Application of 1-Hydroxy-4,5-Dimethyl-Imidazole 3-Oxide as Coformer in Formation of Pharmaceutical Cocrystals


 , ,
, , 
 , and
, and
Abstract
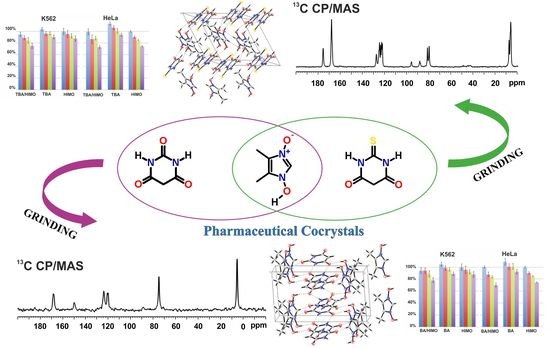
:
1. Introduction
2. Materials and Methods
2.1. Preparation of Thiobarbituric Acid/1-Hydroxy-4,5-Dimethyl-Imidazole 3-Oxide (TBA/HIMO) Cocrystal
Method 1
Method 2
2.2. Preparation of Barbituric Acid/1-Hydroxy-4,5-Dimethyl-Imidazole 3-Oxide (BA/HIMO) Cocrystal
Method 1
Method 2
2.3. NMR Spectroscopy
2.4. Single Crystals X-ray Diffraction Measurements
2.5. X-ray Diffraction of Powders
2.6. Differential Scanning Calorimetry (DSC) Measurements
2.7. Biological Studies of HIMO, TBA/HIMO and BA/HIMO
2.8. Solubility Measurements of BA, TBA, TBA/HIMO and BA/HIMO
3. Results
3.1. Solid State NMR Studies of HIMO
3.2. Formation and Structural Studies of TBA/HIMO and BA/HIMO Cocrystals
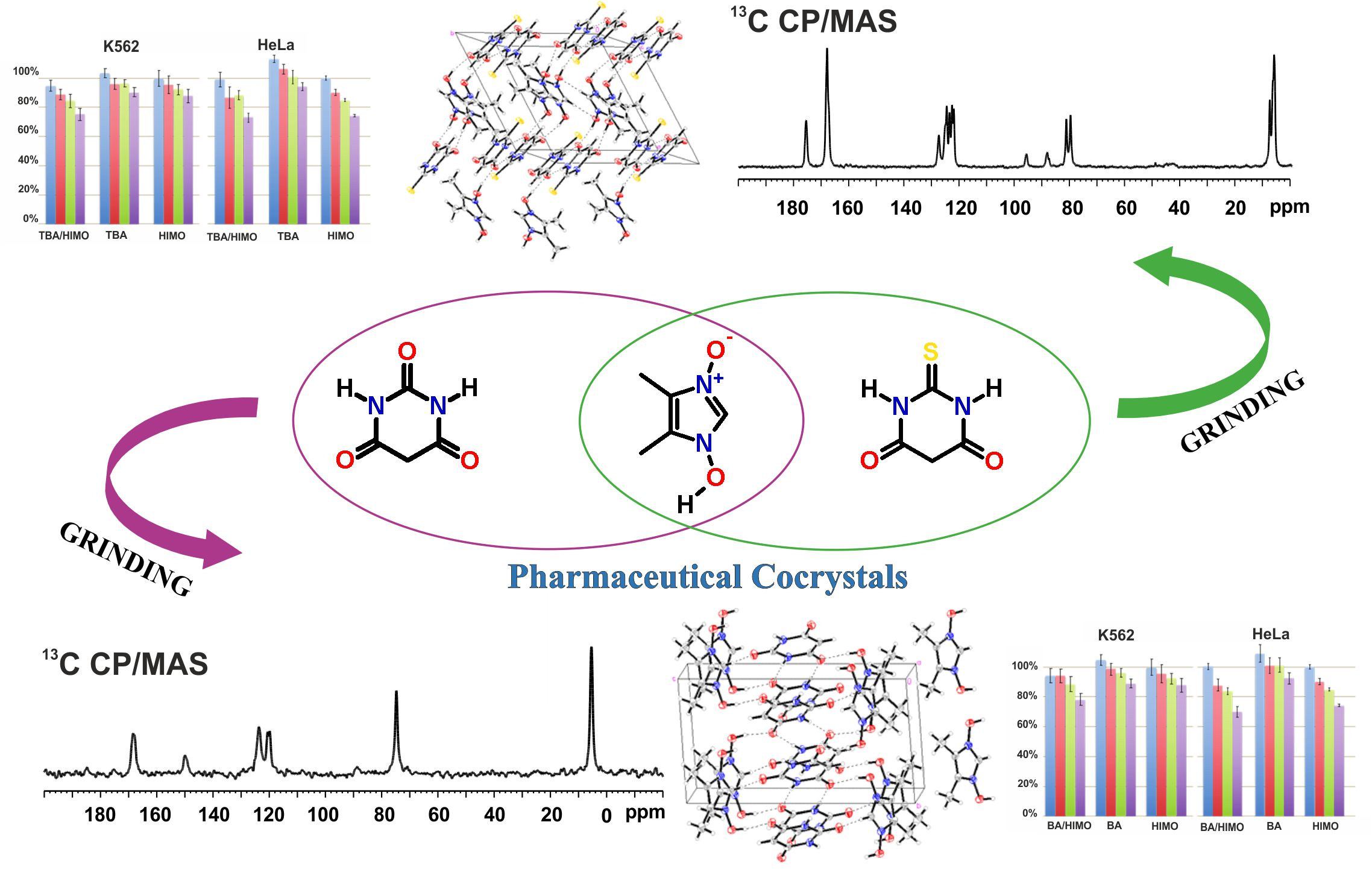
3.1.1. SS NMR Studies of TBA/HIMO Cocrystal
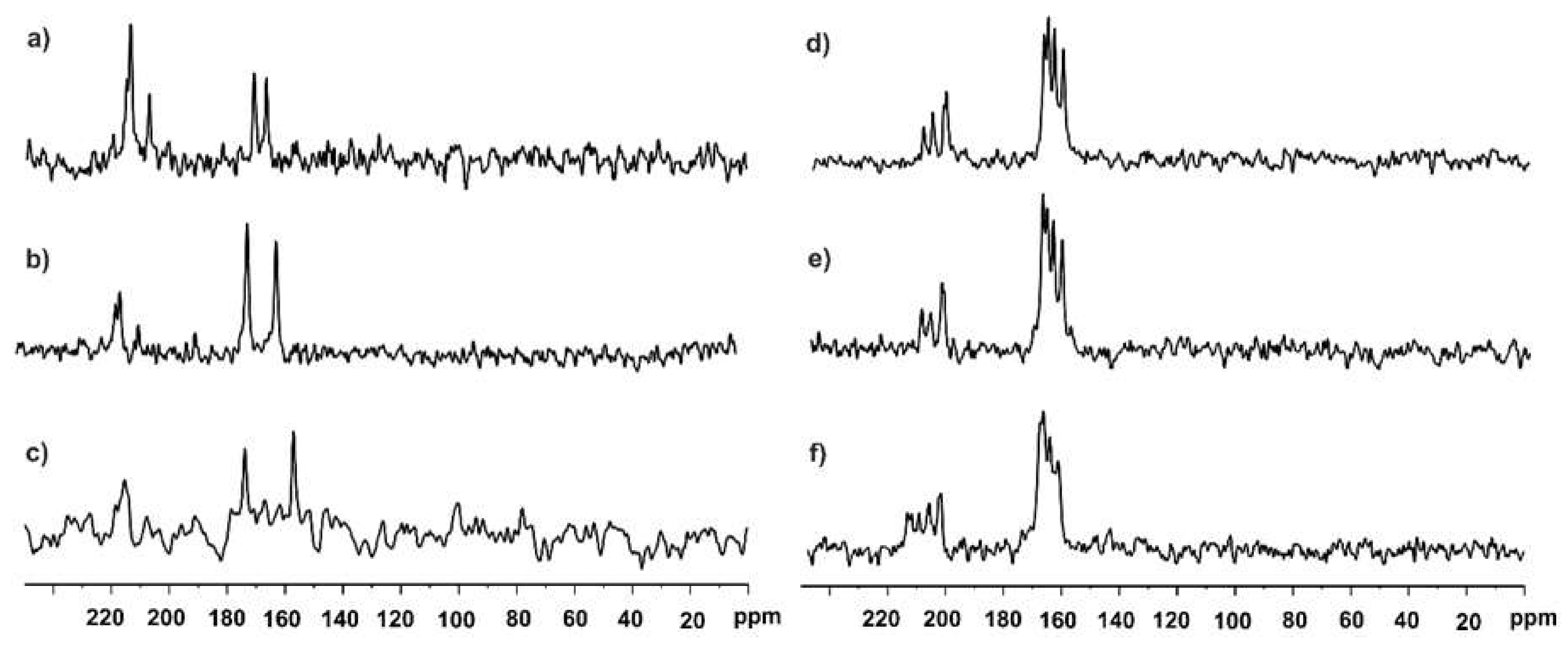
3.1.2. SS NMR Structural Studies of BA/HIMO Cocrystal
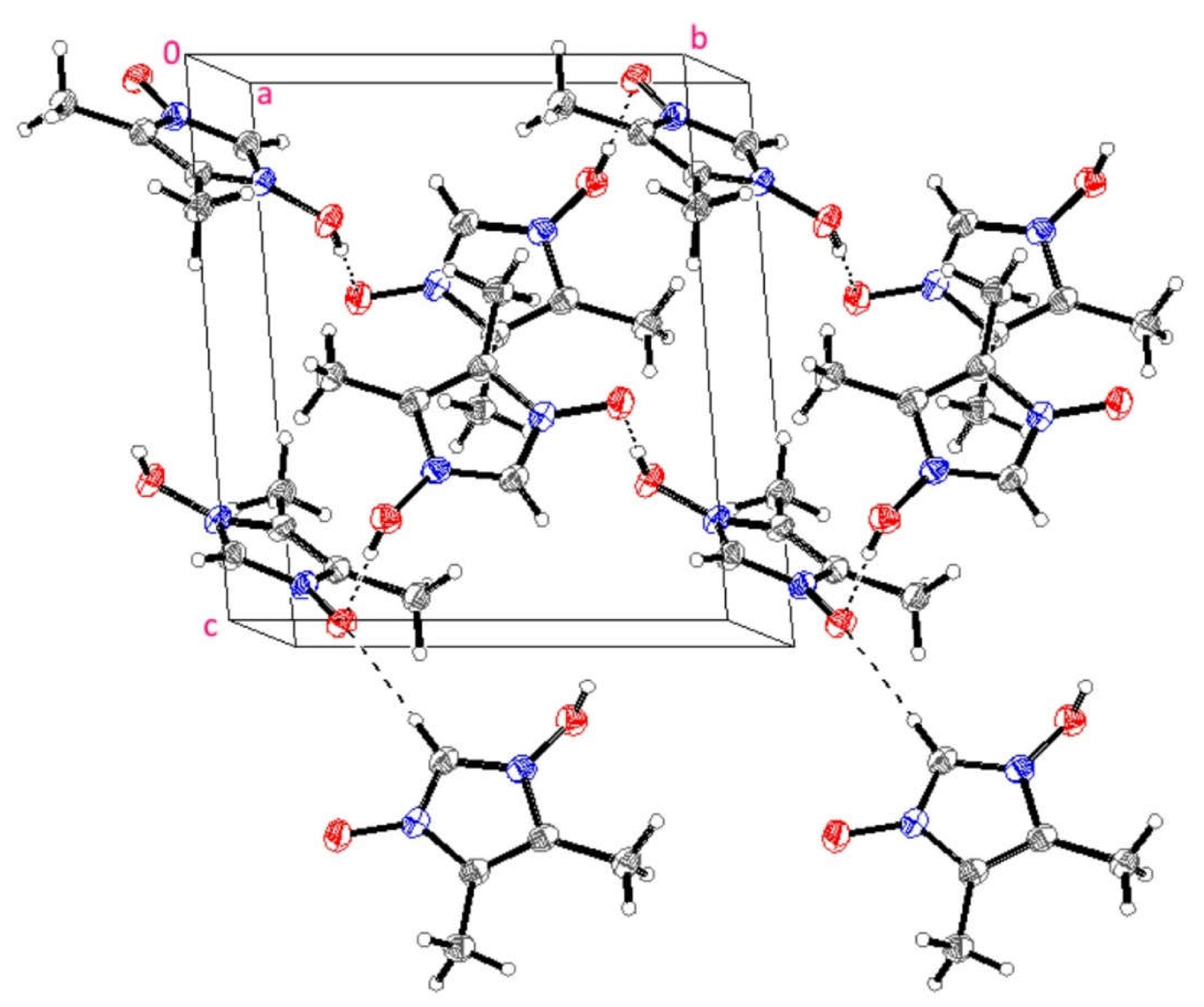
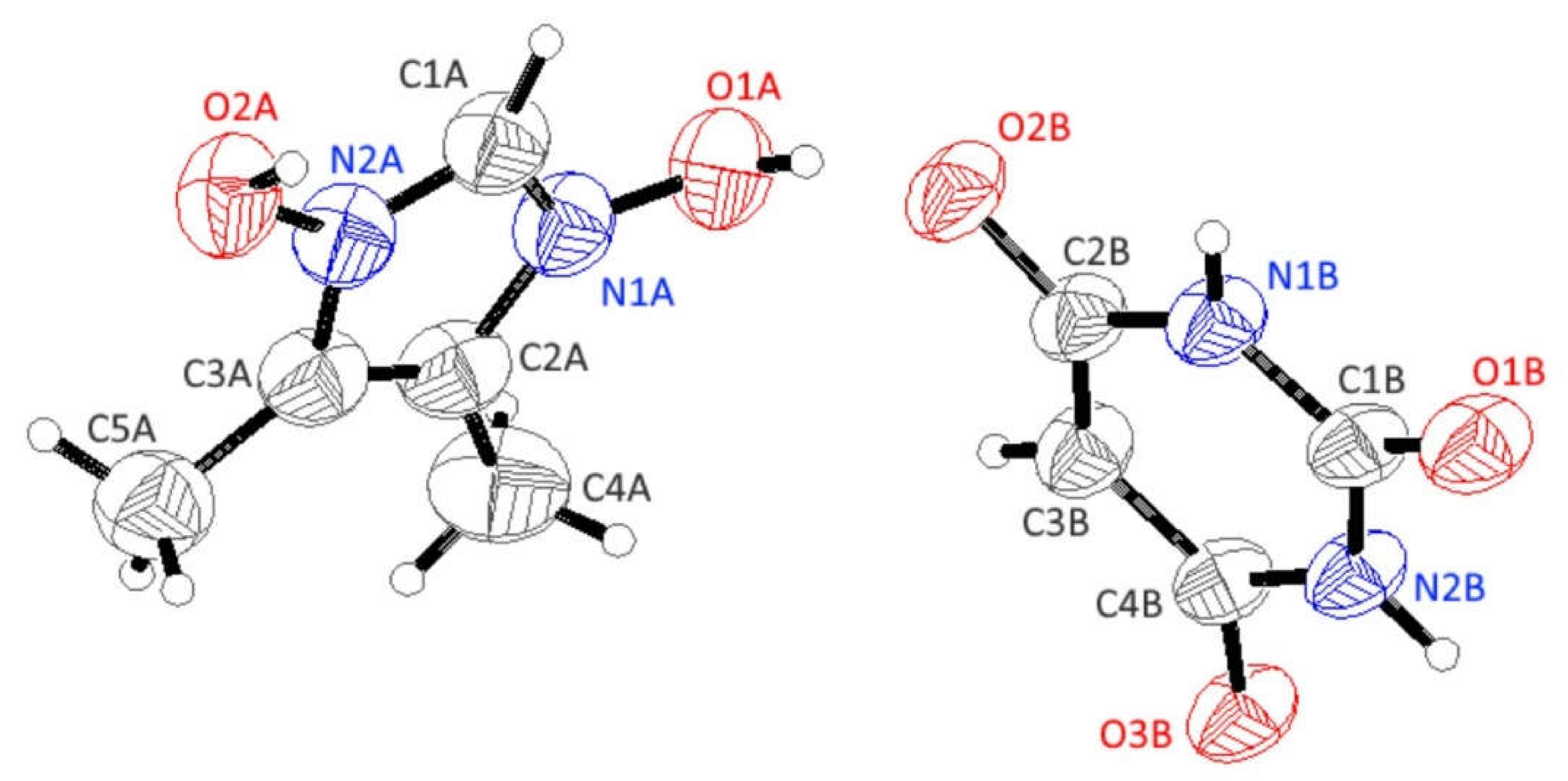
3.3. X-ray Structure of HIMO and TBA/HIMO, BA/HIMO Cocrystals
3.4. Cell Cytotoxicity and Solubility of Tested Compound (HIMO and Its Cocrystals)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Shan, N.; Zaworotko, M.J. The role of cocrystals in pharmaceutical science. Drug Discov. Today 2008, 13, 440–446. [Google Scholar] [CrossRef]
