspended microchannel resonator. Anal. Chem. 2012, 84, 6833–6840. [Google Scholar] [CrossRef]
- Ripple, D.C.; Dimitrova, M.N. Protein particles: What we know and what we do not know. J. Pharm. Sci. 2012, 101, 3568–3579. [Google Scholar] [CrossRef]
- Vasudev, R.; Mathew, S.; Afonina, N. Characterization of Submicron (0.1–1 μm) Particles in Therapeutic Proteins by Nanoparticle Tracking Analysis. J. Pharm. Sci. 2015, 104, 1622–1631. [Google Scholar] [CrossRef]
- Mahler, H.-C.; Friess, W.; Grauschopf, U.; Kiese, S. Protein aggregation: Pathways, induction factors and analysis. J. Pharm. Sci. 2009, 98, 2909–2934. [Google Scholar] [CrossRef]
- Huang, C.-T.; Sharma, D.; Oma, P.; Krishnamurthy, R. Quantitation of protein particles in parenteral solutions using micro-flow imaging. J. Pharm. Sci. 2009, 98, 3058–3071. [Google Scholar] [CrossRef]
- Sharma, D.K.; King, D.; Oma, P.; Merchant, C. Micro-flow imaging: Flow microscopy applied to sub-visible particulate analysis in protein formulations. AAPS J. 2010, 12, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Squire, P.G.; Himmel, M.E. Hydrodynamics and protein hydration. Arch. Biochem. Biophys. 1979, 196, 165–177. [Google Scholar] [CrossRef]
- Fischer, H.; Polikarpov, I.; Craievich, A.F. Average protein density is a molecular-weight-dependent function. Protein Sci. 2004, 13, 2825–2828. [Google Scholar] [CrossRef]
- Godin, M.; Bryan, A.K.; Burg, T.P.; Babcock, K.; Manalis, S.R. Measuring the mass, density, and size of particles and cells using a suspended microchannel resonator. Appl. Phys. Lett. 2007, 91, 123121. [Google Scholar] [CrossRef] [Green Version]
- Lammers, W.T. Density-Gradient Separation of Organic and Inorganic Particles by Centrifugation. Science 1963, 139, 1298–1299. [Google Scholar] [CrossRef]
- Alford, J.R.; Kendrick, B.S.; Carpenter, J.F.; Randolph, T.W. Measurement of the second osmotic virial coefficient for protein solutions exhibiting monomer-dimer equilibrium. Anal. Biochem. 2008, 377, 128–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asherie, N.; Pande, J.; Lomakin, A.; Ogun, O.; Hanson, S.R.; Smith, J.B.; Benedek, G.B. Oligomerization and phase separation in globular protein solutions. Biophys. Chem. 1998, 75, 213–227. [Google Scholar] [CrossRef]
- Fodeke, A.A.; Minton, A.P. Quantitative characterization of temperature-independent and temperature-dependent protein-protein interactions in highly nonideal solutions. J. Phys. Chem. B 2011, 115, 11261–11268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrielson, J.P.; Brader, M.L.; Pekar, A.H.; Mathis, K.B.; Winter, G.; Carpenter, J.F.; Randolph, T.W. Quantitation of aggregate levels in a recombinant humanized monoclonal antibody formulation by size-exclusion chromatography, asymmetrical flow field flow fractionation, and sedimentation velocity. J. Pharm. Sci. 2007, 96, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Le Ru, E.C.; Etchegoin, P.G. Self-limiting aggregation leads to long-lived metastable clusters in colloidal solutions. J. Phys. Chem. B 2006, 110, 6040–6047. [Google Scholar] [CrossRef]
- Gerhardt, A.; Bonam, K.; Bee, J.S.; Carpenter, J.F.; Randolph, T.W. Ionic Strength Affects Tertiary Structure and Aggregation Propensity of a Monoclonal Antibody Adsorbed to Silicone Oil–Water Interfaces. J. Pharm. Sci. 2013, 102, 429–440. [Google Scholar] [CrossRef]
- Turko, I.V. A New Approach to assess mAb Aggregation. Methods Mol. Biol. 2020, 2131, 245–254. [Google Scholar] [CrossRef]
- Kueltzo, L.A.; Wang, W.; Randolph, T.W.; Carpenter, J.F. Effects of solution conditions, processing parameters, and container materials on aggregation of a monoclonal antibody during freeze-thawing. J. Pharm. Sci. 2008, 97, 1801–1812. [Google Scholar] [CrossRef]
- Chi, E.Y.; Weickmann, J.; Carpenter, J.F.; Manning, M.C.; Randolph, T.W. Heterogeneous nucleation-controlled particulate formation of recombinant human platelet-activating factor acetylhydrolase in pharmaceutical formulation. J. Pharm. Sci. 2005, 94, 256–274. [Google Scholar] [CrossRef]
- Wang, W.; Ohtake, S. Science and art of protein formulation development. Int. J. Pharm. 2019, 568, 118505. [Google Scholar] [CrossRef]
