Theory of Structural and Secondary Relaxation in Amorphous Drugs under Compression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Structural Relaxation Time of Amorphous Drugs under Pressure
2.1. Local and Collective Dynamics
2.2. Relaxation Process
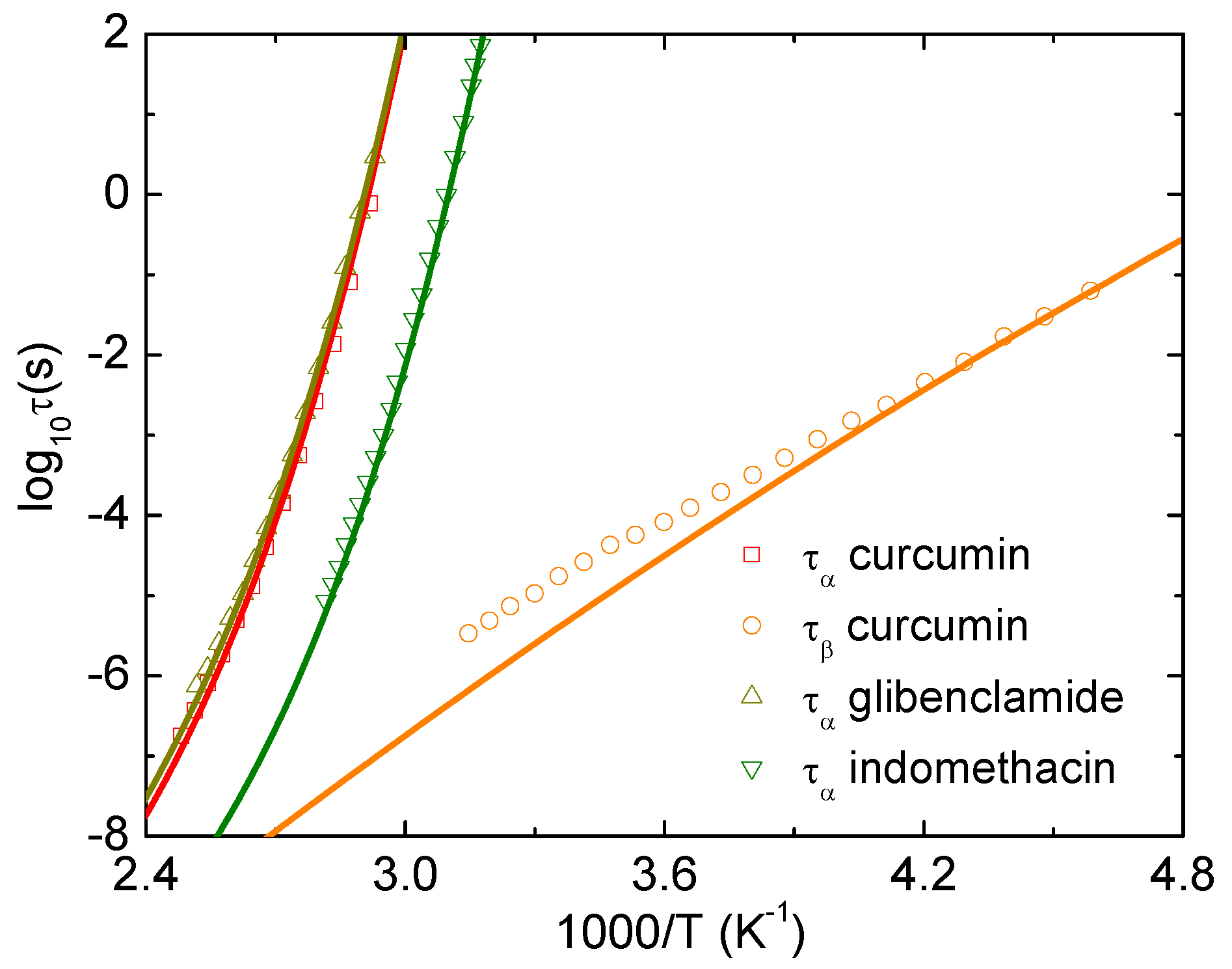
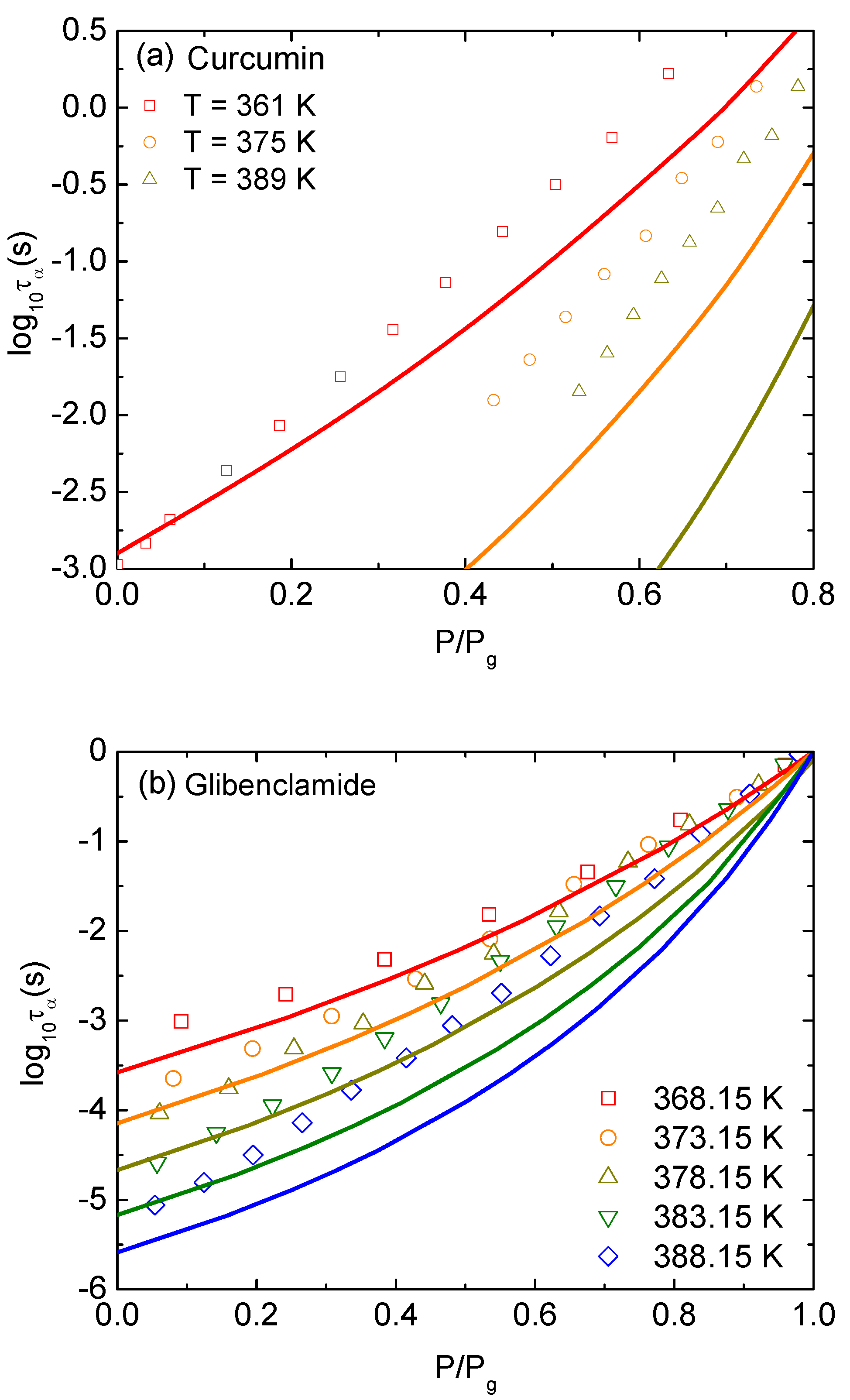
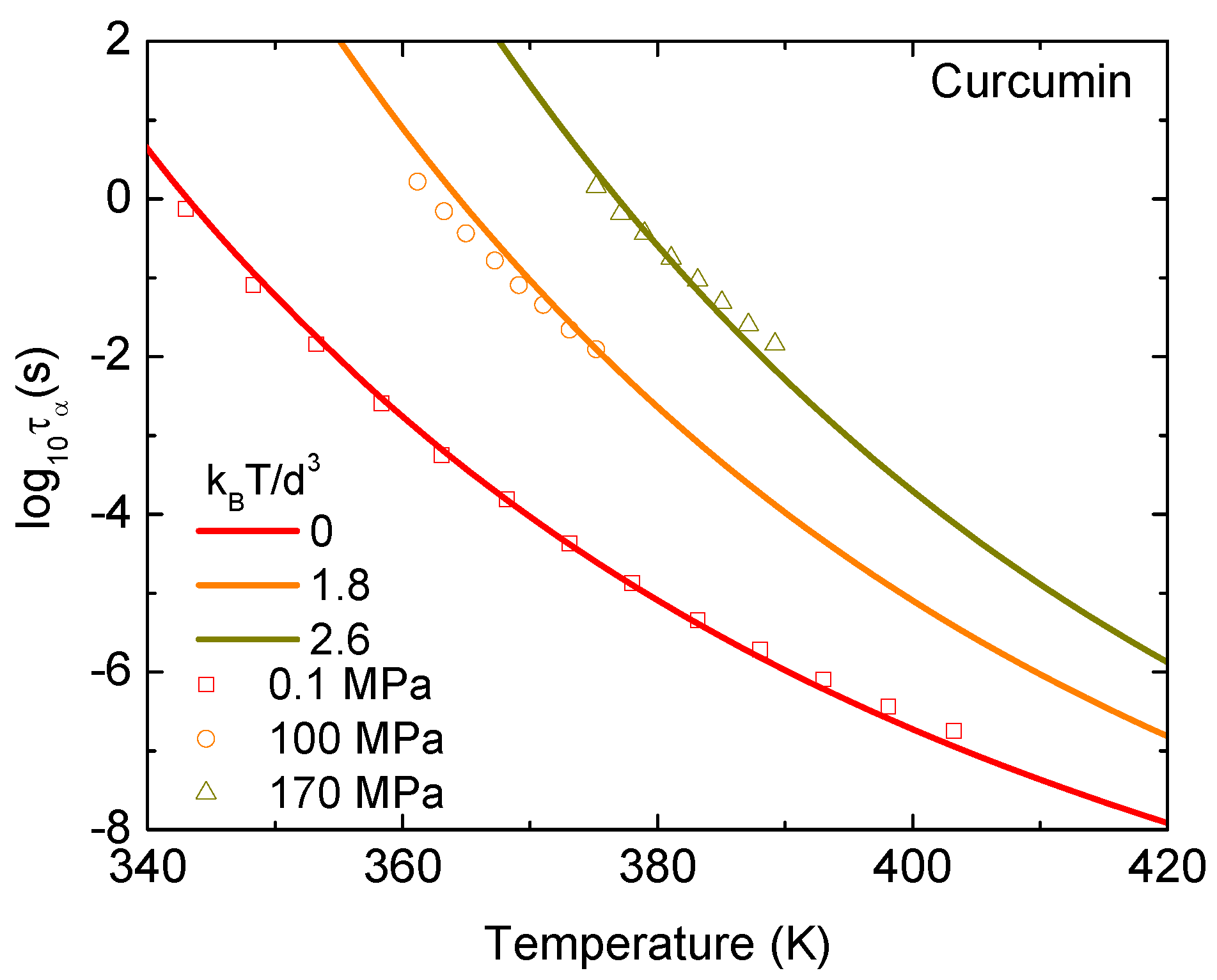
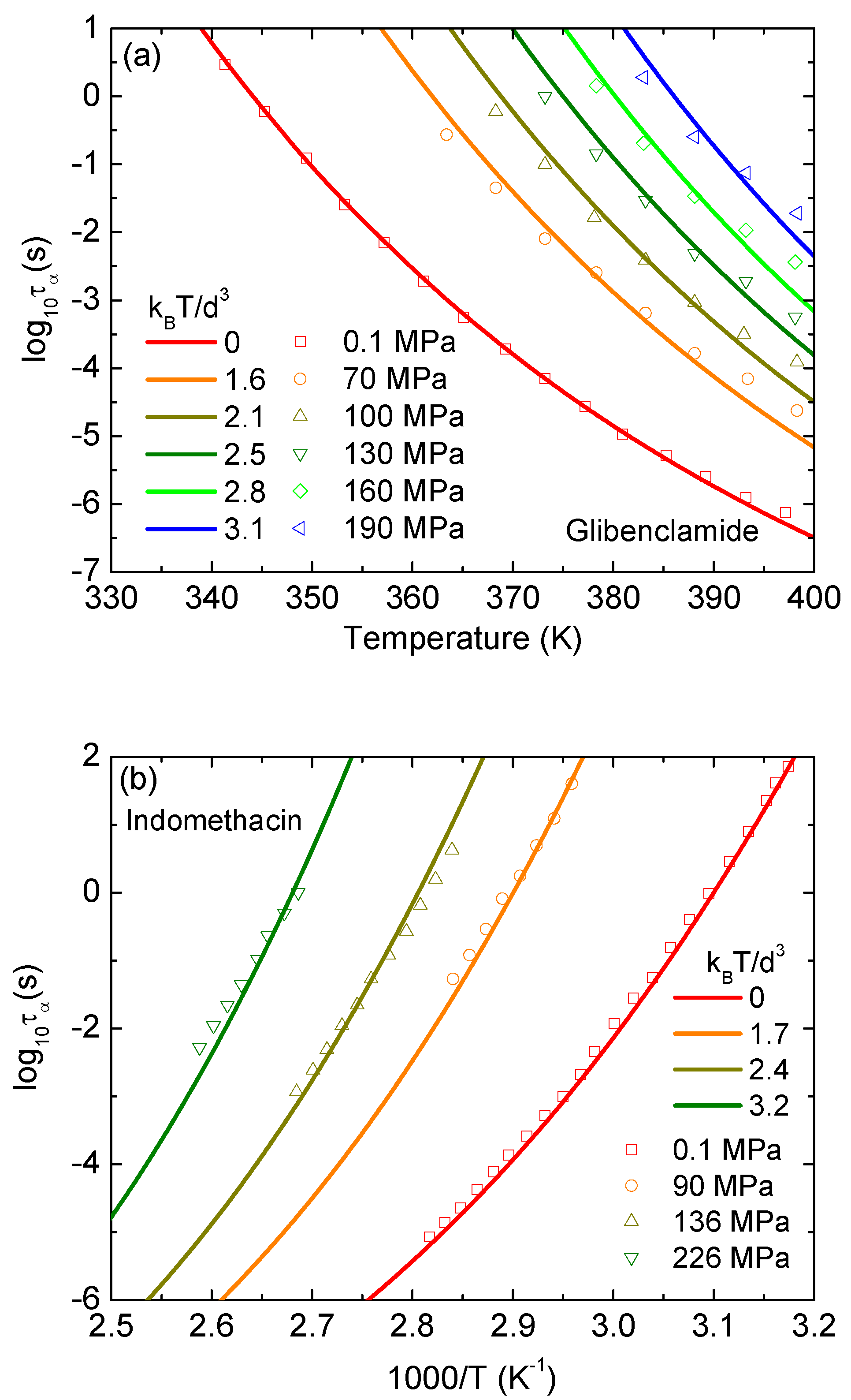
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Williams, R.O., III; Watts, A.B.; Miller, D.A. Formulating Poorly Water Soluble Drugs, 2nd ed.; Springer: New York, NY, USA, 2016. [Google Scholar]
- Rams-Baron, M.; Jachowicz, R.; Boldyreva, E.; Zhou, D.; Jamroz, W.; Paluch, M. Amorphous Drugs; Springer: Heidelberg, Germany, 2018. [Google Scholar]
- Bhardwaj, S.P.; Suryanarayanan, R. Molecular mobility as an effective predictor of the physical stability of amorphous trehalose. Mol. Pharm. 2012, 9, 3209–3217. [Google Scholar] [CrossRef] [PubMed]
- Wojnarowska, Z.; Grzybowska, K.; Hawelek, L.; Dulski, M.; Wrzalik, R.; Gruszka, I.; Paluch, M. Molecular Dynamics, Physical Stability and Solubility Advantage from Amorphous Indapamide Drug. Mol. Pharm. 2013, 10, 3612–3627. [Google Scholar] [CrossRef] [PubMed]
