Dual Peptide-Modified Nanoparticles Improve Combination Chemotherapy of Etoposide and siPIK3CA Against Drug-Resistant Small Cell Lung Carcinoma

Abstract
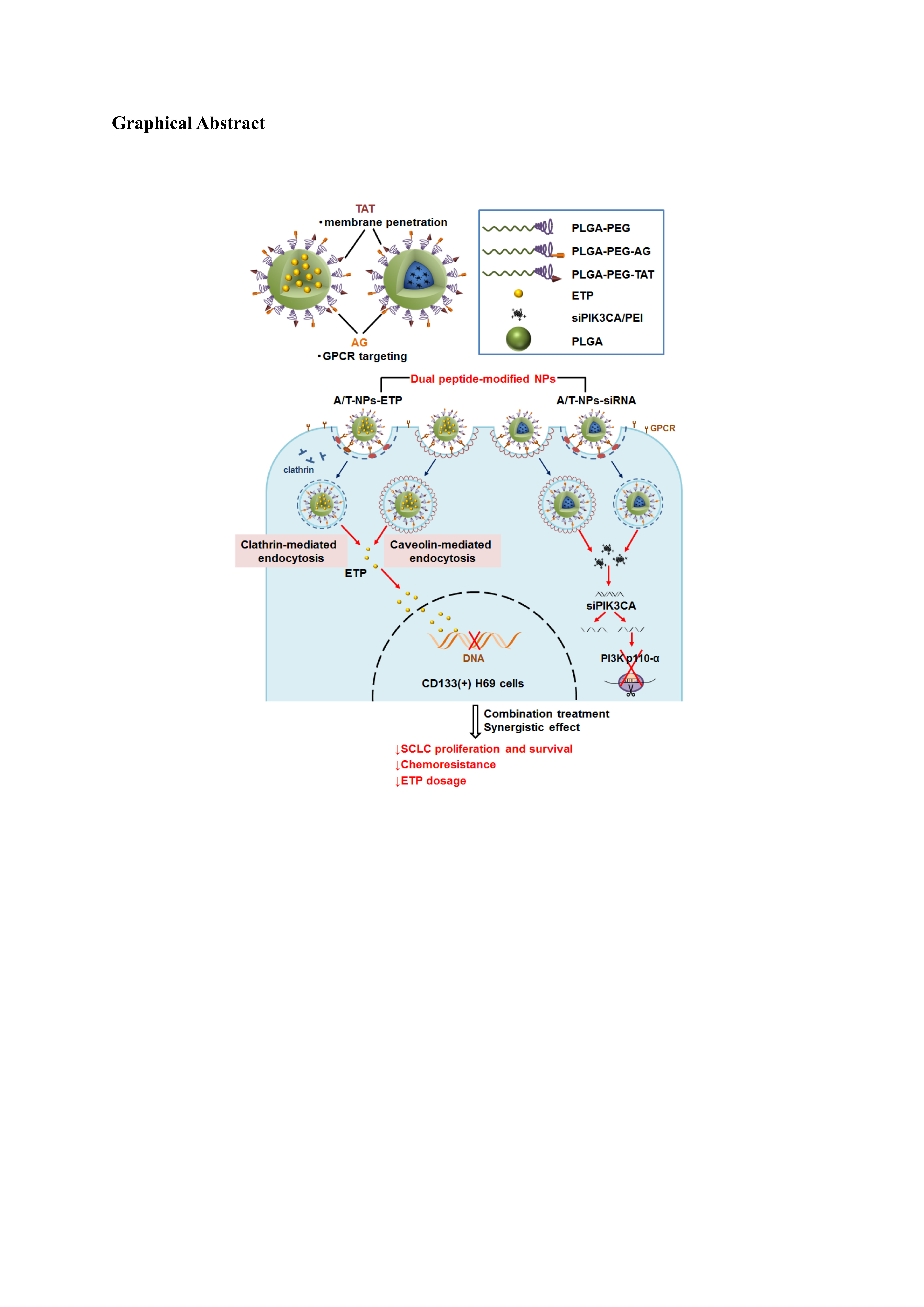
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Peptide-Conjugated Copolymers
2.3. Preparation and Characterization of ETP@NPs and siRNA@NPs