- Kiese, S.; Pappenberger, A.; Friess, W.; Mahler, H.-C. Equilibrium studies of protein aggregates and homogeneous nucleation in protein formulation. J. Pharm. Sci. 2010, 99, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Nicoud, L.; Jagielski, J.; Pfister, D.; Lazzari, S.; Massant, J.; Lattuada, M.; Morbidelli, M. Kinetics of Monoclonal Antibody Aggregation from Dilute toward Concentrated Conditions. J. Phys. Chem. B 2016, 120, 3267–3280. [Google Scholar] [CrossRef] [PubMed]
- Nicoud, L.; Lattuada, M.; Yates, A.; Morbidelli, M. Impact of aggregate formation on the viscosity of protein solutions. Soft Matter 2015, 11, 5513–5522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gühlke, M.; Hecht, J.; Böhrer, A.; Hawe, A.; Nikels, F.; Garidel, P.; Menzen, T. Taking Subvisible Particle Quantitation to the Limit: Uncertainties and Statistical Challenges With Ophthalmic Products for Intravitreal Injection. J. Pharm. Sci. 2020, 109, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Prestrelski, S.J.; Kenney, W.C.; Carpenter, J.F. Factors affecting short-term and long-term stabilities of proteins. Adv. Drug Deliv. Rev. 2001, 46, 307–326. [Google Scholar] [CrossRef]
- Carpenter, J.F.; Manning, M.C.; Randolph, T.W. Long-term storage of proteins. Curr. Protoc. Protein Sci. 2002, 27, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, B.S.; Cleland, J.L.; Lam, X.; Nguyen, T.; Randolph, T.W.; Manning, M.C.; Carpenter, J.F. Aggregation of recombinant human interferon gamma: Kinetics and structural transitions. J. Pharm. Sci. 1998, 87, 1069–1076. [Google Scholar] [CrossRef]
- McDonald, T.A.; Zepeda, M.L.; Tomlinson, M.J.; Bee, W.H.; Ivens, I.A. Subcutaneous administration of biotherapeutics: Current experience in animal models. Curr. Opin. Mol. Ther. 2010, 12, 461–470. [Google Scholar]
- Jackisch, C.; Müller, V.; Maintz, C.; Hell, S.; Ataseven, B. Subcutaneous Administration of Monoclonal Antibodies in Oncology. Geburtshilfe Frauenheilkd. 2014, 74, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Garidel, P.; Kuhn, A.B.; Schäfer, L.V.; Karow-Zwick, A.R.; Blech, M. High-concentration protein formulations: How high is high? Eur. J. Pharm. Biopharm. 2017, 119, 353–360. [Google Scholar] [CrossRef]
- Ellis, R.J.; Minton, A.P. Protein aggregation in crowded environments. Biol. Chem. 2006, 387, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Blaffert, J.; Haeri, H.H.; Blech, M.; Hinderberger, D.; Garidel, P. Spectroscopic methods for assessing the molecular origins of macroscopic solution properties of highly concentrated liquid protein solutions. Anal. Biochem. 2018, 561–562, 70–88. [Google Scholar] [CrossRef] [PubMed]
- Nicoud, L.; Lattuada, M.; Lazzari, S.; Morbidelli, M. Viscosity scaling in concentrated dispersions and its impact on colloidal aggregation. Phys. Chem. Chem. Phys. 2015, 17, 24392–24402. [Google Scholar] [CrossRef] [PubMed]
- Pindrus, M.; Shire, S.J.; Kelley, R.F.; Demeule, B.; Wong, R.; Xu, Y.; Yadav, S. Solubility Challenges in High Concentration Monoclonal Antibody Formulations: Relationship with Amino Acid Sequence and Intermolecular Interactions. Mol. Pharm. 2015, 12, 3896–3907. [Google Scholar] [CrossRef]
- Bee, J.S.; Davis, M.; Freund, E.; Carpenter, J.F.; Randolph, T.W. Aggregation of a monoclonal antibody induced by adsorption to stainless steel. Biotechnol. Bioeng. 2010, 105, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Bee, J.S.; Nelson, S.A.; Freund, E.; Carpenter, J.F.; Randolph, T.W. Precipitation of a monoclonal antibody by soluble tungsten. J. Pharm. Sci. 2009, 98, 3290–3301. [Google Scholar] [CrossRef] [Green Version]
- Bee, J.S.; Randolph, T.W.; Carpenter, J.F.; Bishop, S.M.; Dimitrova, M.N. Effects of surfaces and leachables on the stability of biopharmaceuticals. J. Pharm. Sci. 2011, 100, 4158–4170. [Google Scholar] [CrossRef]
- Britt, K.A.; Schwartz, D.K.; Wurth, C.; Mahler, H.-C.; Carpenter, J.F.; Randolph, T.W. Excipient effects on humanized monoclonal antibody interactions with silicone oil emulsions. J. Pharm. Sci. 2012, 101, 4419–4432. [Google Scholar] [CrossRef]
- Ludwig, D.B.; Carpenter, J.F.; Hamel, J.-B.; Randolph, T.W. Protein adsorption and excipient effects on kinetic stability of silicone oil emulsions. J. Pharm. Sci. 2010, 99, 1721–1733. [Google Scholar] [CrossRef]