- Kumar, S.; Nanda, A. Approaches to design of pharmaceutical cocrystals: A Review. Mol. Cryst. Liq. Cryst. 2018, 667, 54–77. [Google Scholar] [CrossRef]
- Shaikh, R.; Singh, R.; Walker, G.M.; Croker, D.M. Pharmaceutical cocrystal drug products: An outlook on product development. Trends Pharmacol. Sci. 2018, 39, 1033–1048. [Google Scholar] [CrossRef]
- Kulla, H.; Michalchuk, A.A.L.; Emmerling, F. Manipulating the dynamics of mechanochemical ternary cocrystal formation. Chem. Commun. 2019, 55, 9793–9796. [Google Scholar] [CrossRef]
- Sinha, A.S.; Maguire, A.R.; Lawrence, S.E. Cocrystallization of nutraceuticals. Cryst. Growth Des. 2015, 15, 984–1009. [Google Scholar] [CrossRef]
- Kavanagh, O.N.; Croker, D.M.; Walker, G.M.; Zaworotko, M.J. Pharmaceutical cocrystals: From serendipity to design to application. Drug Discov. Today 2019, 24, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Xie, C.; Lu, H.; Guo, N.; Lou, Y.; Su, W.; Hao, H. Cocrystal and its Application in the Field of Active Pharmaceutical Ingredients and Food Ingredients. Curr. Pharm. Des. 2018, 24, 2339–2348. [Google Scholar] [CrossRef]
- Good, D.J.; Rodríguez-Hornedo, N. Solubility advantage of pharmaceutical cocrystals. Cryst. Growth Des. 2009, 9, 2252–2264. [Google Scholar] [CrossRef]
- Wang, Y.; Ren, F.; Cao, D. A dynamic and electrostatic potential prediction of the prototropic tautomerism between imidazole 3-oxide and 1-hydroxyimidazole in external electric field. J. Mol. Model. 2019, 25, 330. [Google Scholar] [CrossRef]
