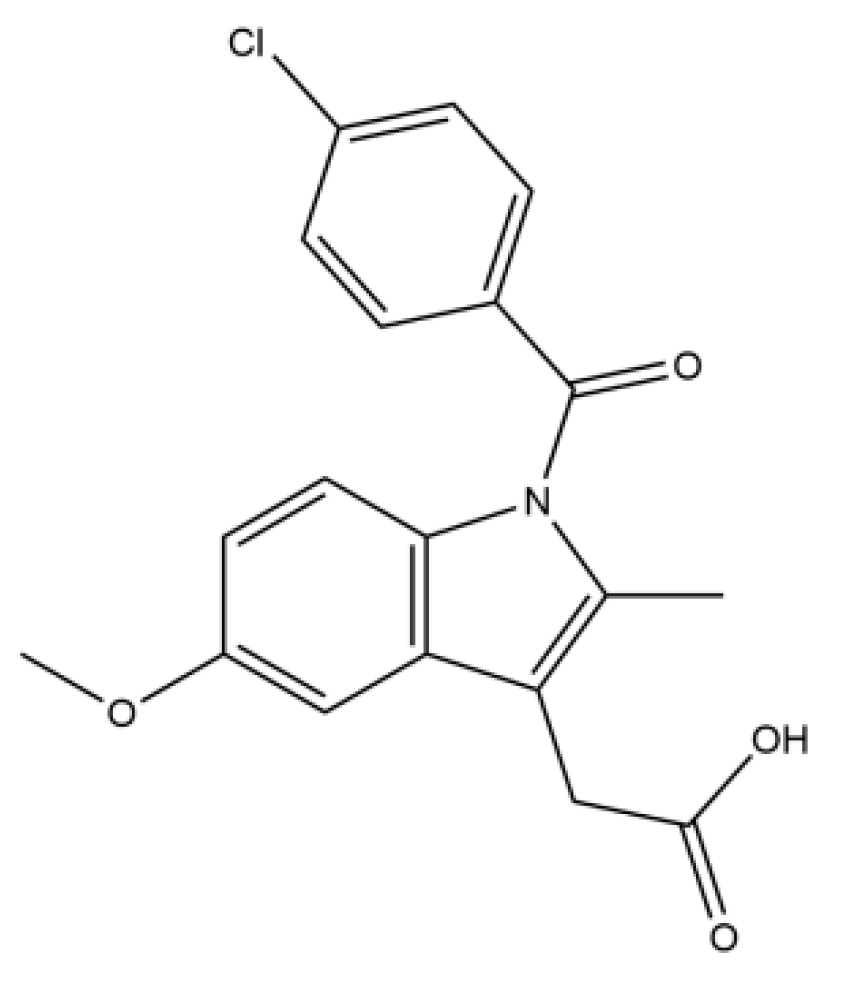
Evaluation of the Solid Dispersion System Engineered from Mesoporous Silica and Polymers for the Poorly Water Soluble Drug Indomethacin: In Vitro and In Vivo


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Mesoporous Silica Nanoparticles (MSNs)
2.3. Preparation of Solid Dispersions
2.4. Characterization of Mesoporous Silica Nanoparticles
2.5. Characterization of Solid Dispersions
2.5.1. Field Emission Scanning Electron Microscope (FESEM)
2.5.2. Differential Scanning Calorimetry (DSC)
2.5.3. Powder X-ray Diffraction (PXRD)
2.5.4. Infrared (IR) Spectroscopy
2.6. In Vitro Studies
2.6.1. Liquid–Liquid Phase Separation (LLPS) Concentration Determination
2.6.2. Solubility Studies
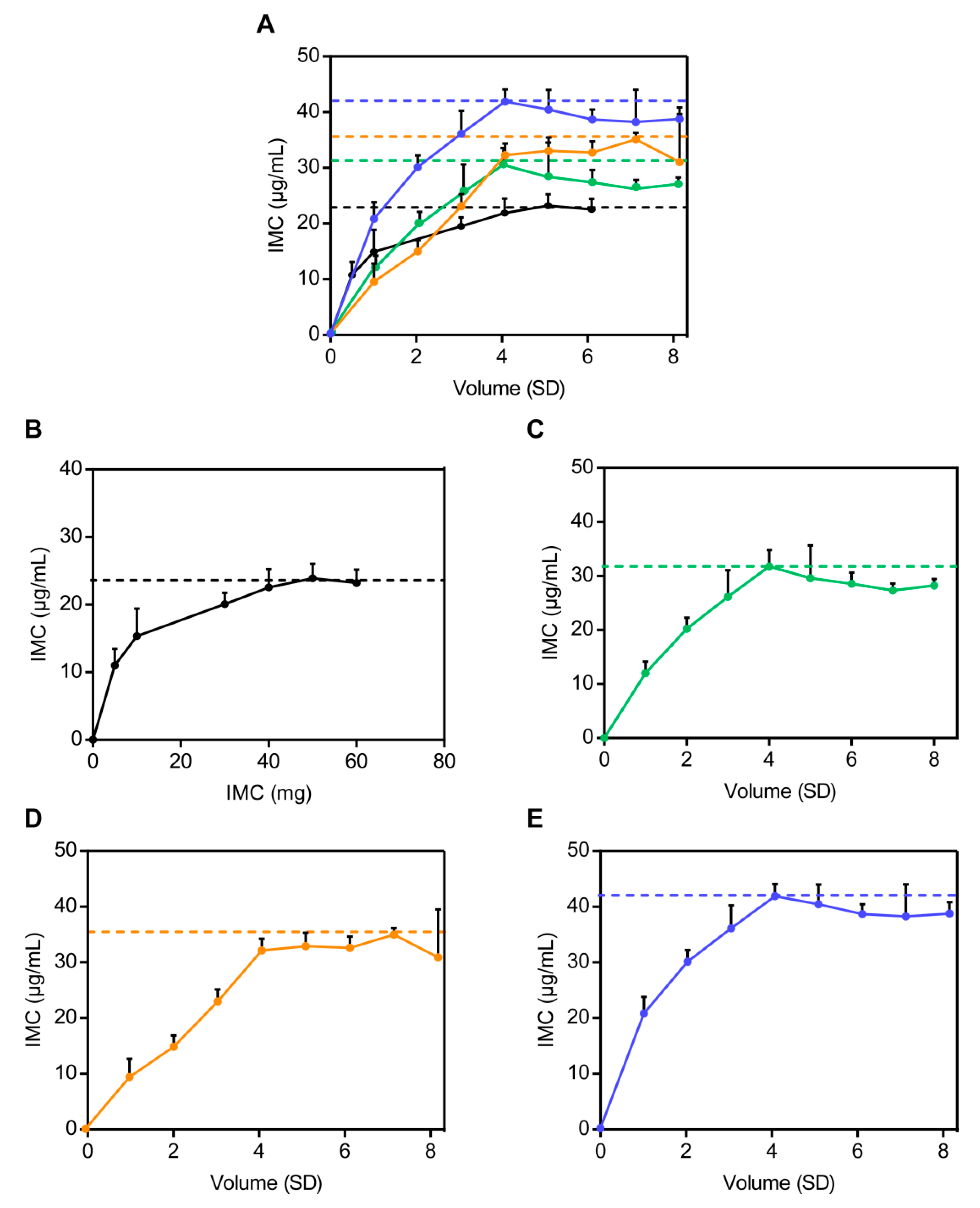
2.6.3. In Vitro Dissolution Studies and Determination of the Optimum Proportion of Solid Dispersion Carriers
2.6.4. Moisture Sorption Analysis
2.6.5. Stability Studies
2.6.6. Parallel Artificial Membrane Penetration (PAMPA)
2.7. In Vivo Studies
2.7.1. In Vivo Pharmacokinetic Study
2.7.2. In Vivo Oral Delivery Imaging Studies
2.8. Statistical Analysis
3. Results and Discussion
3.1. Characterization of Mesoporous Silica Nanoparticles (MSNs)
3.2. Characterization of Solid Dispersions
3.2.1. Field Emission Scanning Electron Microscopy (FESEM)
3.2.2. Powder X-ray Diffraction (PXRD), Differential Scanning Calorimetry (DSC), and Thermal Analysis
3.2.3. Infrared (IR) Spectroscopy
3.3. In Vitro Studies
3.3.1. Solubility Analysis
Crystalline Solubility
Amorphous Solubility
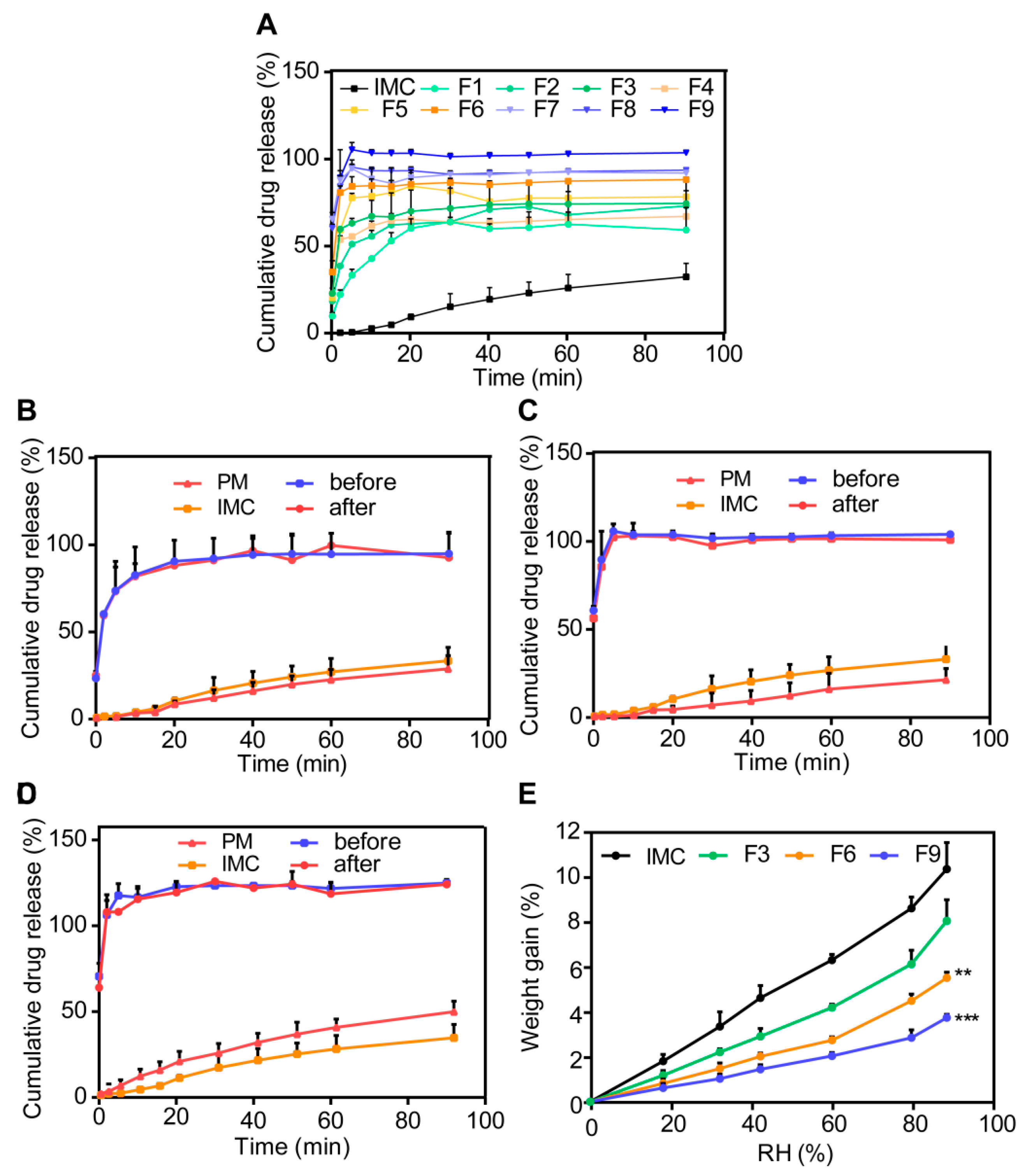
3.3.2. In Vitro Dissolution Studies
3.3.3. Moisture Sorption Analysis
3.3.4. Stability Studies
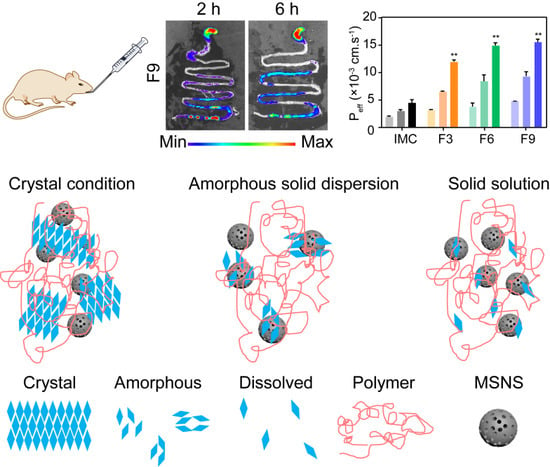
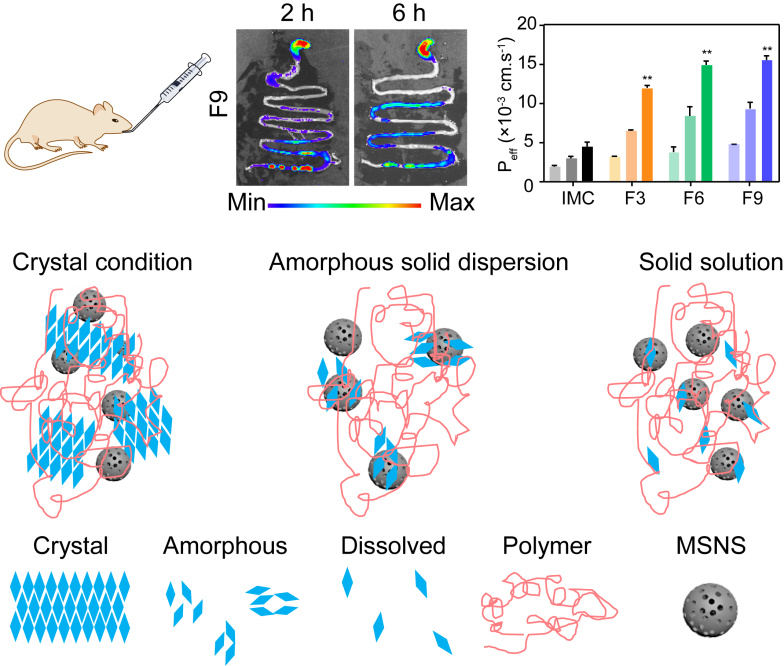
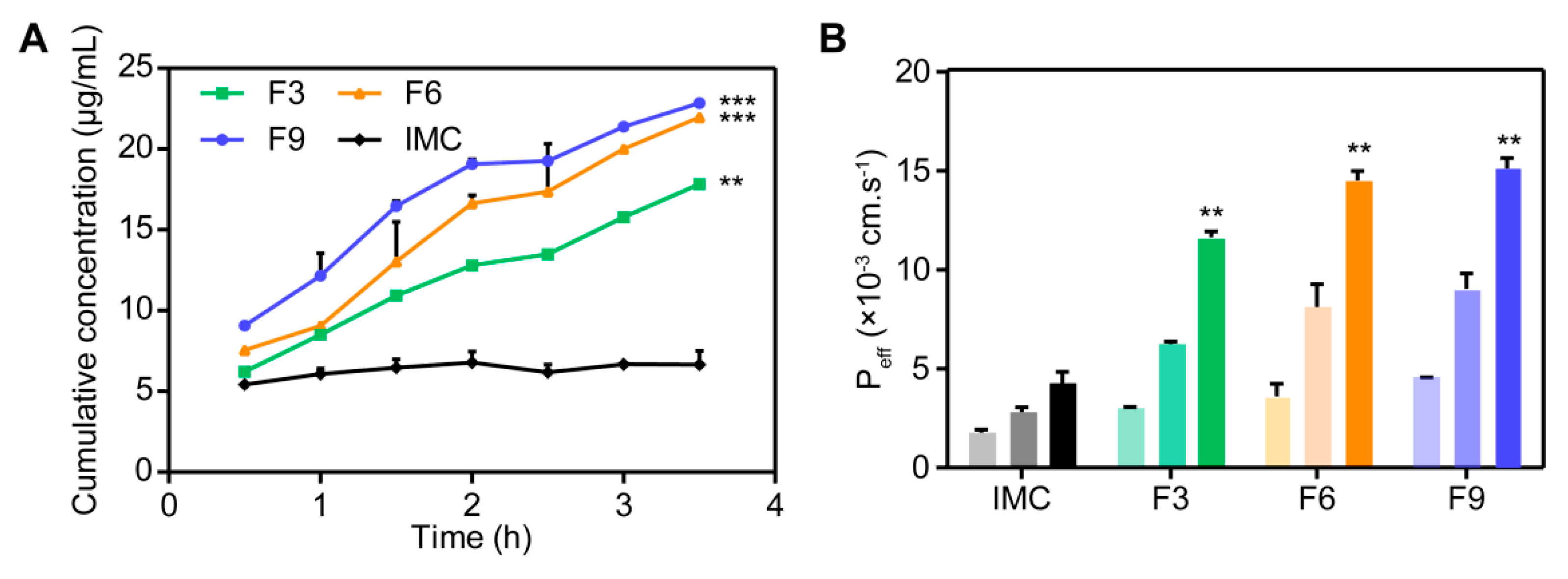
3.3.5. Permeability and Bioavailability Studies
3.4. In Vivo Studies
3.4.1. In Vivo Pharmacokinetic Study
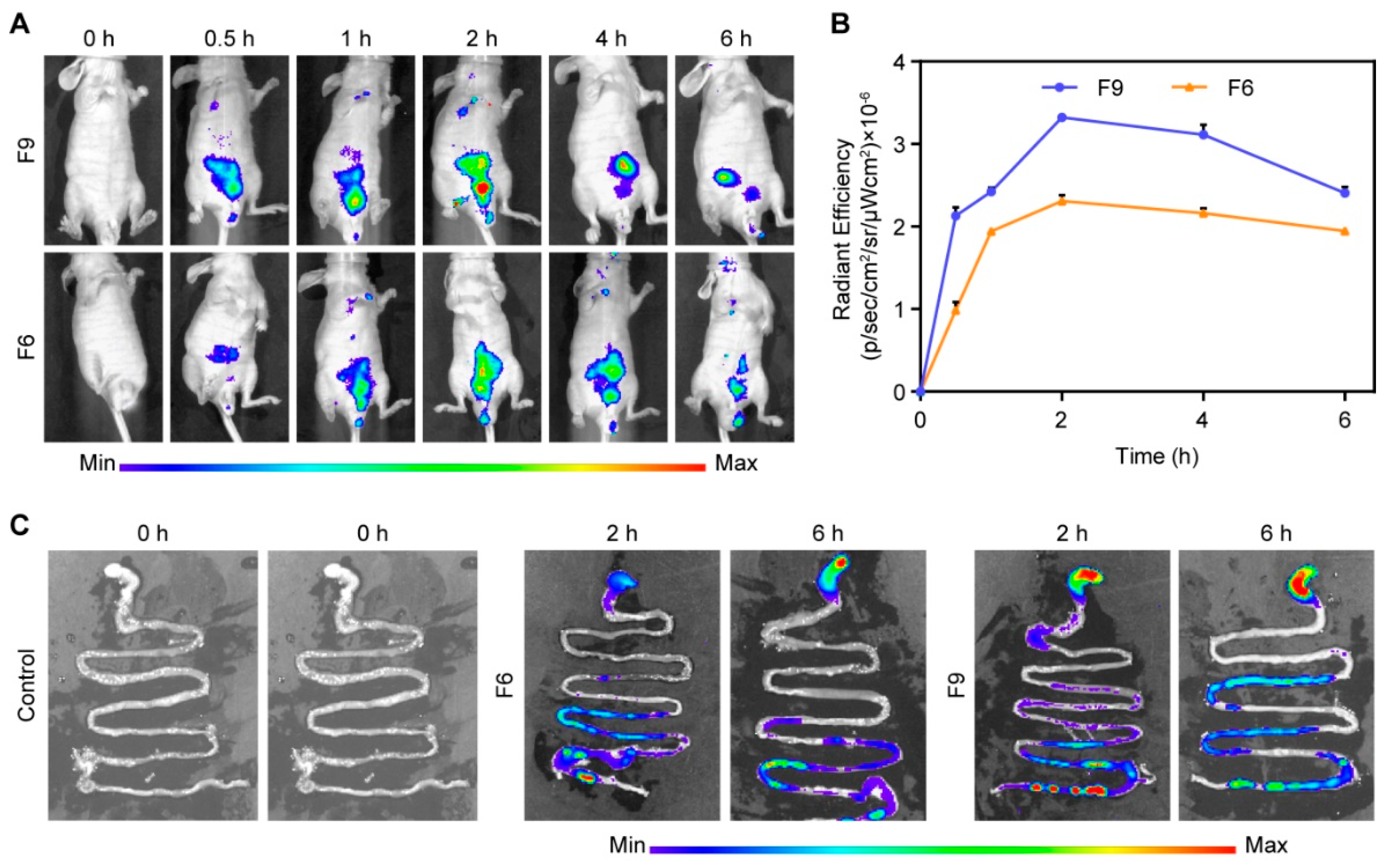