- Floudas, G.; Paluch, M.; Grzybowski, A.; Ngai, K.L. Molecular Dynamics of Glass-Forming Systems: Effects of Pressure; Springer: Heidelberg, Germany, 2011. [Google Scholar]
- Roland, C.M.; Hensel-Bielowka, S.; Paluch, M.; Casalini, R. Supercooled dynamics of glass-forming liquids and polymers under hydrostatic pressure. Rep. Prog. Phys. 2005, 68, 1405–1478. [Google Scholar] [CrossRef] [Green Version]
- Paluch, M.; Ziolo, J.; Rzoska, S.J.; Habdas, P. High-pressure and temperature dependence of dielectric relaxation in supercooled di-isobutyl phthalate. Phys. Rev. E 1996, 54, 4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paluch, M.; Rzoska, S.J.; Habdas, P.; Ziolo, J. On the isothermal pressure behaviour of the relaxation times for supercooled glass-forming liquids. J. Phys. Condens. Matter 1998, 10, 4131. [Google Scholar] [CrossRef]
- Phan, A.D.; Schweizer, K.S. Elastically Collective Nonlinear Langevin Equation Theory of Glass-Forming Liquids: Transient Localization, Thermodynamic Mapping, and Cooperativity. J. Phys. Chem. B 2018, 122, 8451–8461. [Google Scholar] [CrossRef] [PubMed]
- Mirigian, S.; Schweizer, K.S. Elastically cooperative activated barrier hopping theory of relaxation in viscous fluids. I. General formulation and application to hard sphere fluids. J. Chem. Phys. 2014, 140, 194506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirigian, S.; Schweizer, K.S. Unified Theory of Activated Relaxation in Liquids over 14 Decades in Time. J. Phys. Chem. Lett. 2013, 4, 3648–3653. [Google Scholar] [CrossRef] [Green Version]