- Gadade, D.D.; Pekamwar, S.S. Pharmaceutical cocrystals: Regulatory and strategic aspects, design and development. Adv. Pharm. Bull. 2016, 6, 479–494. [Google Scholar] [CrossRef]
- Sathisaran, I.; Dalvi, S.V. Engineering cocrystals of poorly water-soluble drugs to enhance dissolution in aqueous medium. Pharmaceutics 2018, 10, 108. [Google Scholar] [CrossRef] [Green Version]
- Ross, S.A.; Lamprou, D.A.; Douroumis, D. Engineering and manufacturing of pharmaceutical cocrystals: A review of solvent-free manufacturing technologies. Chem. Commun. 2016, 52, 8772–8786. [Google Scholar] [CrossRef] [Green Version]
- Karagianni, A.; Malamatari, M.; Kachrimanis, K. Pharmaceutical cocrystals: New solid phase modification approaches for the formulation of APIs. Pharmaceutics 2018, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.; Desiraju, G.R. From a binary to a quaternary cocrystal: An unusual supramolecular synthon. Angew. Chem. Int. Ed. 2019, 58, 12027–12031. [Google Scholar] [CrossRef] [Green Version]
- Fukte, S.R.; Wagh, M.P.; Rawat, S. Coformer selection: An important tool in cocrystal formation. Int. J. Pharm. Pharm. Sci. 2014, 6, 9–14. [Google Scholar]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.M.; Croker, D.M. Creating cocrystals: A review of pharmaceutical cocrystal preparation routes and applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef]