- Weinbuch, D.; Zölls, S.; Wiggenhorn, M.; Friess, W.; Winter, G.; Jiskoot, W.; Hawe, A. Micro-flow imaging and resonant mass measurement (Archimedes)—Complementary methods to quantitatively differentiate protein particles and silicone oil droplets. J. Pharm. Sci. 2013, 102, 2152–2165. [Google Scholar] [CrossRef]
- Ripple, D.C.; Montgomery, C.B.; Hu, Z. An interlaboratory comparison of sizing and counting of subvisible particles mimicking protein aggregates. J. Pharm. Sci. 2015, 104, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Ripple, D.C.; Wayment, J.R.; Carrier, M.J. Standards for Optical Detection of Protein Particles. Am. Pharm. Rev. 2012, 14, 90–96. [Google Scholar]
- Varenne, F.; Makky, A.; Gaucher-Delmas, M.; Violleau, F.; Vauthier, C. Multimodal Dispersion of Nanoparticles: A Comprehensive Evaluation of Size Distribution with 9 Size Measurement Methods. Pharm. Res. 2016, 33, 1220–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stetefeld, J.; McKenna, S.A.; Patel, T.R. Dynamic light scattering: A practical guide and applications in biomedical sciences. Biophys. Rev. 2016, 8, 409–427. [Google Scholar] [CrossRef]
- United States Pharmacopeia Convention. USP<776>: Optical Microscopy; United States Pharmacopeia Convention: Washington, DC, USA, 2016. [Google Scholar]
- United States Pharmacopeia Convention. USP<790>: Visible Particulates in Injections; United States Pharmacopeia Convention: Washington, DC, USA, 2016. [Google Scholar]
- Hamrang, Z.; Rattray, N.J.W.; Pluen, A. Proteins behaving badly: Emerging technologies in profiling biopharmaceutical aggregation. Trends Biotechnol. 2013, 31, 448–458. [Google Scholar] [CrossRef]
- Roberts, G.S.; Kozak, D.; Anderson, W.; Broom, M.F.; Vogel, R.; Trau, M. Tunable nano/micropores for particle detection and discrimination: Scanning ion occlusion spectroscopy. Small 2010, 6, 2653–2658. [Google Scholar] [CrossRef]
- Krueger, A.B.; Carnell, P.; Carpenter, J.F. Characterization of Factors Affecting Nanoparticle Tracking Analysis Results With Synthetic and Protein Nanoparticles. J. Pharm. Sci. 2016, 105, 1434–1443. [Google Scholar] [CrossRef]
- Gross, J.; Sayle, S.; Karow, A.R.; Bakowsky, U.; Garidel, P. Nanoparticle tracking analysis of particle size and concentration detection in suspensions of polymer and protein samples: Influence of experimental and data evaluation parameters. Eur. J. Pharm. Biopharm. 2016, 104, 30–41. [Google Scholar] [CrossRef]
- Kathe, N.; Henriksen, B.; Chauhan, H. Physicochemical characterization techniques for solid lipid nanoparticles: Principles and limitations. Drug Dev. Ind. Pharm. 2014, 40, 1565–1575. [Google Scholar] [CrossRef]
- Tuchin, V.V. Laser light scattering in biomedical diagnostics and therapy. J. Laser Appl. 1993, 5, 43–60. [Google Scholar] [CrossRef]
- Tuchin, V.V. Polarized light interaction with tissues. J. Biomed. Opt. 2016, 21, 71114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Some, D. Light-scattering-based analysis of biomolecular interactions. Biophys. Rev. 2013, 5, 147–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naruse, M.; Tate, N.; Aono, M.; Ohtsu, M. Information physics fundamentals of nanophotonics. Rep. Prog. Phys. 2013, 76, 56401. [Google Scholar] [CrossRef] [PubMed]
- Mishchenko, M.I.; Travis, L.D.; Lacis, A.A. Scattering, Absorption, and Emission of Light by Small Particles; Cambridge Univ. Press: Cambridge, UK, 2002; ISBN 052178252x. [Google Scholar]
- Lebedev, A.D.; Ivanova, M.A.; Lomakin, A.V.; Noskin, V.A. Heterodyne quasi-elastic light-scattering instrument for biomedical diagnostics. Appl. Opt. 1997, 36, 7518–7522. [Google Scholar] [CrossRef] [PubMed]
- Kokhanovsky, A.A. Light Scattering Reviews 7. In Radiative Transfer and Optical Properties of Atmosphere and Underlying Surface; Springer: Berlin/Heidelberg, Germany, 2013; ISBN 9783642219061. [Google Scholar]
- Kerker, M.; Loebl, E.M. The Scattering of Light and Other Electromagnetic Radiation. Physical Chemistry: A Series of Monographs; Elsevier Science: Burlington, NJ, USA, 1969; ISBN 9780124045507. [Google Scholar]
- Jonasz, M.; Fournier, G. Light Scattering by Particles in Water. Theoretical and Experimental Foundations, 1st ed.; Elsevier/Academic Press: Amsterdam, The Netherlands, 2007; ISBN 9780123887511. [Google Scholar]