- Mirigian, S.; Schweizer, K.S. Elastically cooperative activated barrier hopping theory of relaxation in viscous fluids. II. Thermal liquids. J. Chem. Phys. 2014, 140, 194507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, S.-J.; Schweizer, K.S. Nonuniversal Coupling of Cage Scale Hopping and Collective Elastic Distortion as the Origin of Dynamic Fragility Diversity in Glass-Forming Polymer Liquids. Macromolecules 2016, 49, 9655–9664. [Google Scholar] [CrossRef]
- Phan, A.D.; Knapik-Kowalczuk, J.; Paluch, M.; Hoang, T.X.; Wakabayashi, K. Theoretical Model for the Structural Relaxation Time in Coamorphous Drugs. Mol. Pharm. 2019, 16, 2992–2998. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.D.; Wakabayashi, K.; Paluch, M.; Lam, V.D. Effects of cooling rate on structural relaxation in amorphous drugs: Elastically collective nonlinear langevin equation theory and machine learning study. RSC Adv. 2019, 9, 40214–40221. [Google Scholar] [CrossRef] [Green Version]
- Minecka, A.; Kaminska, E.; Heczko, D.; Jurkiewicz, K.; Wolnica, K.; Dulski, M.; Hachula, B.; Pisarski, W.; Tarnacka, M.; Talik, A.; et al. Studying structural and local dynamics in model H-bonded active ingredient—Curcumin in the supercooled and glassy states at various thermodynamic conditions. Eur. J. Pharm. Sci. 2019, 135, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Wojnarowska, Z.; Adrjanowicz, K.; Kaminski, K.; Hawelek, L.; Paluch, M. Effect of Pressure on Tautomers’ Equilibrium in Supercooled Glibenclamide Drug: Analysis of Fragility Behavior. J. Phys. Chem. B 2010, 114, 14815–14820. [Google Scholar] [CrossRef] [PubMed]
- Adrjanowicz, K.; Grzybowski, A.; Grzybowska, K.; Pionteck, J.; Paluch, M. Toward Better Understanding Crystallization of Supercooled Liquids under Compression: Isochronal Crystallization Kinetics Approach. Cryst. Growth Des. 2013, 11, 4648–4654. [Google Scholar] [CrossRef]
- Schweizer, K.S.; Saltzman, E.J. Entropic barriers, activated hopping, and the glass transition in colloidal suspensions. J. Chem. Phys. 2003, 119, 1181. [Google Scholar] [CrossRef] [Green Version]
- Saltzman, E.J.; Schweizer, K.S. Activated hopping and dynamical fluctuation effects in hard sphere suspensions and fluids. J. Chem. Phys. 2006, 125, 044509. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.-P.; McDonald, I.R. Theory of Simple Liquids; Academic Press: London, UK, 2006. [Google Scholar]