- Bartz, S.; Blumenröder, B.; Kern, A.; Fleckenstein, J.; Frohnapfel, S.; Schatz, J.; Wagner, A. Hydroxy-1H-imidazole-3-oxides—Synthesis, kinetic acidity, and application in catalysis and supramolecular anion recognition. Z. Naturforsch. 2009, 64b, 629–638. [Google Scholar] [CrossRef]
- Zheng, Z.; Wu, T.; Zheng, R.; Wu, Y.; Zhou, X. Study on the synthesis of quaternary ammonium salts using imidazolium ionic liquid as catalyst. Catal. Commun. 2007, 8, 39–42. [Google Scholar] [CrossRef]
- Zhang, L.; Peng, X.-M.; Damu, G.L.V.; Geng, R.-X.; Zhou, C.-H. Comprehensive Review in current developments of imidazole-based medicinal chemistry. Med. Res. Rev. 2013, 34, 340–437. [Google Scholar] [CrossRef]
- Vekariya, R.L. A review of ionic liquids: Applications towards catalytic organic transformations. J. Mol. Liq. 2017, 227, 44–60. [Google Scholar] [CrossRef]
- Zheng, D.; Dong, L.; Huang, W.; Wu, X.; Nie, N. A review of imidazolium ionic liquids research and development towards working pair of absorption cycle. Renew. Sustain. Energy Rev. 2014, 37, 47–68. [Google Scholar] [CrossRef]
- Pieczonka, A.M.; Strzelczyk, A.; Sadowska, B.; Mlostoń, G.; Stączek, P. Synthesis and evaluation of antimicrobial activity of hydrazones derived from 3-oxido-1H-imidazole-4-carbohydrazides. Eur. J. Med. Chem. 2013, 64, 389–395. [Google Scholar] [CrossRef]