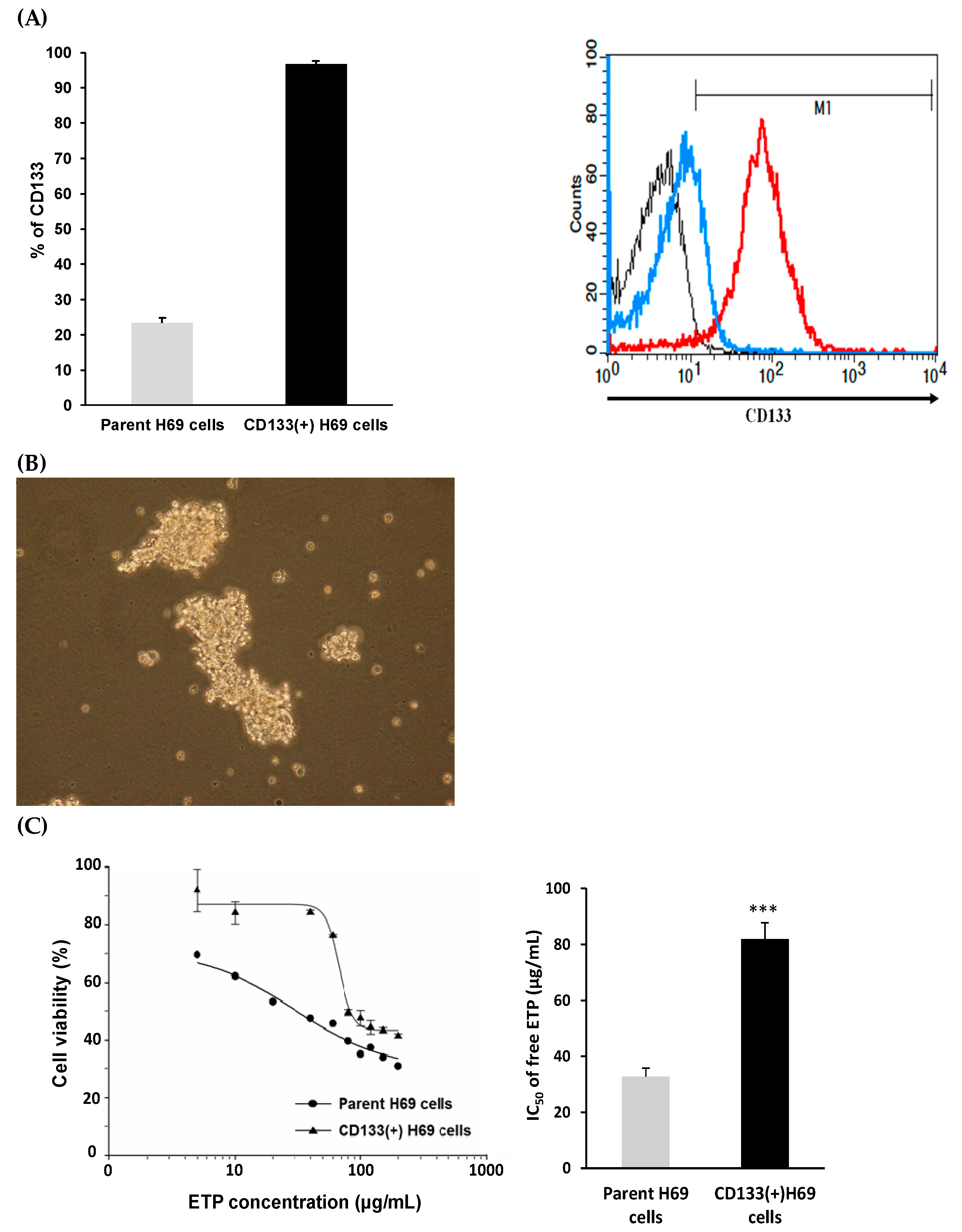
2.4. Sorting of CD133(+) H69 Cells
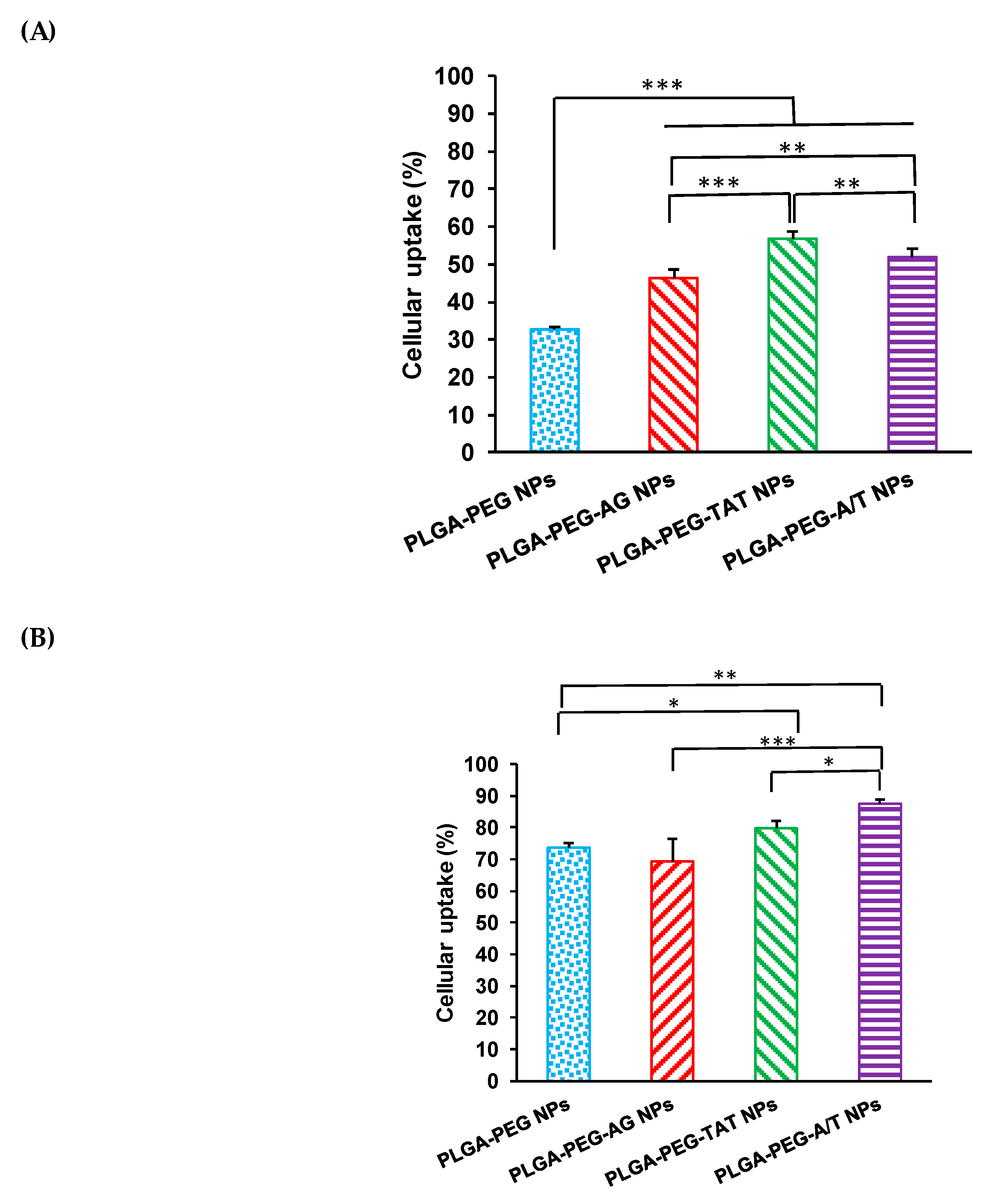
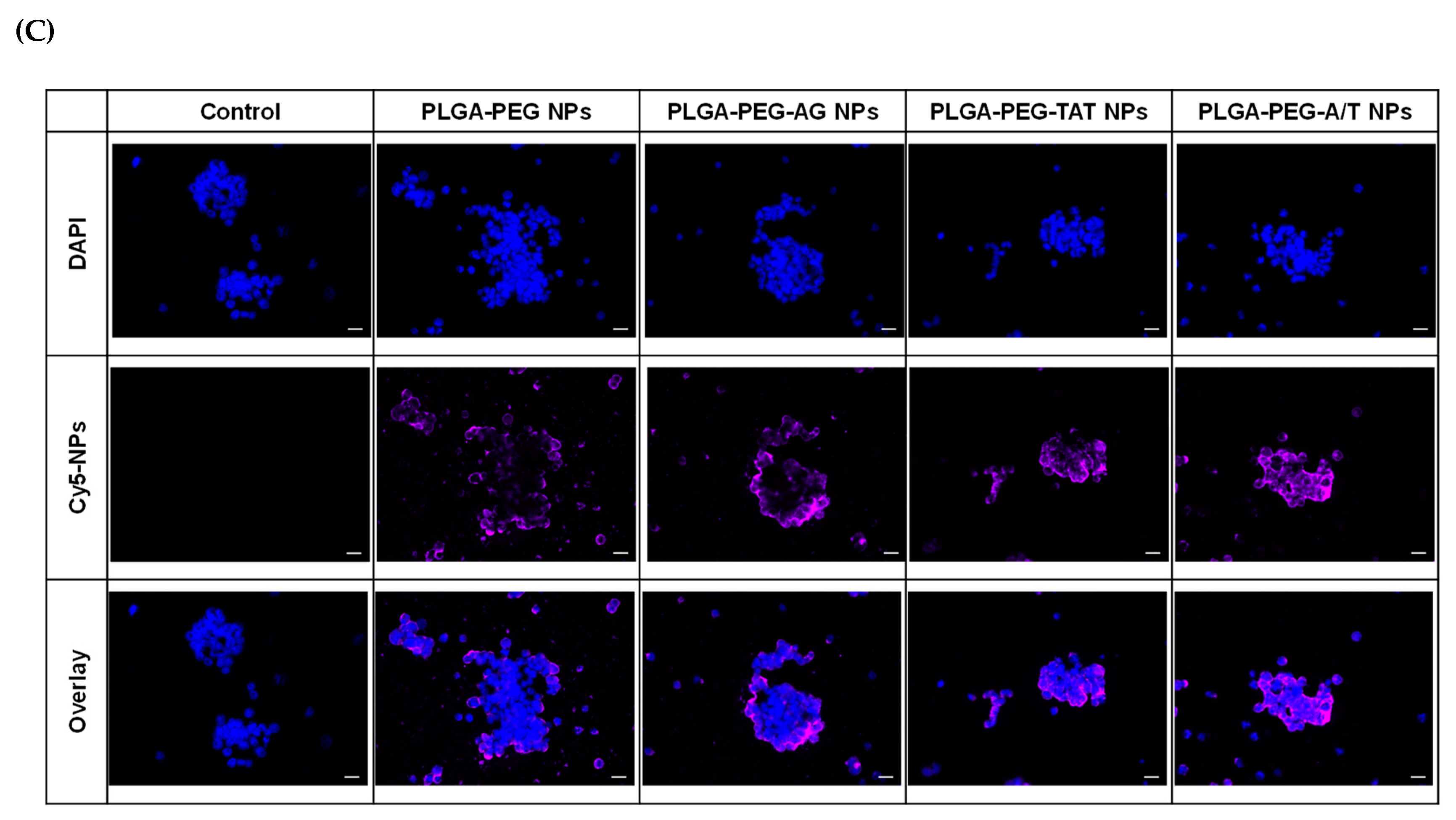
2.5. Cellular Uptake
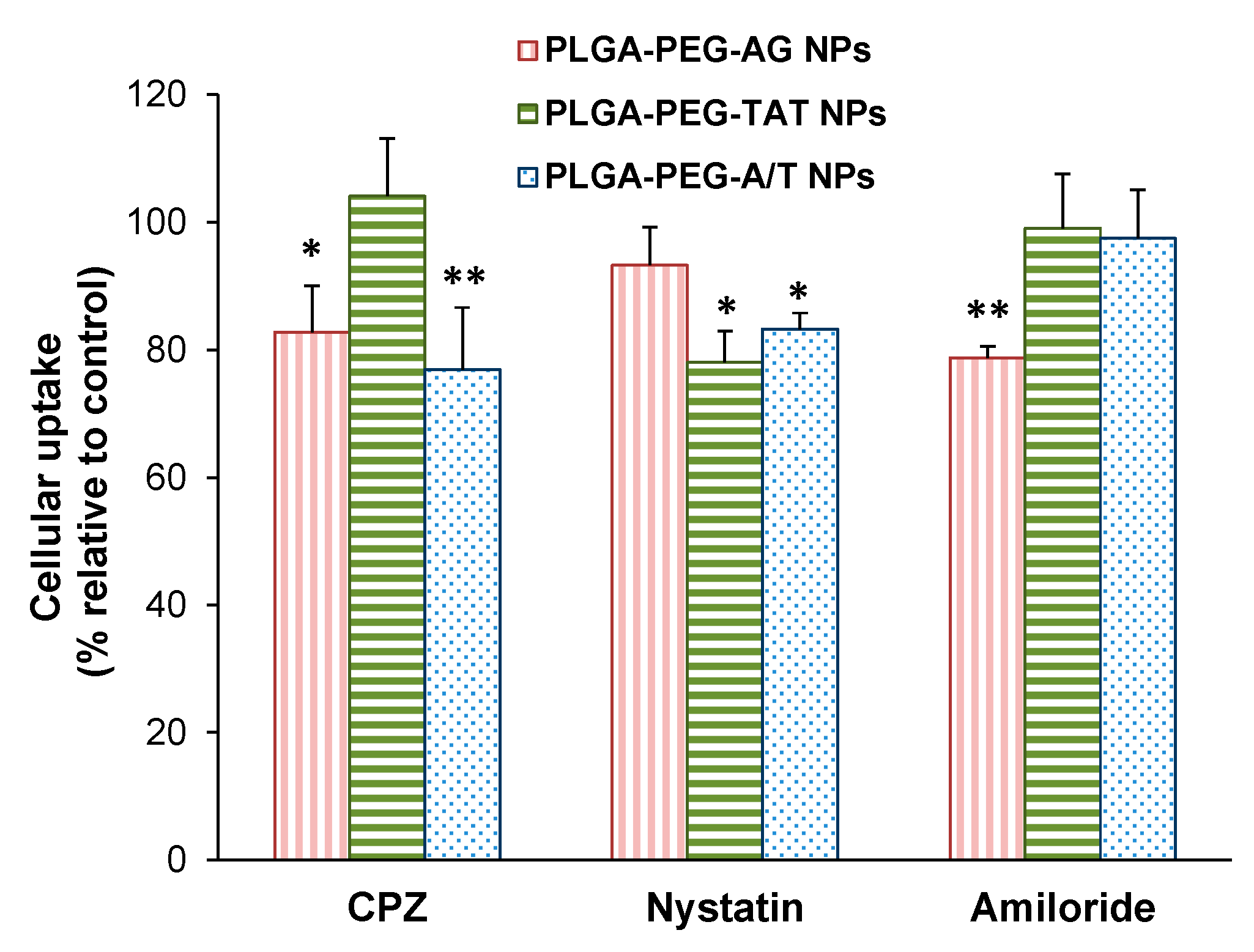
2.6. Endocytosis Mechanism
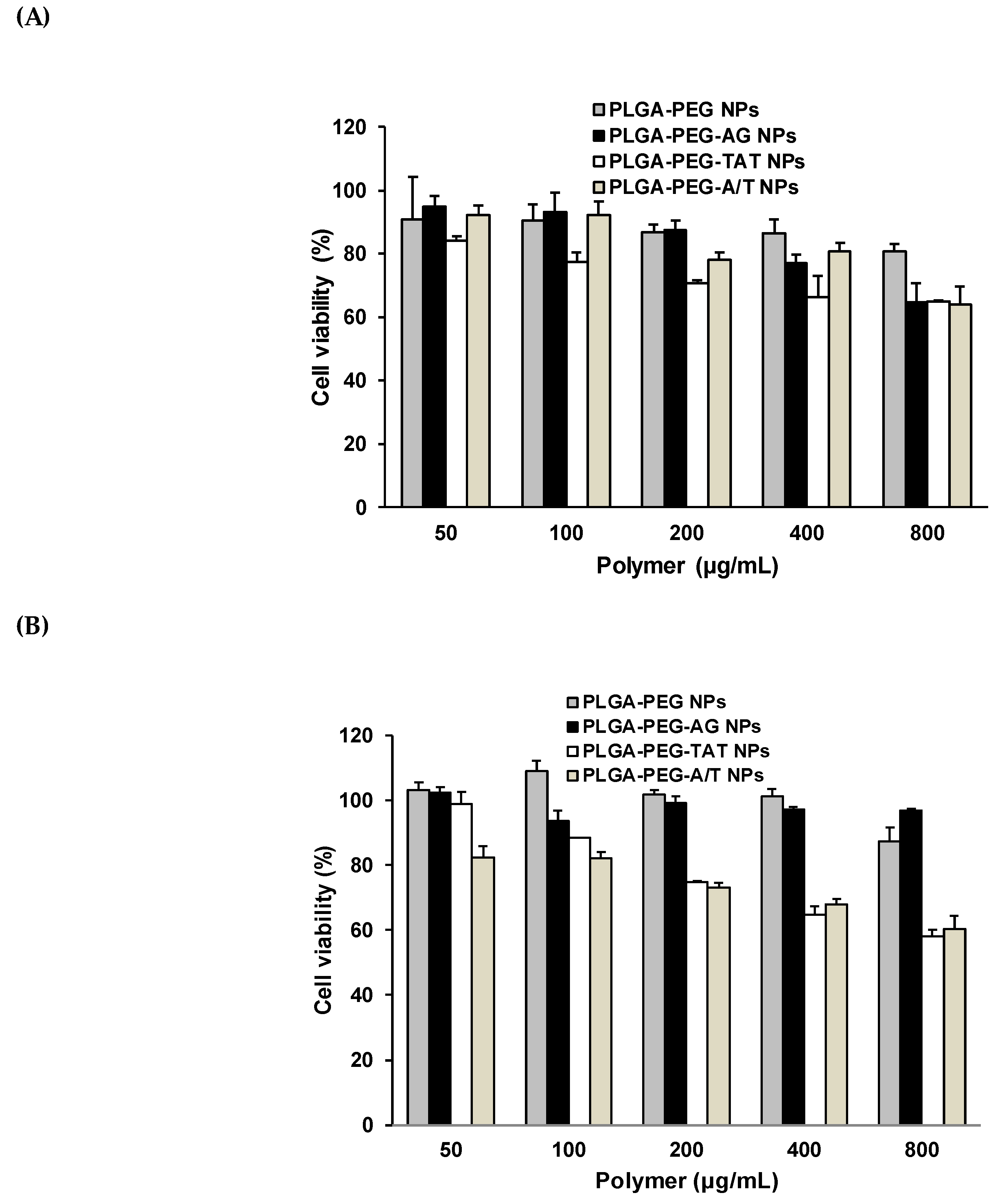
2.7. Cytotoxicity Study
2.8. Statistical Analysis