3.4.2. In Vivo Gas Intestine Tract Absorption Studies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Asare-Addo, K.; Alshafiee, M.; Walton, K.; Ward, A.; Totea, A.M.; Taheri, S.; Mawla, N.; Adebisi, A.O.; Elawad, S.; Diza, C.; et al. Effect of preparation method on the surface properties and UV imaging of indomethacin solid dispersions. Eur. J. Pharm. Biopharm. 2019, 137, 148–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; Lalonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Guzman, H.R.; Tawa, M.; Zhang, Z.; Ratanabanangkoon, P.; Shaw, P.; Gardner, C.R.; Chen, H.; Moreau, J.P.; Almarsson, O.; Remenar, J.F. Combined use of crystalline salt forms and precipitation inhibitors to improve oral absorption of celecoxib from solid oral formulations. J. Pharm. Sci. 2007, 96, 2686–2702. [Google Scholar] [CrossRef] [PubMed]
- Laine, A.L.; Price, D.; Davis, J.; Roberts, D.; Hudson, R.; Back, K.; Bungay, P.; Flanagan, N. Enhanced oral delivery of celecoxib via the development of a supersaturable amorphous formulation utilising mesoporous silica and co-loaded HPMCAS. Int. J. Pharm. 2016, 512, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Van Speybroeck, M.; Mols, R.; Mellaerts, R.; Thi, T.D.; Martens, J.A.; Van Humbeeck, J.; Annaert, P.; Van den Mooter, G.; Augustijns, P. Combined use of ordered mesoporous silica and precipitation inhibitors for improved oral absorption of the poorly soluble weak base itraconazole. Eur. J. Pharm. Biopharm. 2010, 75, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Abreu-Villela, R.; Schonenberger, M.; Caraballo, I.; Kuentz, M. Early stages of drug crystallization from amorphous solid dispersion via fractal analysis based on chemical imaging. Eur. J. Pharm. Biopharm. 2018, 133, 122–130. [Google Scholar] [CrossRef]
- Kaminska, E.; Adrjanowicz, K.; Zakowiecki, D.; Milanowski, B.; Tarnacka, M.; Hawelek, L.; Dulski, M.; Pilch, J.; Smolka, W.; Kaczmarczyk-Sedlak, I.; et al. Enhancement of the physical stability of amorphous indomethacin by mixing it with octaacetylmaltose. inter and intra molecular studies. Pharm. Res. 2014, 31, 2887–2903. [Google Scholar] [CrossRef]