- Landau, L.D.; Lifshitz, E.M. Theory of Elasticity, 3rd ed.; Permagon Press: London, UK, 1975. [Google Scholar]
- Mirigian, S.; Schweizer, K.S. Dynamical Theory of Segmental Relaxation and Emergent Elasticity in Supercooled Polymer Melts. Macromolecules 2015, 48, 1901–1913. [Google Scholar] [CrossRef]
- Forrest, J.A.; Dalnoki-Veress, K. When Does a Glass Transition Temperature Not Signify a Glass Transition? ACS Macro Lett. 2014, 3, 310–314. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phan, A.D.; Wakabayashi, K. Theory of Structural and Secondary Relaxation in Amorphous Drugs under Compression. Pharmaceutics 2020, 12, 177. https://doi.org/10.3390/pharmaceutics12020177
Phan AD, Wakabayashi K. Theory of Structural and Secondary Relaxation in Amorphous Drugs under Compression. Pharmaceutics. 2020; 12(2):177. https://doi.org/10.3390/pharmaceutics12020177
Chicago/Turabian StylePhan, Anh D., and Katsunori Wakabayashi. 2020. "Theory of Structural and Secondary Relaxation in Amorphous Drugs under Compression" Pharmaceutics 12, no. 2: 177. https://doi.org/10.3390/pharmaceutics12020177
APA StylePhan, A. D., & Wakabayashi, K. (2020). Theory of Structural and Secondary Relaxation in Amorphous Drugs under Compression. Pharmaceutics, 12(2), 177. https://doi.org/10.3390/pharmaceutics12020177