- Haina, D.; Landthaler, M. Fundamentals of laser light interaction with human tissue, especially in the cardiovascular system. Thorac. Cardiovasc. Surg. 1988, 36, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Fernández, C.; Minton, A.P. Automated measurement of the static light scattering of macromolecular solutions over a broad range of concentrations. Anal. Biochem. 2008, 381, 254–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohren, C.F.; Huffman, D.R. Absorption and Scattering of Light by Small Particles; Wiley-VCH: Weinheim, Germany, 2008; ISBN 9780471293408. [Google Scholar]
- Mie, G. Beiträge zur Optik trüber Medien, speziell kolloidaler Metallösungen. Ann. Phys. 1908, 330, 377–445. [Google Scholar] [CrossRef]
- Hergert, W.; Wriedt, T. The Mie Theory. Basics and Applications; Springer: Berlin/Heidelberg, Germany, 2012; ISBN 978-3-642-28737-4. [Google Scholar]
- Krishna, R.; Unsworth, T.J.; Edge, R. Raman Spectroscopy and Microscopy. In Reference Module in Materials Science and Materials Engineering; Hashmi, S., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; ISBN 9780128035818. [Google Scholar]
- Einstein, A. Investigations on the Theory of Brownian Motion; Dover: New York, NY, USA, 1956. [Google Scholar]
- Aleandri, S.; Vaccaro, A.; Armenta, R.; Völker, A.C.; Kuentz, M. Dynamic Light Scattering of Biopharmaceutics-Can Analytical Performance Be Enhanced by Laser Power? Pharmaceutics 2018, 10, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evennett, P.J.; Hammond, C. MICROSCOPY|Overview. In Encyclopedia of Analytical Science; Worsfold, P.J., Townshend, A., Poole, C.F., Eds.; Elsevier: Amsterdam, The Netherlands, 2010; pp. 32–41. ISBN 9780123693976. [Google Scholar]
- Hammond, C.; Evennett, P.J. MICROSCOPY TECHNIQUES|Light Microscopy. In Encyclopedia of Analytical Science; Worsfold, P.J., Townshend, A., Poole, C.F., Eds.; Elsevier: Amsterdam, The Netherlands, 2010; pp. 100–106. ISBN 9780123693976. [Google Scholar]
- Stewart, P.L. Cryo-electron microscopy and cryo-electron tomography of nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9. [Google Scholar] [CrossRef]
- Zölls, S.; Weinbuch, D.; Wiggenhorn, M.; Winter, G.; Friess, W.; Jiskoot, W.; Hawe, A. Flow imaging microscopy for protein particle analysis—A comparative evaluation of four different analytical instruments. AAPS J. 2013, 15, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.K.; Oma, P.; Pollo, M.J.; Sukumar, M. Quantification and characterization of subvisible proteinaceous particles in opalescent mAb formulations using micro-flow imaging. J. Pharm. Sci. 2010, 99, 2628–2642. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.S.; Persson, M. Unmasking Translucent Protein Particles by Improved Micro-Flow Imaging™ Algorithms. J. Pharm. Sci. 2014, 103, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Coulter, W.H. Means for Counting Particles Suspended in a Fluid. U.S. Patent 2,656,508 (A), 27 August 1949. [Google Scholar]
- DeBlois, R.W.; Bean, C.P. Counting and Sizing of Submicron Particles by the Resistive Pulse Technique. Rev. Sci. Instrum. 1970, 41, 909–916. [Google Scholar] [CrossRef]
- Kasianowicz, J.J.; Brandin, E.; Branton, D.; Deamer, D.W. Characterization of individual polynucleotide molecules using a membrane channel. Proc. Natl. Acad. Sci. USA 1996, 93, 13770–13773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riordon, J.; Mirzaei, M.; Godin, M. Microfluidic cell volume sensor with tunable sensitivity. Lab Chip 2012, 12, 3016–3019. [Google Scholar] [CrossRef]
- Trujillo, N.J.; Baxamusa, S.H.; Gleason, K.K. Multi-Scale Grafted Polymeric Nanostructures Patterned Bottom-Up by Colloidal Lithography and Initiated Chemical Vapor Deposition (iCVD). MRS Proc. 2008, 1134. [Google Scholar] [CrossRef]
- Burg, T.P.; Manalis, S.R. Suspended microchannel resonators for biomolecular detection. Appl. Phys. Lett. 2003, 83, 2698–2700. [Google Scholar] [CrossRef] [Green Version]
- Burg, T.P.; Godin, M.; Knudsen, S.M.; Shen, W.; Carlson, G.; Foster, J.S.; Babcock, K.; Manalis, S.R. Weighing of biomolecules, single cells and single nanoparticles in fluid. Nature 2007, 446, 1066–1069. [Google Scholar] [CrossRef] [Green Version]
- Dextras, P.; Burg, T.P.; Manalis, S.R. Integrated measurement of the mass and surface charge of discrete microparticles using a suspended microchannel resonator. Anal. Chem. 2009, 81, 4517–4523. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Park, K.; Lee, J. Note: Precision viscosity measurement using suspended microchannel resonators. Rev. Sci. Instrum. 2012, 83, 116106. [Google Scholar] [CrossRef]