- Bertoni, S.; Albertini, B.; Ferraro, L.; Beggiato, S.; Dalpiaz, A.; Passerini, N. Exploring the use of spray congealing to produce solid dispersions with enhanced indomethacin bioavailability: In vitro characterization and in vivo study. Eur. J. Pharm. Biopharm. 2019, 139, 132–141. [Google Scholar] [CrossRef]
- Dening, T.J.; Taylor, L.S. Supersaturation Potential of Ordered Mesoporous Silica Delivery Systems. Part 1: Dissolution Performance and Drug Membrane Transport Rates. Mol. Pharm. 2018, 15, 3489–3501. [Google Scholar] [CrossRef]
- Le, T.T.; Elzhry Elyafi, A.K.; Mohammed, A.R.; Al-Khattawi, A. Delivery of Poorly Soluble Drugs via Mesoporous Silica: Impact of Drug Overloading on Release and Thermal Profiles. Pharmaceutics 2019, 11, 269. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sun, Y.; Wang, J.; Yang, Y.; Li, Y.; Yuan, Y.; Liu, C. Charge-Reversal APTES-Modified Mesoporous Silica Nanoparticles with High Drug Loading and Release Controllability. ACS Appl. Mater. Interfaces 2016, 8, 17166–17175. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Z.; Wei, F.; Luo, Z. Combining autophagy-inducing peptides and brefeldin A delivered by perinuclear-localized mesoporous silica nanoparticles: A manipulation strategy for ER-phagy. Nanoscale 2018, 10, 8796–8805. [Google Scholar] [CrossRef]
- Li, N.; Taylor, L.S. Tailoring supersaturation from amorphous solid dispersions. J. Control. Release 2018, 279, 114–125. [Google Scholar] [CrossRef]
- Lobmann, K.; Strachan, C.; Grohganz, H.; Rades, T.; Korhonen, O.; Laitinen, R. Co-amorphous simvastatin and glipizide combinations show improved physical stability without evidence of intermolecular interactions. Eur. J. Pharm. Biopharm. 2012, 81, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Mosquera-Giraldo, L.I.; Taylor, L.S. Glass-liquid phase separation in highly supersaturated aqueous solutions of telaprevir. Mol. Pharm. 