3. Results and Discussion
3.1. Characterization of Peptide-Conjugated Copolymers
3.2. Characterization of Peptide-Conjugated Nanoparticles
3.3. Characterization of ETP@NPs and siRNA@NPs
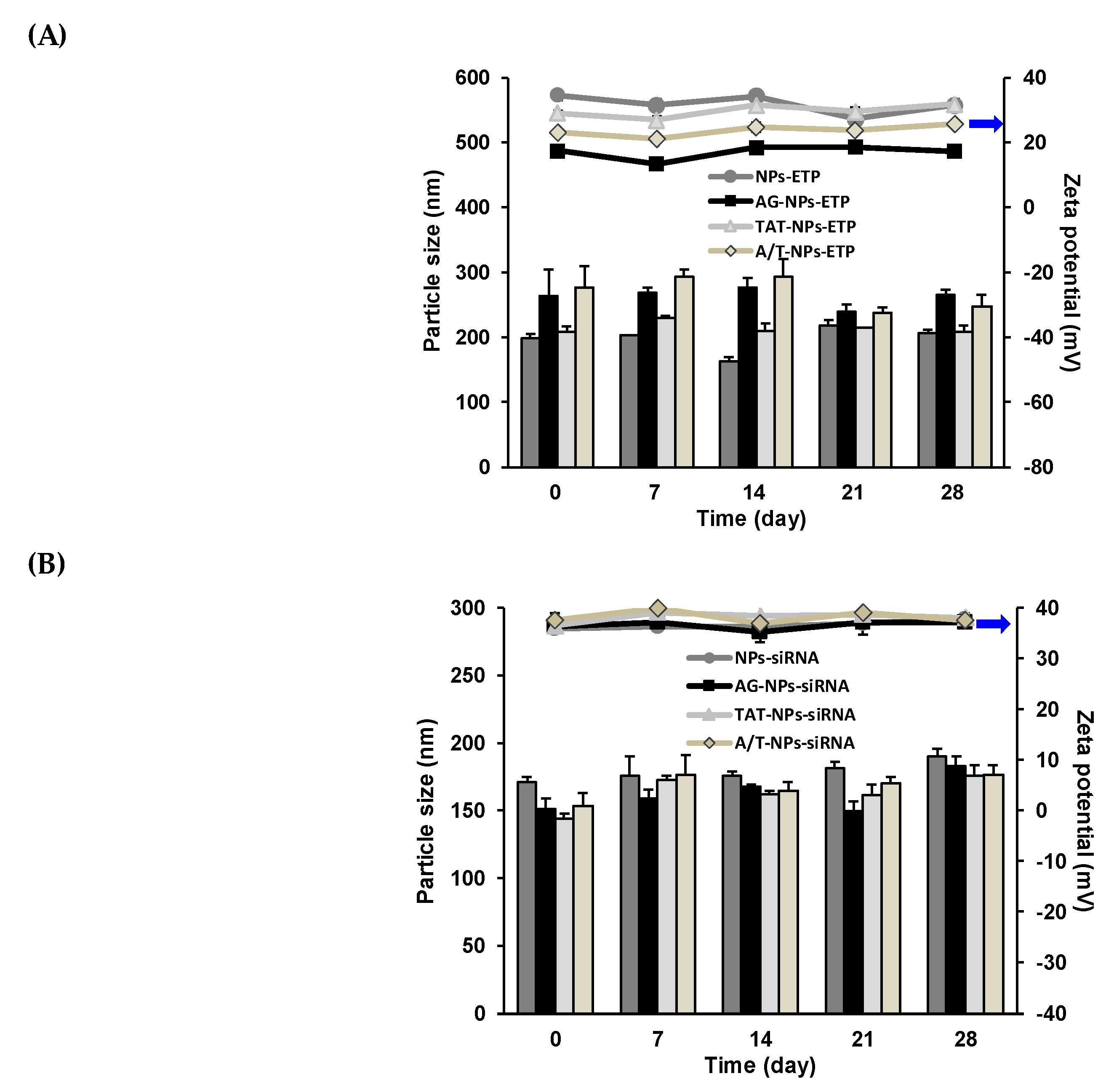
3.4. Stability of ETP@NPs and siRNA@NPs
3.5. Cellular Uptake
3.6. Endocytosis Mechanism