- Panchal, J.; Kotarek, J.; Marszal, E.; Topp, E.M. Analyzing subvisible particles in protein drug products: A comparison of dynamic light scattering (DLS) and resonant mass measurement (RMM). AAPS J. 2014, 16, 440–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan, A.K.; Hecht, V.C.; Shen, W.; Payer, K.; Grover, W.H.; Manalis, S.R. Measuring single cell mass, volume, and density with dual suspended microchannel resonators. Lab Chip 2014, 14, 569–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godin, M.; Delgado, F.F.; Son, S.; Grover, W.H.; Bryan, A.K.; Tzur, A.; Jorgensen, P.; Payer, K.; Grossman, A.D.; Kirschner, M.W.; et al. Using buoyant mass to measure the growth of single cells. Nat. Methods 2010, 7, 387–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, S.; Hecht, V.C.; Manalis, S.R. Characterizing Cellular Biophysical Responses to Stress by Relating Density, Deformability, and Size. Biophys. J. 2015, 109, 1565–1573. [Google Scholar] [CrossRef] [Green Version]
- Nejadnik, M.R.; Jiskoot, W. Measurement of the average mass of proteins adsorbed to a nanoparticle by using a suspended microchannel resonator. J. Pharm. Sci. 2015, 104, 698–704. [Google Scholar] [CrossRef]
- Son, S.; Grover, W.H.; Burg, T.P.; Manalis, S.R. Suspended microchannel resonators for ultralow volume universal detection. Anal. Chem. 2008, 80, 4757–4760. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Modena, M.M.; Platen, M.; Schaap, I.A.T.; Burg, T.P. Label-free measurement of amyloid elongation by suspended microchannel resonators. Anal. Chem. 2015, 87, 1821–1828. [Google Scholar] [CrossRef]
- Bryan, A.K.; Goranov, A.; Amon, A.; Manalis, S.R. Measurement of mass, density, and volume during the cell cycle of yeast. Proc. Natl. Acad. Sci. USA 2010, 107, 999–1004. [Google Scholar] [CrossRef] [Green Version]
- Olcum, S.; Cermak, N.; Wasserman, S.C.; Christine, K.S.; Atsumi, H.; Payer, K.R.; Shen, W.; Lee, J.; Belcher, A.M.; Bhatia, S.N.; et al. Weighing nanoparticles in solution at the attogram scale. Proc. Natl. Acad. Sci. USA 2014, 111, 1310–1315. [Google Scholar] [CrossRef] [Green Version]
- Olcum, S.; Cermak, N.; Wasserman, S.C.; Manalis, S.R. High-speed multiple-mode mass-sensing resolves dynamic nanoscale mass distributions. Nat. Commun. 2015, 6, 7070. [Google Scholar] [CrossRef] [Green Version]
- Sajeesh, P.; Sen, A.K. Particle separation and sorting in microfluidic devices: A review. Microfluid Nanofluid 2014, 17, 1–52. [Google Scholar] [CrossRef]
- Sirkar, K.K. Separation of Molecules, Macromolecules and Particles. Principles, Phenomena and Processes; Cambridge Univ. Press: Cambridge, UK, 2014; ISBN 9780521895736. [Google Scholar]
- Underberg, W.J.; Hoitink, M.A.; Reubsaet, J.L.; Waterval, J.C. Separation and detection techniques for peptides and proteins in stability research and bioanalysis. J. Chromatogr. B Biomed. Sci. Appl. 2000, 742, 401–409. [Google Scholar] [CrossRef]
- Poole, C. (Ed.) Handbook of Methods and Instrumentation in Separation Science: Volume 1; Elsevier: Amsterdam, The Netherlands, 2009; ISBN 0123750954. [Google Scholar]
- Van Nieuwenhuijzen, A. (Ed.) Handbook on Particle Separation Processes: 2008 Summer School on Particle Separation; IWA Publishing: London, UK, 2011; ISBN 978-1-84-339277-4. [Google Scholar]
- Fraunhofer, W.; Winter, G. The use of asymmetrical flow field-flow fractionation in pharmaceutics and biopharmaceutics. Eur. J. Pharm. Biopharm. 2004, 58, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Malloy, A. Count, size and visualize nanoparticles. Mater. Today 2011, 14, 170–173. [Google Scholar] [CrossRef]
- Filipe, V.; Hawe, A.; Jiskoot, W. Critical evaluation of Nanoparticle Tracking Analysis (NTA) by NanoSight for the measurement of nanoparticles and protein aggregates. Pharm. Res. 2010, 27, 796–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Pol, E.; Coumans, F.A.W.; Sturk, A.; Nieuwland, R.; van Leeuwen, T.G. Refractive index determination of nanoparticles in suspension using nanoparticle tracking analysis. Nano Lett. 2014, 14, 6195–6201. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.proteinsimple.com/mfi_overview.html (accessed on 23 June 2016).