2015, 12, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, Z.; Subramaniam, S.; Khan, G.M.; Abeer, M.M.; Qu, Z.; Janjua, T.; Kumeria, T.; Batra, J.; Popat, A. Encapsulation and Controlled Release of Resveratrol Within Functionalized Mesoporous Silica Nanoparticles for Prostate Cancer Therapy. Front. Bioeng. Biotechnol. 2019, 7, 225. [Google Scholar] [CrossRef] [PubMed]
- Peruzynska, M.; Cendrowski, K.; Barylak, M.; Roginska, D.; Tarnowski, M.; Tkacz, M.; Kurzawski, M.; Machalinski, B.; Mijowska, E.; Drozdzik, M. Study on size effect of the silica nanospheres with solid core and mesoporous shell on cellular uptake. Biomed. Mater. 2015, 10, 065012. [Google Scholar] [CrossRef]
- Djuris, J.; Milovanovic, S.; Medarevic, D.; Dobricic, V.; Dapcevic, A.; Ibric, S. Selection of the suitable polymer for supercritical fluid assisted preparation of carvedilol solid dispersions. Int. J. Pharm. 2019, 554, 190–200. [Google Scholar] [CrossRef]
- Zhang, Q.; Polyakov, N.E.; Chistyachenko, Y.S.; Khvostov, M.V.; Frolova, T.S.; Tolstikova, T.G.; Dushkin, A.V.; Su, W. Preparation of curcumin self-micelle solid dispersion with enhanced bioavailability and cytotoxic activity by mechanochemistry. Drug Deliv. 2018, 25, 198–209. [Google Scholar] [CrossRef]
- Bujard, A.; Petit, C.; Carrupt, P.A.; Rudaz, S.; Schappler, J. HDM-PAMPA to predict gastrointestinal absorption, binding percentage, equilibrium and kinetics constants with human serum albumin and using 2 end-point measurements. Eur. J. Pharm. Sci. 2017, 97, 143–150. [Google Scholar] [CrossRef]
- Shekhawat, P.; Pokharkar, V. Risk assessment and QbD based optimization of an Eprosartan mesylate nanosuspension: In-vitro characterization, PAMPA and in-vivo assessment. Int. J. Pharm. 2019, 567, 118415. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.S.; Yu, J.S.; Lee, J.; Ji, Y.S.; Joung, H.J.; Han, Y.M.; Yoo, H.H.; Kang, K.S. A Hydroxypropyl Methylcellulose-Based Solid Dispersion of Curcumin with Enhanced Bioavailability and its Hepatoprotective Activity. Biomolecules 2019, 9, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Yuan, M.; Deng, Y.; Ke, X.; Ci, T. Different effects of silica added internal or external on in vitro dissolution of indomethacin hot-melt extrudates. Int. J. Pharm. 2017, 534, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Price, D.; Davis, J.; Roberts, D.; Hudson, R.; Back, K.; Bungay, P.; Flanagan, N. Enhanced Oral Bioavailability of Celecoxib Nanocrystalline Solid Dispersion based on Wet Media Milling Technique: Formulation, Optimization and In Vitro/In Vivo Evaluation. Pharmaceutics 2019, 11, 328. [Google Scholar] [CrossRef] [Green Version]
- Doreth, M.; Lobmann, K.; Priemel, P.; Grohganz, H.; Taylor, R.; Holm, R.; Lopez de Diego, H.; Rades, T. Influence of PVP molecular weight on the microwave assisted in situ amorphization of indomethacin. Eur. J. Pharm. Biopharm. 2018, 122, 62–69. [Google Scholar] [CrossRef]
- Alleso, M.; Alleso, M.; Chieng, N.; Rehder, S.; Rantanen, J.; Rades, T.; Aaltonen, J. Enhanced dissolution rate and synchronized release of drugs in binary systems through formulation: Amorphous naproxen-cimetidine mixtures prepared by mechanical activation. J. Control Release 2009, 136, 45–53. [Google Scholar] [CrossRef]
- Surwase, S.A.; Boetker, J.P.; Saville, D.; Boyd, B.J.; Gordon, K.C.; Peltonen, L.; Strachan, C.J. Indomethacin: New polymorphs of an old drug. Mol. Pharm. 2013, 10, 4472–4480. [Google Scholar] [CrossRef]
- Van Duong, T.; Lüdeker, D.; Van Bockstal, P.-J.; De Beer, T.; Van Humbeeck, J.; Van den Mooter, G. Polymorphism of Indomethacin in Semicrystalline Dispersions: Formation, Transformation, and Segregation. Mol. Pharm. 2018, 15, 1037–1051. [Google Scholar] [CrossRef]
- Madgulkar, A.; Bandivadekar, M.; Shid, T.; Rao, S. Sugars as solid dispersion carrier to improve solubility and dissolution of the BCS class II drug: Clotrimazole. Drug Dev. Ind. Pharm. 2016, 42, 28–38. [Google Scholar] [CrossRef]