3.7. Cytotoxicity of Peptide-Free and Peptide-Conjugated NPs
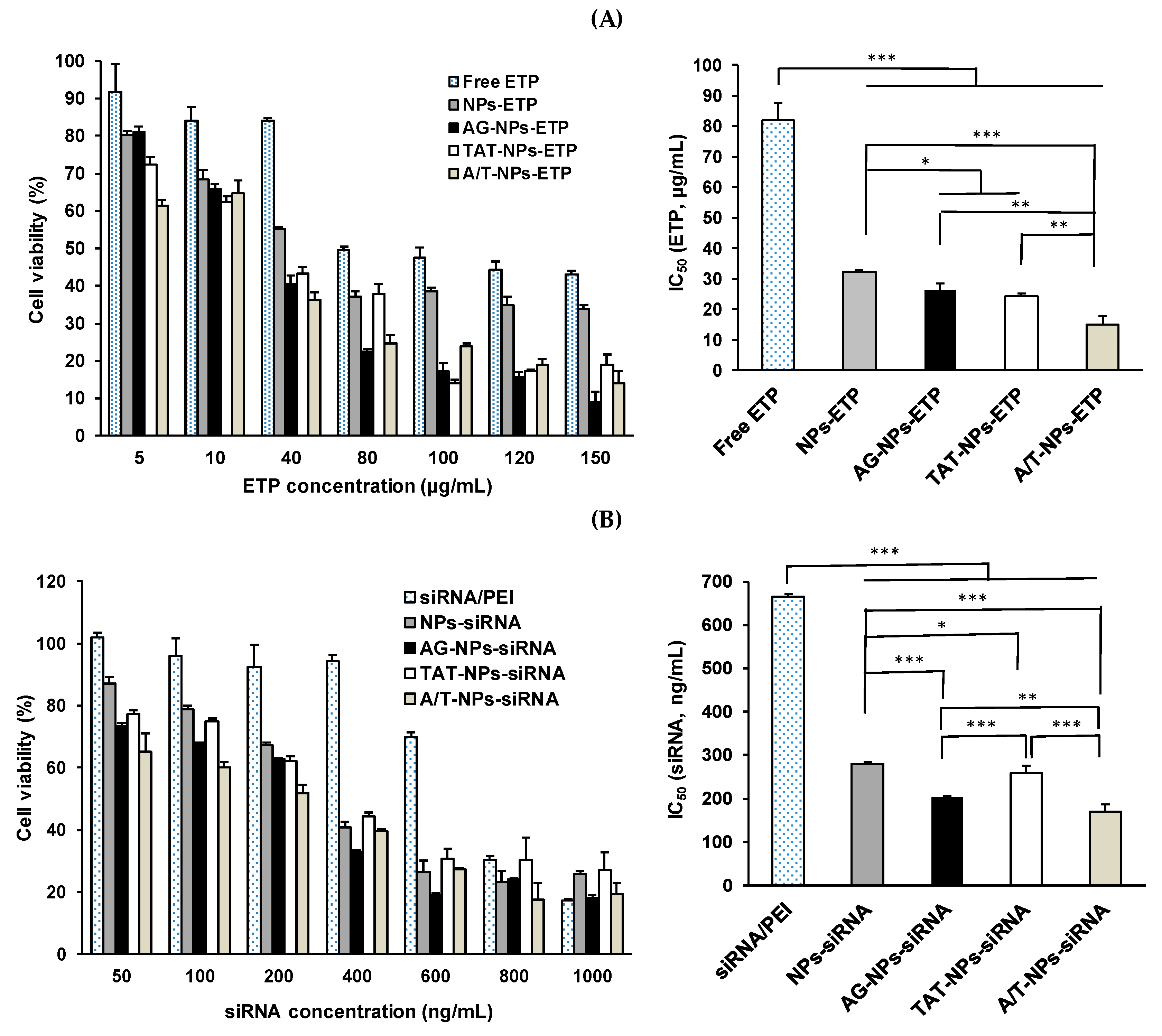
3.8. Cytotoxicity of ETP@NPs and siRNA@NPs
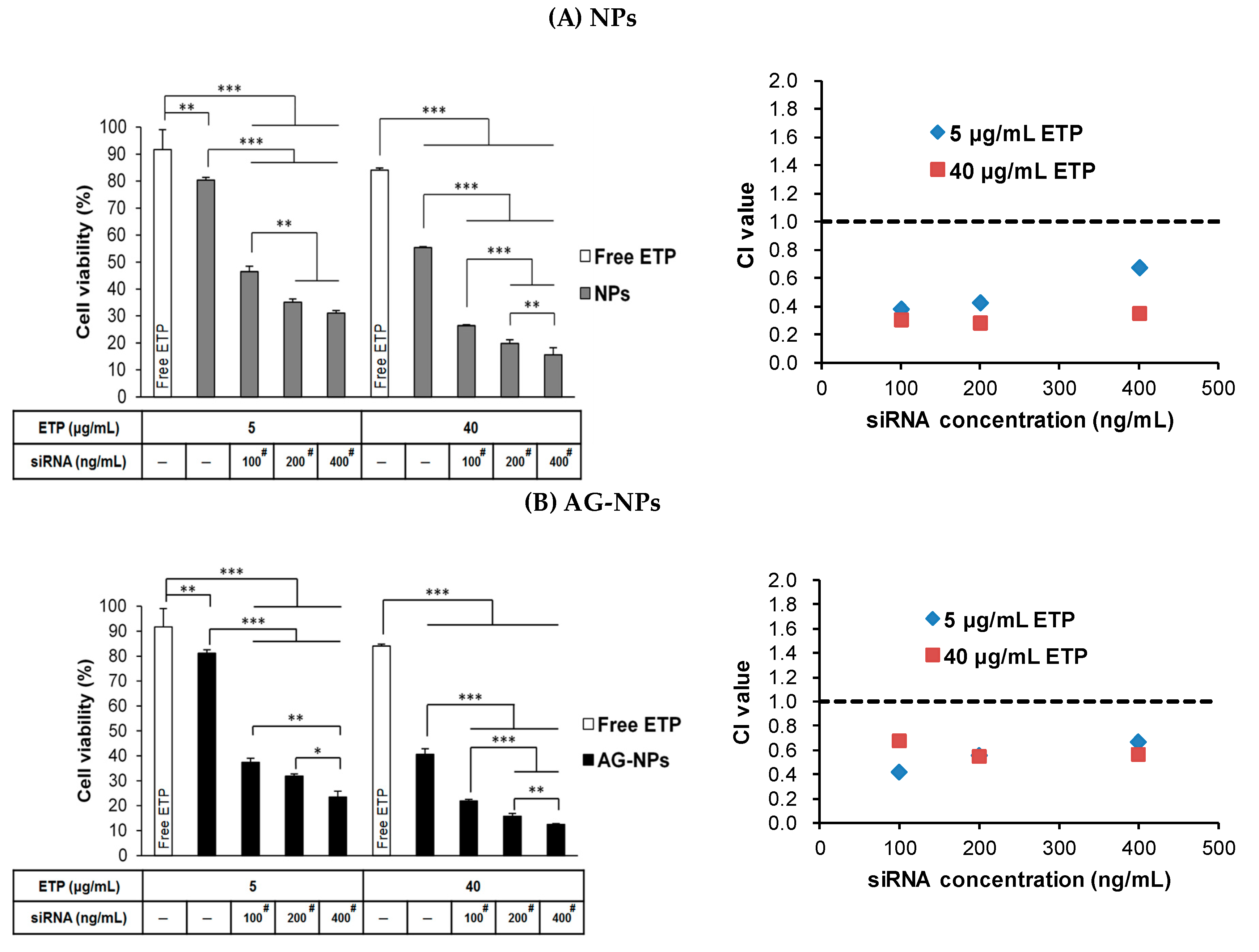
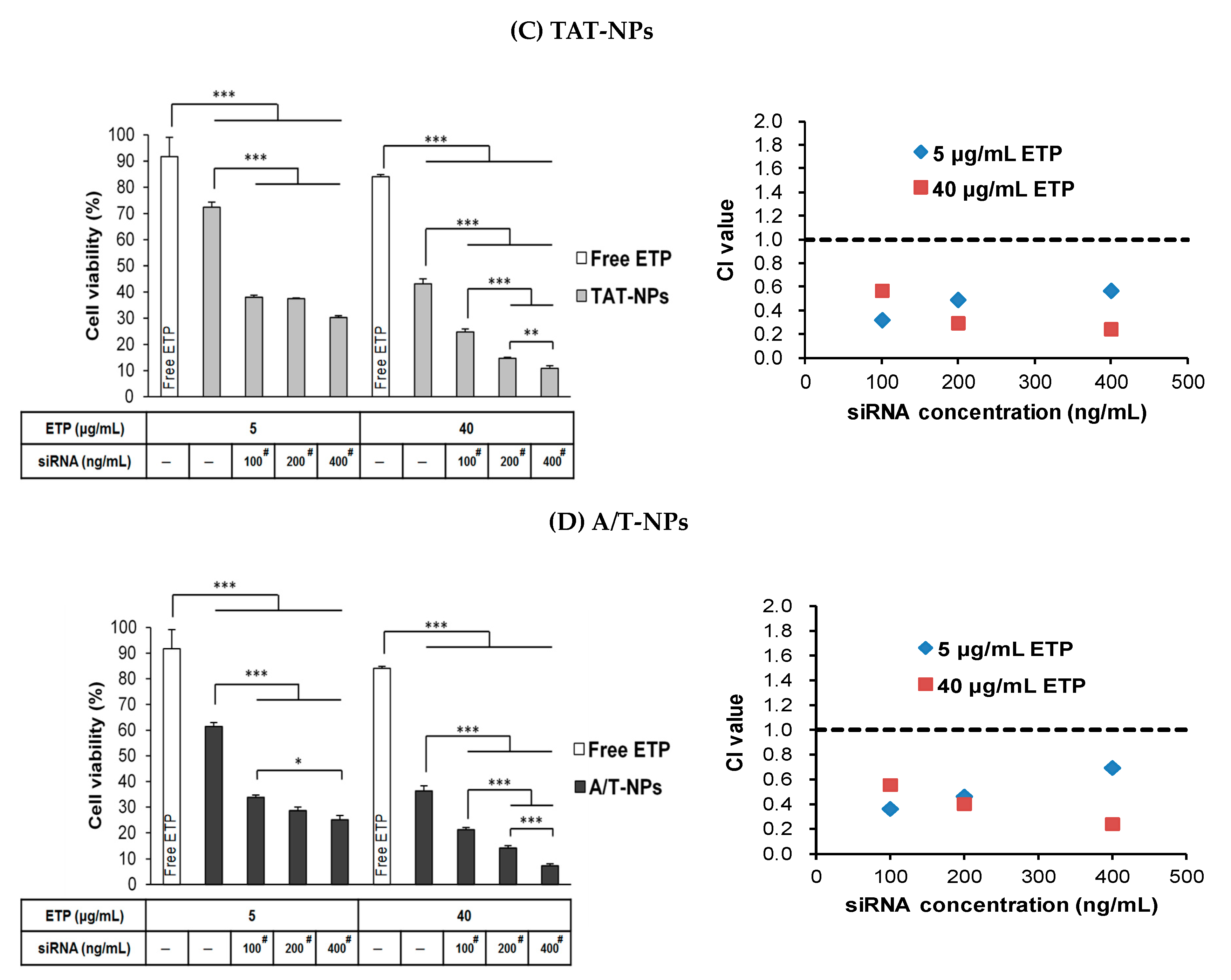
3.9. Combination Treatment of ETP@NPs and siRNA@NPs
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Van Meerbeeck, J.P.; Fennell, D.A.; De Ruysscher, D.K. Small-cell lung cancer. Lancet 2011, 378, 1741–1755. [Google Scholar] [CrossRef]
- Planchard, D.; Le Pechoux, C. Small cell lung cancer: New clinical recommendations and current status of biomarker assessment. Eur. J. Cancer 2011, 47, S272–S283. [Google Scholar] [CrossRef]
- Travis, W.D. Advances in neuroendocrine lung tumors. Ann. Oncol. 2010, 21, vii65–vii71. [Google Scholar] [CrossRef] [PubMed]
- Karen, K. Extensive stage small cell lung cancer: Initial management. UpToDate. 2019. Available online: https://www.uptodate.com/contents/extensive-stage-small-cell-lung-cancer-initial-management (accessed on 15 January 2020).