- Ghandi, K.A. A review of ionic liquids, their limits and applications. Green Sustain. Chem. 2014, 4, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Irge, D.D. Ionic liquids: A review on greener chemistry applications, quality ionic liquid synthesis and economical viability in a chemical processes. Am. J. Phys. Chem. 2016, 5, 74–79. [Google Scholar] [CrossRef]
- Welton, T. Ionic liquids in catalysis. Coord. Chem. Rev. 2004, 248, 2459–2477. [Google Scholar] [CrossRef]
- Olivier-Bourbigou, H.; Magna, L.; Morvan, D. Ionic liquids and catalysis: Recent progress from knowledge to applications. Appl. Catal. Gen. 2010, 373, 1–56. [Google Scholar] [CrossRef]
- Zhao, D.; Wu, M.; Kou, Y.; Min, E. Ionic liquids: Applications in catalysis. Catal. Today 2002, 74, 157–189. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef]
- Fèvre, M.; Pinaud, J.; Gnanou, Y.; Vignolle, J.; Taton, D. N-Heterocyclic carbenes (NHCs) as organocatalysts and structural components in metal-free polymer synthesis. Chem. Soc. Rev. 2013, 42, 2142–2172. [Google Scholar] [CrossRef]
- Nikitina, P.A.; Koldaeva, T.Y.; Mityanov, V.S.; Miroshnikov, V.S.; Basanova, E.I.; Perevalov, V.P. Prototropic tautomerism and some features of the IR spectra of 2-(3-Chromenyl)-1-hydroxyimidazoles. Aust. J. Chem. 2019, 72, 699–708. [Google Scholar]
- Visbal, R.; Gimeno, M.C. N-heterocyclic carbene metal complexes: Photoluminescence and applications. Chem. Soc. Rev. 2014, 43, 3551–3574. [Google Scholar] [CrossRef] [PubMed]