- Kojo, Y.; Suzuki, H.; Kato, K.; Kaneko, Y.; Yuminoki, K.; Hashimoto, N.; Sato, H.; Seto, Y.; Onoue, S. Enhanced biopharmaceutical effects of tranilast on experimental colitis model with use of self-micellizing solid dispersion technology. Int. J. Pharm. 2018, 545, 19–26. [Google Scholar] [CrossRef]
- Hancock, B.C.; Parks, M. What is the True Solubility Advantage for Amorphous Pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Kalaitzaki, A.; Papanikolaou, N.E.; Karamaouna, F.; Dourtoglou, V.; Xenakis, A.; Papadimitriou, V. Biocompatible colloidal dispersions as potential formulations of natural pyrethrins: A structural and efficacy study. Langmuir 2015, 31, 5722–5730. [Google Scholar] [CrossRef] [PubMed]
- Raina, S.A.; Zhang, G.Z.; Alonzo, D.E.; Wu, J.; Zhu, D.; Catron, N.D.; Gao, Y.; Taylor, L.S. Impact of Solubilizing Additives on Supersaturation.and Membrane Transport of Drugs. Pharm. Res. 2015, 2015, 1–15. [Google Scholar]
- Lavra, Z.M.M.; Santana, D.P.D.; Ré, M.I. Solubility and dissolution performances of spray dried solid dispersion of Efavirenz in Soluplus. Drug Dev. Ind. Pharm. 2016, 43, 42–54. [Google Scholar] [CrossRef]
- Mokhtarpour, M.; Shekaari, H.; Martinez, F.; Zafarani-Moattar, M.T. Effect of Tetrabutylammonium Bromide-Based Deep Eutectic Solvents on the Aqueous Solubility of Indomethacin at Various Temperatures: Measurement, Modeling, and Prediction with Three-Dimensional Hansen Solubility Parameters. AAPS PharmSciTech 2019, 20, 204. [Google Scholar] [CrossRef]
- Wlodarskia, K.; Sawicki, W.; Haber, K.; Knapik, J.; Wojnarowsk, Z.; Paluch, M.; Lepek, P.; Hawelek, L.; Tajber, L. Physicochemical properties of tadalafil solid dispersions—Impact of polymer on the apparent solubility and dissolution rate of tadalafil. Eur. J. Pharm. Biopharm. 2015, 94, 106–115. [Google Scholar] [CrossRef]
- Archer, W.L. Hansen solubility parameters for selected cellulose ether derivatives and their use in the pharmaceutical industry. Drug Dev. Ind. Pharm. 1992, 18, 599–616. [Google Scholar] [CrossRef]