- United States Pharmacopeia Convention. USP<789>: Particulate Matter in Ophthalmic Solutions; United States Pharmacopeia Convention: Washington, DC, USA, 2013. [Google Scholar]
- Yang, H.; Liu, G.-J.; Lu, M.-D.; Xu, H.-X.; Xie, X.-Y. Evaluation of the vascular architecture of focal liver lesions using micro flow imaging. J. Ultrasound Med. 2013, 32, 1157–1171. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Martin, T.; Li, Y.; Yang, L.; Halpenny, M.; Giulivi, A.; Allan, D.S. Cell aggregation in thawed haematopoietic stem cell products visualised using micro-flow imaging. Transfus. Med. 2012, 22, 218–220. [Google Scholar] [CrossRef]
- Sugimoto, K.; Shiraishi, J.; Moriyasu, F.; Ichimura, S.; Metoki, R.; Doi, K. Analysis of intrahepatic vascular morphological changes of chronic liver disease for assessment of liver fibrosis stages by micro-flow imaging with contrast-enhanced ultrasound: Preliminary experience. Eur. Radiol. 2010, 20, 2749–2757. [Google Scholar] [CrossRef]
- Sharma, D.K.; King, D.; Merchant, C. Reference material development for calibration and verification of image-based particle analyzers. Int. J. Pharm. 2011, 416, 293–295. [Google Scholar] [CrossRef]
- Kozak, D.; Anderson, W.; Vogel, R.; Chen, S.; Antaw, F.; Trau, M. Simultaneous size and ζ-potential measurements of individual nanoparticles in dispersion using size-tunable pore sensors. ACS Nano 2012, 6, 6990–6997. [Google Scholar] [CrossRef] [PubMed]
- Vogel, R.; Willmott, G.; Kozak, D.; Roberts, G.S.; Anderson, W.; Groenewegen, L.; Glossop, B.; Barnett, A.; Turner, A.; Trau, M. Quantitative sizing of nano/microparticles with a tunable elastomeric pore sensor. Anal. Chem. 2011, 83, 3499–3506. [Google Scholar] [CrossRef] [PubMed]
- Sowerby, S.J.; Broom, M.F.; Petersen, G.B. Dynamically resizable nanometre-scale apertures for molecular sensing. Sens. Actuators B Chem. 2007, 123, 325–330. [Google Scholar] [CrossRef]
- Willmott, G.R.; Moore, P.W. Reversible mechanical actuation of elastomeric nanopores. Nanotechnology 2008, 19, 475504. [Google Scholar] [CrossRef] [PubMed]
- Kozak, D.; Anderson, W.; Vogel, R.; Trau, M. Advances in Resistive Pulse Sensors: Devices bridging the void between molecular and microscopic detection. Nano Today 2011, 6, 531–545. [Google Scholar] [CrossRef] [Green Version]
- Weatherall, E.; Willmott, G.R. Applications of tunable resistive pulse sensing. Analyst 2015, 140, 3318–3334. [Google Scholar] [CrossRef] [PubMed]
- Willmott, G.R.; Vogel, R.; Yu, S.S.C.; Groenewegen, L.G.; Roberts, G.S.; Kozak, D.; Anderson, W.; Trau, M. Use of tunable nanopore blockade rates to investigate colloidal dispersions. J. Phys. Condens. Matter 2010, 22, 454116. [Google Scholar] [CrossRef]
- Blundell, E.L.C.J.; Vogel, R.; Platt, M. Particle-by-Particle Charge Analysis of DNA-Modified Nanoparticles Using Tunable Resistive Pulse Sensing. Langmuir 2016, 32, 1082–1090. [Google Scholar] [CrossRef] [Green Version]
- Blundell, E.L.C.J.; Mayne, L.J.; Billinge, E.R.; Platt, M. Emergence of tunable resistive pulse sensing as a biosensor. Anal. Methods 2015, 7, 7055–7066. [Google Scholar] [CrossRef] [Green Version]
- Homepage iZon. Available online: http://www.izon.com/products/qnano/ (accessed on 10 October 2016).
- Bell, N.C.; Minelli, C.; Tompkins, J.; Stevens, M.M.; Shard, A.G. Emerging techniques for submicrometer particle sizing applied to Stöber silica. Langmuir 2012, 28, 10860–10872. [Google Scholar] [CrossRef]
- Roberts, G.S.; Yu, S.; Zeng, Q.; Chan, L.C.L.; Anderson, W.; Colby, A.H.; Grinstaff, M.W.; Reid, S.; Vogel, R. Tunable pores for measuring concentrations of synthetic and biological nanoparticle dispersions. Biosens. Bioelectron. 2012, 31, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, E.; Coumans, F.A.W.; Grootemaat, A.E.; Gardiner, C.; Sargent, I.L.; Harrison, P.; Sturk, A.; van Leeuwen, T.G.; Nieuwland, R. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. J. Thromb. Haemost. 2014, 12, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Böing, A.N.; van der Pol, E.; Grootemaat, A.E.; Coumans, F.A.W.; Sturk, A.; Nieuwland, R. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef]
- Picot, J.; Guerin, C.L.; van Kim, C.; Boulanger, C.M. Flow cytometry: Retrospective, fundamentals and recent instrumentation. Cytotechnology 2012, 64, 109–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosland-Taylor, P.J. A device for counting small particles suspended in a fluid through a tube. Nature 1953, 171, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Van Dilla, M.A.; Trujillo, T.T.; Mullaney, P.F.; Coulter, J.R. Cell microfluorometry: A method for rapid fluorescence measurement. Science 1969, 163, 1213–1214. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, D.B.; Trotter, J.T.; Gabrielson, J.P.; Carpenter, J.F.; Randolph, T.W. Flow cytometry: A promising technique for the study of silicone oil-induced particulate formation in protein formulations. Anal. Biochem. 2011, 410, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Mach, H.; Bhambhani, A.; Meyer, B.K.; Burek, S.; Davis, H.; Blue, J.T.; Evans, R.K. The use of flow cytometry for the detection of subvisible particles in therapeutic protein formulations. J. Pharm. Sci. 2011, 100, 1671–1678. [Google Scholar] [CrossRef]