- Luca, L.D. Bacterial symbionts: Prospects for the sustainable production of invertebrate-derived pharmaceuticals. Curr. Med. Chem. 2006, 13, 1–50. [Google Scholar] [PubMed]
- Verma, A.; Joshi, S.; Singh, D. Imidazole: Having versatile biological activities. J. Chem. 2013, 2013, 1–12. [Google Scholar] [CrossRef]
- Menon, K.; Mousa, A.; de Courten, B. Effects of supplementation with carnosine and other histidine-containing dipeptides on chronic disease risk factors and outcomes: Protocol for a systematic review of randomized controlled trials. BMJ Open 2018, 8, e020623. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Peters, F.B.; Yang, P.-Y.; Reed, S.; Chittuluru, J.R.; Schultz, P.G. genetic incorporation of histidine derivatives using an engineered pyrrolysyl-tRNA synthetase. ACS Chem. Biol. 2014, 9, 1092–1096. [Google Scholar] [CrossRef]
- Nikitina, G.V.; Pevzner, M.S. Imidazole and benzimidazole N-oxides (a review). Chem. Heterocycl. Compd. 1993, 29, 127–151. [Google Scholar] [CrossRef]
- Mlostoń, G.; Jasiński, M.; Wróblewska, A.; Heimgartner, H. Recent progress in the chemistry of 2-unsubstituted 1H-Imidazole 3-Oxides. Curr. Org. Chem. 2016, 20, 1359–1369. [Google Scholar] [CrossRef] [Green Version]
- Mlostoń, G.; Jasiński, M. First synthesis of the N(1)-bulky substituted imidazole 3-oxides and their complexation with hexafluoroacetone hydrate. ARKIVOC 2011, 6, 162–175. [Google Scholar]