- Thakral, N.K.; Ray, A.R.; Bar-Shalom, D.; Eriksson, A.H.; Majumdar, D.K. Soluplus-solubilized citrated camptothecin--a potential drug delivery strategy in colon cancer. AAPS PharmSciTech 2012, 13, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, H.; Wang, S.; Liu, C.; Qian, F. A Single Hydrogen to Fluorine Substitution Reverses the Trend of Surface Composition Enrichment of Sorafenib Amorphous Solid Dispersion upon Moisture Exposure. Pharm. Res. 2019, 36, 105. [Google Scholar] [CrossRef]
- Ueda, H.; Aikawa, S.; Kashima, Y.; Kikuchi, J.; Ida, Y.; Tanino, T.; Kadota, K.; Tozuka, Y. Anti-plasticizing Effect of Amorphous Indomethacin Induced by Specific Intermolecular Interactions with PVA Copolymer. J. Pharm. Sci. 2014, 103, 2829–2838. [Google Scholar] [CrossRef]
- Ma, X.; Huang, S.; Lowinger, M.B.; Liu, X.; Lu, X.; Su, Y.; Williams, R.O., 3rd. Influence of mechanical and thermal energy on nifedipine amorphous solid dispersions prepared by hot melt extrusion: Preparation and physical stability. Int. J. Pharm. 2019, 561, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rantanen, J.; Yang, M.; Bohr, A. Molecular structure and impact of amorphization strategies on intrinsic dissolution of spray dried indomethacin. Eur. J. Pharm. Sci. 2019, 129, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Matziouridou, C.; Rocha, S.D.C.; Haabeth, O.A.; Rudi, K.; Carlsen, H.; Kielland, A. iNOS- and NOX1-dependent ROS production maintains bacterial homeostasis in the ileum of mice. Mucosal Immunol. 2018, 11, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, M.; Muraki, Y.; Nishimoto, Y.; Murakawa, Y.; Matsuo, T. Fluorescence-labeled liposome accumulation in injured colon of a mouse model of T-cell transfer-mediated inflammatory bowel disease. Biochem. Biophys. Res. Commun. 2017, 494, 188–193. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | IMC | MSNs | Kollicoat IR | HPMC |
|---|---|---|---|---|
| F1 | 1 | 1 | - | - |
| F2 | 1 | 3 | - | - |