- Filipe, V.; Poole, R.; Oladunjoye, O.; Braeckmans, K.; Jiskoot, W. Detection and characterization of subvisible aggregates of monoclonal IgG in serum. Pharm. Res. 2012, 29, 2202–2212. [Google Scholar] [CrossRef] [Green Version]
- Nishi, H.; Mathäs, R.; Fürst, R.; Winter, G. Label-free flow cytometry analysis of subvisible aggregates in liquid IgG1 antibody formulations. J. Pharm. Sci. 2014, 103, 90–99. [Google Scholar] [CrossRef]
- Telikepalli, S.; Shinogle, H.E.; Thapa, P.S.; Kim, J.H.; Deshpande, M.; Jawa, V.; Middaugh, C.R.; Narhi, L.O.; Joubert, M.K.; Volkin, D.B. Physical characterization and in vitro biological impact of highly aggregated antibodies separated into size-enriched populations by fluorescence-activated cell sorting. J. Pharm. Sci. 2015, 104, 1575–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaz, S.H. Flow Cytometry for Automated Particle Detection in Formulation Development. Master’s Thesis, Biberach University of Applied Science, Biberach an der Riss, Germany, 2015. [Google Scholar]
- Golabgir, A.; Ehgartner, D.; Neutsch, L.; Posch, A.E.; Sagmeister, P.; Herwig, C. Imaging Flow Cytometry and High-Throughput Microscopy for Automated Macroscopic Morphological Analysis of Filamentous Fungi. In Genetic Transformation Systems in Fungi: Volume 2; van den Berg, M.A., Maruthachalam, K., Eds.; Springer International Publishing: Cham, Germany, 2015; pp. 201–210. ISBN 978-3-319-10502-4. [Google Scholar]
- Walter, J.; Thajudeen, T.; Süss, S.; Segets, D.; Peukert, W. New possibilities of accurate particle characterisation by applying direct boundary models to analytical centrifugation. Nanoscale 2015, 7, 6574–6587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, J.; Peukert, W. Dynamic range multiwavelength particle characterization using analytical ultracentrifugation. Nanoscale 2016, 8, 7484–7495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, J.; Löhr, K.; Karabudak, E.; Reis, W.; Mikhael, J.; Peukert, W.; Wohlleben, W.; Cölfen, H. Multidimensional analysis of nanoparticles with highly disperse properties using multiwavelength analytical ultracentrifugation. ACS Nano 2014, 8, 8871–8886. [Google Scholar] [CrossRef] [PubMed]
- Bharti, B.; Meissner, J.; Klapp, S.H.L.; Findenegg, G.H. Bridging interactions of proteins with silica nanoparticles: The influence of pH, ionic strength and protein concentration. Soft Matter 2014, 10, 718–728. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Particle Light Interactions | Microscopy and Imaging | Resistive Pulse Sensing | Suspended Microchannel Resonators | Sedimentation and Centrifugation | Further Separation Approaches | |
|---|---|---|---|---|---|---|
| Physical phenomenon | Absorption, diffraction, refraction, scattering, fluorescence, polarization, etc. | Space-resolved imaging of particles coded by colors and intensity | Current blockage of specific intensity and duration | Change in the resonator frequency density and size dependent | Sedimentation velocity, separation | Separation in fractions due to a particle property |
| Output particle properties (examples) | Size and size distribution, molecular weight, material and structural properties, concentration, interaction parameter | Size and size distribution, shape, optical properties, concentration, etc. | Size and size distribution, shape, concentration, charge, etc. | Size and size distribution, concentration, density, material differentiation, etc. | Size and size distribution, shape, density, equilibrium parameters, etc. | Depending on the investigated property and the used detector |
| Main advantage(s) | Variable approaches possible | Direct measurement and imaging | Independent of optical/material properties | Differentiation between materials | Variable approaches, no destructive preparation expected | Analysis of separated fractions, advantageous for polydisperse/polymodal samples |
| Main challenge(s) | Complex physical processes and calculation, polydispersity | Resolution, preparation | Conductivity, polydispersity | Channel blockage due to interactions | Material properties necessary; no concentration determination | Separation conditions |
| Techniques (examples) | LO, SLS, DLS, turbidimetry, UV/Vis spectroscopy and spectrometry, Raman spectroscopy, flow cytometers, etc. | Various types of microscopes, e.g., optical, REM, TEM, AFM, flow imaging devices, e.g., MFI | Coulter counters and some FACS instruments | Chip-based SMRs, commercial instrument: Archimedes | All types of preparative and analytical centrifuges, e.g., AUC, DISC | Chromatographic methods, e.g., HP-SEC; AF4; SDS-PAGE |