- Taylor, W.F.; Moghadam, S.E.; Moridi Farimani, M.; Ebrahimi, S.N.; Tabefam, M.; Jabbarzadeh, E. A multi-targeting natural compound with growth inhibitory and anti-angiogenic properties re-sensitizes chemotherapy resistant cancer. PLoS ONE 2019, 14, e0218125. [Google Scholar] [CrossRef] [Green Version]
- Offerman, S.C.; Kadirvel, M.; Abusara, O.H.; Bryant, J.L.; Telfer, B.A.; Brown, G.; Freeman, S.; White, A.; Williams, K.J.; Aojula, H.S. N-tert-Prenylation of the indole ring improves the cytotoxicity of a short antagonist G analogue against small cell lung cancer. Medchemcomm 2017, 8, 551–558. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Xiong, Y.; Lee, L.T.O. Application of Nanoparticles for Targeting G Protein-Coupled Receptors. Int. J. Mol. Sci. 2018, 19, 2006. [Google Scholar] [CrossRef] [Green Version]
- Moreira, J.N.; Gaspar, R.; Allen, T.M. Targeting Stealth liposomes in a murine model of human small cell lung cancer. Biochim. Biophys. Acta (BBA)-Biomembr. 2001, 1515, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Moreira, J.N.; Gaspar, R. Antagonist G-mediated targeting and cytotoxicity of liposomal doxorubicin in NCI-H82 variant small cell lung cancer. Braz. J. Med. Biol. Res. 2004, 37, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Futaki, S.; Nakase, I.; Tadokoro, A.; Takeuchi, T.; Jones, A.T. Arginine-rich peptides and their internalization mechanisms. Biochem. Soc. Trans. 2007, 35, 784–787. [Google Scholar] [CrossRef]
- Letoha, T.; Keller-Pinter, A.; Kusz, E.; Kolozsi, C.; Bozso, Z.; Toth, G.; Vizler, C.; Oláh, Z.; Szilák, L. Cell-penetrating peptide exploited syndecans. Biochim. Biophys. Acta (BBA)-Biomembr. 2010, 1798, 2258–2265. [Google Scholar] [CrossRef] [Green Version]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, U. Cell-penetrating peptides: Design, synthesis, and applications. ACS Nano 2014, 8, 1972–1994. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.B.; Pereira, M.P.; Kelley, S.O. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv. Drug Deliv. Rev. 2009, 61, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Rizzuti, M.; Nizzardo, M.; Zanetta, C.; Ramirez, A.; Corti, S. Therapeutic applications of the cell-penetrating HIV-1 Tat peptide. Drug Discov. Today 2015, 20, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Levina, V.; Marrangoni, A.M.; DeMarco, R.; Gorelik, E.; Lokshin, A.E. Drug-selected human lung cancer stem cells: Cytokine network, tumorigenic and metastatic properties. PLoS ONE 2008, 3, e3077. [Google Scholar] [CrossRef] [Green Version]
- Codony-Servat, J.; Verlicchi, A.; Rosell, R. Cancer stem cells in small cell lung cancer. Transl. Lung Cancer Res. 2016, 5, 16–25. [Google Scholar]
- Sarvi, S.; Mackinnon, A.C.; Avlonitis, N.; Bradley, M.; Rintoul, R.C.; Rassl, D.M.; Wang, W.; Forbes, S.J.; Gregory, C.D.; Sethi, T. CD133+ cancer stem-like cells in small cell lung cancer are highly tumorigenic and chemoresistant but sensitive to a novel neuropeptide antagonist. Cancer Res. 2014, 74, 1554–1565. [Google Scholar] [CrossRef] [Green Version]
- Walls, M.; Baxi, S.M.; Mehta, P.P.; Liu, K.K.; Zhu, J.; Estrella, H.; Li, C.; Zientek, M.; Zong, Q.; Smeal, T.; et al. Targeting small cell lung cancer harboring PIK3CA mutation with a selective oral PI3K inhibitor PF-4989216. Clin. Cancer Res. 2014, 20, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Wojtalla, A.; Fischer, B.; Kotelevets, N.; Mauri, F.A.; Sobek, J.; Rehrauer, H.; Wotzkow, C.; Tschan, M.P.; Seck, M.J.; Zangemeister-Wittke, U.; et al. Targeting the phosphoinositide 3-kinase p110-alpha isoform impairs cell proliferation, survival, and tumor growth in small cell lung cancer. Clin. Cancer Res. 2013, 19, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Iranpur Mobarakeh, V.; Modarressi, M.H.; Rahimi, P.; Bolhassani, A.; Arefian, E.; Atyabi, F.; Vahabpour, R. Optimization of chitosan nanoparticles as an anti-HIV siRNA delivery vehicle. Int. J. Biol. Macromol. 2019, 129, 305–315. [Google Scholar] [CrossRef]
- Jyotsana, N.; Sharm, A.; Chaturvedi, A.; Budida, R.; Scherr, M.; Kuchenbauer, F.; Lindner, R.; Noyan, F.; Sühs, K.W.; Stangel, M.; et al. Lipid nanoparticle-mediated siRNA delivery for safe targeting of human CML in vivo. Ann. Hematol. 2019, 98, 1905–1918. [Google Scholar] [CrossRef]
- Rios de la Rosa, J.M.; Pingrajai, P.; Pelliccia, M.; Spadea, A.; Lallana, E.; Gennari, A.; Stratford, I.J.; Rocchia, W.; Tirella, A.; Tirelli, N. Binding and Internalization in Receptor-Targeted Carriers: The Complex Role of CD44 in the Uptake of Hyaluronic Acid-Based Nanoparticles (siRNA Delivery). Adv. Healthc. Mater. 2019, 8, e1901182. [Google Scholar] [CrossRef] [PubMed]
- Glisson, B.; Besse, B.; Dols, M.C.; Dubey, S.; Schupp, M.; Jain, R.; Jiang, Y.; Menon, H.; Nackaerts, K.; Orlov, S.; et al. Randomized, Placebo-Controlled, Phase 1b/2 Study of Rilotumumab or Ganitumab in Combination with Platinum-Based Chemotherapy as First-Line Treatment for Extensive-Stage Small-Cell Lung Cancer. Clin. Lung Cancer 2017, 18, 615–625.e8. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Liu, C.; Lin, Y.; Fu, J.; Lu, G.; Lu, Z. pH sensitive peptide functionalized nanoparticles for co-delivery of erlotinib and DAPT to restrict the progress of triple negative breast cancer. Drug Deliv. 2019, 26, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.E.; Ciampa, O.C.; Wong, M.; Fletcher, J.I.; Kamili, A.; Chen, J.; Ho, N.; Sun, Y.; Carter, D.R.; Cheung, B.B.; et al. Combination therapy with the CDK7 inhibitor and the tyrosine kinase inhibitor exerts synergistic anticancer effects against MYCN-amplified neuroblastoma. Int. J. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Flont, M.; Jastrzębska, E.; Brzózka, Z. Synergistic effect of the combination therapy on ovarian cancer cells under microfluidic conditions. Anal. Chim. Acta 2020, 1100, 138–148. [Google Scholar] [CrossRef]
- Feng, C.; Zhang, H.; Chen, J.; Wang, S.; Xin, Y.; Qu, Y.; Zhang, Q.; Ji, W.; Yamashita, F.; Rui, M.; et al. Ratiometric co-encapsulation and co-delivery of doxorubicin and paclitaxel by tumor-targeted lipodisks for combination therapy of breast cancer. Int. J. Pharm. 2019, 560, 191–204. [Google Scholar] [CrossRef]