| F3 | 1 | 5 | - | - |
| F4 | 1 | 1 | 3 | - |
| F5 | 1 | 3 | 3 | - |
| F6 | 1 | 5 | 3 | - |
| F7 | 1 | 1 | - | 3 |
| F8 | 1 | 3 | - | 3 |
| F9 | 1 | 5 | - | 3 |
| Formulation | Cumulative Drug Dissolution Rate (%) | Folds |
|---|---|---|
| IMC | 27.43 ± 0.96 | 1.00 ± 0.28 |
| F3 | 74.96 ± 12.43 | 2.73 ± 0.45 |
| F6 | 83.54 ± 0.39 | 3.23 ± 0.02 |
| F9 | 104.00 ± 1.39 | 3.79 ± 0.05 |
| Formulation | Cmax (μg/mL) | Tmax (h) | AUC0→t (μg·mL−1·h) | Frel (%) |
|---|---|---|---|---|
| IMC | 124.6 ± 3.8 | 4.0 | 1236.5 ± 188.8 | - |
| F3 | 223.1 ± 44.1 | 4.0 | 2010.5 ± 441.9 | 162.6 ± 35.7 |
| F6 | 392.5 ± 47.0 | 2.0 | 2976.0 ± 207.9 | 240.7 ± 16.8 |
| F9 | 516.8 ± 18.1 | 2.0 | 4086.5 ± 485.8 | 330.5 ± 39.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xi, Z.; Zhang, W.; Fei, Y.; Cui, M.; Xie, L.; Chen, L.; Xu, L. Evaluation of the Solid Dispersion System Engineered from Mesoporous Silica and Polymers for the Poorly Water Soluble Drug Indomethacin: In Vitro and In Vivo. Pharmaceutics 2020, 12, 144. https://doi.org/10.3390/pharmaceutics12020144
Xi Z, Zhang W, Fei Y, Cui M, Xie L, Chen L, Xu L. Evaluation of the Solid Dispersion System Engineered from Mesoporous Silica and Polymers for the Poorly Water Soluble Drug Indomethacin: In Vitro and In Vivo. Pharmaceutics. 2020; 12(2):144. https://doi.org/10.3390/pharmaceutics12020144
Chicago/Turabian StyleXi, Ziyue, Wei Zhang, Yali Fei, Mingshu Cui, Luyao Xie, Lu Chen, and Lu Xu. 2020. "Evaluation of the Solid Dispersion System Engineered from Mesoporous Silica and Polymers for the Poorly Water Soluble Drug Indomethacin: In Vitro and In Vivo" Pharmaceutics 12, no. 2: 144. https://doi.org/10.3390/pharmaceutics12020144
APA StyleXi, Z., Zhang, W., Fei, Y., Cui, M., Xie, L., Chen, L., & Xu, L. (2020). Evaluation of the Solid Dispersion System Engineered from Mesoporous Silica and Polymers for the Poorly Water Soluble Drug Indomethacin: In Vitro and In Vivo. Pharmaceutics, 12(2), 144. https://doi.org/10.3390/pharmaceutics12020144