| Light Obscuration (LO) | Micro- Flow Imaging (MFI) | (Nano-) Particle Tracking Analysis (NTA) | Tunable Resistive Pulse Sensing (TRPS) | Space and Time Resolved Extinction Profile (STEP) | Flow Cytometry (FC) | |
|---|---|---|---|---|---|---|
| Operating principle | Particle–light interactions (static), particle-by-particle | Microscopy and imaging, particle-by-particle | Particle–light interactions (dyn.), particle-by-particle | Electrozone sensing/Coulter principle, particle-by-particle | Sedimentation and centrifugation, ensemble | Particle–light interactions (static), particle-by-particle |
| Size range * | 1–600 µm | 2–100 µm | 20–1000 nm | 50 nm–10 µm | 20 nm–100 µm | 100 nm–100 µm |
| Input/analysis required information | No | No | Measurement and evaluation settings | Choose pore size, set stretch, pressure, voltage | RI and density for PSD, centrifugation protocol | Size standard for size estimations, settings |
| Output/provided information | ||||||
| Particle concentration | Yes | Yes | Yes | Yes | No | Yes |
| Particle size | Yes | Yes | Yes | Yes | Yes | Limited |
| Size distribution (PSD) | Yes | Yes | Yes | Yes | Yes | Limited |
| Shape | - | Circularity, aspect ratio | - | Yes (duration) | - | Estimations |
| Structure | - | Related to contrast | - | - | - | Estimations |
| Identity | - | Due to shape/structure | - | - | - | - |
| Other | - | particle images | Scatter intensities and RI | Particle charge | sedimentation velocities, suspension stability | polydispersity, (physical properties) |
| Equivalent particle diameter | Projected area | Projected area | Hydrodynamic | Volume | Volume | Scatter profile |
| Material differentiation | No | Yes; due to shape and gray scale | No; potential differentiation due to scatter intensity | No; potential differentiation due to charge | No | Estimations can be made based on calibration standards |
| Detection of translucent particles | + | ++ | ++ | +++ | + | ++ |
| Destructive | No/yes (dilution) | No/yes (dilution) | No | No/yes by voltage | No/yes by crowding | No |
| Particle separation required | No | No | No/yes for highly polydisperse suspensions | No/yes for highly polydisperse suspensions | No | No |
| Sample handling and preparation | ++ | ++ | ++ | ++ | +++ | ++ |
| Sample volume (minimal) | 2 mL/25 mL (Ph.Eur.) | 0.5–1.0 mL | 0.5–1.0 mL | 40–120 µL | 0.4–1.5 mL | 100–200 µL |
| Particle concentration | ≤18,000 counts/mL | ≤850,000 counts/mL | 107–109 particle/mL | 105–1011 particle/mL | 0.00015–90 Vol % | n/a |
| Calibration Yes/No | No | No | No | Yes | No/yes | No/yes |
| Speed/sample | 8 min | 12 min | 10 min | Depending on sample | 1 min | |
| High throughput/autosampler | Yes/no autosampler | Yes/autosampler | No/no autosampler | Yes/no autosampler | Yes/12 samples simultaneously | Yes/autosampler |
| Provided Software | ||||||
| Measurement | Yes | Yes | Yes | Yes | Yes | Yes |
| Data evaluation | Yes | Yes | Yes | Yes | Yes | Yes |
| Report | Yes | Yes | Yes | Yes | Yes | - |
| Data export | ||||||
| Raw data | CSV | CSV, video | CSV, | CSV, PDF | CSV | |
| Graph/processed | PDF, jpeg | rtf, jpeg | CSV, PDF | Images | ||
| Particle size distribution | ||||||
| Accuracy | ++ | ++ | ++ | +++ | ++ | ++ |
| Repeatability | ++ | +++ | ++ | +++ | +++ | ++ |
| Resolution | ++ | ++ | ++ | +++ | ++ | ++ |
| Protein applicability | ||||||
| BSA standard | +++ | +++ | +++ | +++ | +++ | +++ |
| mAB solution | +++ | +++ | ++ | + | + | ++ |
| Portability/space | ++ | ++ | + (NS500), ++ later versions | +++ | ++ | ++ (depending on the system) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gross-Rother, J.; Blech, M.; Preis, E.; Bakowsky, U.; Garidel, P. Particle Detection and Characterization for Biopharmaceutical Applications: Current Principles of Established and Alternative Techniques. Pharmaceutics 2020, 12, 1112. https://doi.org/10.3390/pharmaceutics12111112
Gross-Rother J, Blech M, Preis E, Bakowsky U, Garidel P. Particle Detection and Characterization for Biopharmaceutical Applications: Current Principles of Established and Alternative Techniques. Pharmaceutics. 2020; 12(11):1112. https://doi.org/10.3390/pharmaceutics12111112
Chicago/Turabian StyleGross-Rother, Julia, Michaela Blech, Eduard Preis, Udo Bakowsky, and Patrick Garidel. 2020. "Particle Detection and Characterization for Biopharmaceutical Applications: Current Principles of Established and Alternative Techniques" Pharmaceutics 12, no. 11: 1112. https://doi.org/10.3390/pharmaceutics12111112
APA StyleGross-Rother, J., Blech, M., Preis, E., Bakowsky, U., & Garidel, P. (2020). Particle Detection and Characterization for Biopharmaceutical Applications: Current Principles of Established and Alternative Techniques. Pharmaceutics, 12(11), 1112. https://doi.org/10.3390/pharmaceutics12111112