- Lin, W.J.; Chien, W.H. Peptide Conjugated Micelles as a Targeting Nanocarrier for Gene Delivery. J. Nanoparticle Res. 2015, 17, 349. [Google Scholar] [CrossRef]
- Lin, W.J.; Kao, L.T. Cytotoxic enhancement of hexapeptide-conjugated micelles in epidermal growth factor receptor high-expressed cancer cells. Expert Opin. Drug Deliv. 2014, 11, 1–14. [Google Scholar] [CrossRef]
- Saadati, R.; Dadashzadeh, S. Marked effects of combined TPGS and PVA emulsifiers in the fabrication of etoposide-loaded PLGA-PEG nanoparticles: In vitro and in vivo evaluation. Int. J. Pharm. 2014, 464, 135–144. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Dördelmann, G.; Kozlova, D.; Karczewski, S.; Lizio, R.; Knauer, S.; Epple, M. Calcium phosphate increases the encapsulation efficiency of hydrophilic drugs (proteins, nucleic acids) into poly(D,L-lactide-co-glycolide acid) nanoparticles for intracellular delivery. J. Mater. Chem. B 2014, 2, 7250–7259. [Google Scholar] [CrossRef] [Green Version]
- Adiseshaiah, P.P.; Hall, J.B.; McNeil, S.E. Nanomaterial standards for efficacy and toxicity assessment. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2010, 2, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Z.; Dou, S.; Sun, T.M.; Mao, C.Q.; Wang, H.X.; Wang, J. Systemic delivery of siRNA with cationic lipid assisted PEG-PLA nanoparticles for cancer therapy. J. Control. Release 2011, 156, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Ocejo-Garcia, M.; Ahmed, S.I.; Coulson, J.M.; Woll, P.J. Use of RT-PCR to detect co-expression of neuropeptides and their receptors in lung cancer. Lung Cancer 2001, 33, 1–9. [Google Scholar] [CrossRef]
- Gullotti, E.; Yeo, Y. Beyond the imaging: Limitations of cellular uptake study in the evaluation of nanoparticles. J. Control. Release 2012, 164, 170–176. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Tang, W.; Luo, X.; Zhang, X.; Zhang, C.; Li, H.; Gao, D.; Luo, H.; Jiang, Q.; Liu, J. Dual-ligand modified polymer-lipid hybrid nanoparticles for docetaxel targeting delivery to Her2/neu overexpressed human breast cancer cells. J. Biomed. Nanotechnol. 2015, 11, 1401–1417. [Google Scholar] [CrossRef]
- Das, M.; Solanki, A.; Joshi, A.; Devkar, R.; Seshadri, S.; Thakore, S. β-cyclodextrin based dual-responsive multifunctional nanotheranostics for cancer cell targeting and dual drug delivery. Carbohydr. Polym. 2019, 206, 694–705. [Google Scholar] [CrossRef]
- Babincová, N.; Sourivong, P.; Babinec, P.; Bergemann, C.; Babincová, M.; Durdík, S. Applications of magnetoliposomes with encapsulated doxorubicin for integrated chemotherapy and hyperthermia of rat C6 glioma. Z. Nat. C 2018, 73, 265–271. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, C.; Zeng, F.; Zhou, H.; Li, D.; He, X.; Shen, S.; Yang, X.; Wang, J. A polymeric nanocarrier with a tumor acidity-activatable arginine-rich (R9) peptide for enhanced drug delivery. Biomater. Sci. 2020. [Google Scholar] [CrossRef]
- Tan, T.; Wang, Y.; Wang, J.; Wang, Z.; Wang, H.; Cao, H.; Li, J.; Li, Y.; Zhang, Z.; Wang, S. Targeting peptide-decorated biomimetic lipoproteins improve deep penetration and cancer cells accessibility in solid tumor. Acta Pharm. Sin. B 2020, 10, 529–545. [Google Scholar] [CrossRef]
- Zhao, T.; Zhou, H.; Lei, L.; Guo, C.; Yang, Q.; Gong, T.; Sun, X.; Song, X.; Gong, T.; Zhang, Z. A new tandem peptide modified liposomal doxorubicin for tumor “ecological therapy”. Nanoscale 2020, 12, 3359–3369. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NPs | PLGA-PEG | PLGA-PEG-AG | PLGA-PEG-TAT | PLGA-PEG-A/T |
|---|---|---|---|---|
| Particle size (nm) | 166.9 ± 5.3 | 167.8 ± 9.8 | 174.0 ± 8.4 | 169.2 ± 7.6 |
| PDI | 0.15 ± 0.06 | 0.13 ± 0.05 | 0.13 ± 0.03 | 0.11 ± 0.01 |
| Zeta potential (mV) | 26.7 ± 1.8 | 23.1 ± 4.3 | 35.3 ± 2.2 | 24.1 ± 0.5 |
| Yield (%) | 75.3 ± 2.9 | 79.5 ± 10.3 | 85.3 ± 3.6 | 81.1 ± 2.5 |
| ETP@NPs | NPs-ETP | AG-NPs-ETP | TAT-NPs-ETP | A/T-NPs-ETP |
|---|---|---|---|---|
| Particle size (nm) | 201.0 ± 1.9 | 201.3 ± 4.6 | 202.3 ± 3.5 | 206.5 ± 0.7 |
| PDI | 0.13 ± 0.01 | 0.08 ± 0.04 | 0.08 ± 0.07 | 0.05 ± 0.05 |
| Zeta potential (mV) | 36.3 ± 2.4 | 36.3 ± 1.0 | 39.6 ± 1.0 | 36.6 ± 1.5 |
| EE (%) | 61.6 ± 2.6 | 61.4 ± 12.9 | 61.1 ± 6.0 | 79.3 ± 15.3 |
| Drug loading (%) | 10.1 ± 0.8 | 10.2 ± 2.3 | 10.2 ± 1.2 | 12.3 ± 1.8 |
| Yield (%) | 87.2 ± 4.2 | 86.7 ± 1.7 | 86.1 ± 1.9 | 91.7 ± 5.0 |
| siRNA@NPs | NPs-siRNA | AG-NPs-siRNA | TAT-NPs-siRNA | A/T-NPs-siRNA |
|---|---|---|---|---|
| Particle size (nm) | 166.2 ± 6.2 | 162.6 ± 21.0 | 155.3 ± 12.4 | 169.1 ± 11.2 |
| PDI | 0.16 ± 0.06 | 0.13 ± 0.01 | 0.10 ± 0.03 | 0.12 ± 0.03 |
| Zeta potential (mV) | 34.1 ± 1.4 | 31.0 ± 2.7 | 33.2 ± 0.9 | 32.9 ± 2.3 |
| EE (%) | 60.7 ± 5.4 | 63.2 ± 7.4 | 58.6 ± 5.0 | 71.6 ± 15.6 |
| Drug loading (%) | 0.26 ± 0.01 | 0.24 ± 0.03 | 0.27 ± 0.03 | 0.29 ± 0.05 |
| Yield (%) | 57.0 ± 2.8 | 64.9 ± 2.0 | 54.5 ± 2.9 | 61.6 ± 3.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, H.-L.; Lin, W.J. Dual Peptide-Modified Nanoparticles Improve Combination Chemotherapy of Etoposide and siPIK3CA Against Drug-Resistant Small Cell Lung Carcinoma. Pharmaceutics 2020, 12, 254. https://doi.org/10.3390/pharmaceutics12030254
Huang H-L, Lin WJ. Dual Peptide-Modified Nanoparticles Improve Combination Chemotherapy of Etoposide and siPIK3CA Against Drug-Resistant Small Cell Lung Carcinoma. Pharmaceutics. 2020; 12(3):254. https://doi.org/10.3390/pharmaceutics12030254
Chicago/Turabian StyleHuang, Hsin-Lin, and Wen Jen Lin. 2020. "Dual Peptide-Modified Nanoparticles Improve Combination Chemotherapy of Etoposide and siPIK3CA Against Drug-Resistant Small Cell Lung Carcinoma" Pharmaceutics 12, no. 3: 254. https://doi.org/10.3390/pharmaceutics12030254
APA StyleHuang, H. -L., & Lin, W. J. (2020). Dual Peptide-Modified Nanoparticles Improve Combination Chemotherapy of Etoposide and siPIK3CA Against Drug-Resistant Small Cell Lung Carcinoma. Pharmaceutics, 12(3), 254. https://doi.org/10.3390/pharmaceutics12030254