- Chierotti, M.R.; Ferrero, L.; Garino, N.; Gobetto, R.; Pellegrino, L.; Braga, D.; Grepioni, F.; Maini, L. The richest collection of tautomeric polymorphs: The Case of 2-thiobarbituric acid. Chem. Eur. J. 2010, 16, 4347–4358. [Google Scholar] [CrossRef]
- Lewis, T.C.; Tocher, D.A.; Price, S.L. An experimental and theoretical search for polymorphs of barbituric acid: The challenges of even limited conformational flexibility. Cryst. Growth Des. 2004, 4, 979–987. [Google Scholar] [CrossRef]
- Zuccarello, F.; Buemi, G.; Gandolfo, C.; Contino, A. Barbituric and thiobarbituric acids: A conformational and spectroscopic study. Spectrochim. Acta Part A 2003, 59, 139–151. [Google Scholar] [CrossRef]
- Golovnev, N.N.; Molokeev, M.S.; Lesnikov, M.K.; Atuchin, V.V. Two salts and the salt cocrystal of ciprofloxacin with thiobarbituric and barbituric acids: The structure and properties. J. Phys. Org. Chem. 2018, 31, e3773. [Google Scholar] [CrossRef] [Green Version]
- Ziarani, G.M.; Alealia, F.; Lashgari, N. Recent applications of barbituric acid in multicomponent reactions. RSC Adv. 2016, 6, 50895–50922. [Google Scholar] [CrossRef]
- Chierotti, M.R.; Gobetto, R.; Pellergrino, L.; Milone, L.; Venturello, P. Mechanically induced phase change in Barbituric acid. Cryst. Growth Des. 2008, 8, 1454–1457. [Google Scholar] [CrossRef]
- Schmidt, M.U.; Brning, J.; Glinnemann, J.; Htzler, M.W.; Mçrschel, P.; Ivashevskaya, S.N.; van de Streek, J.; Braga, D.; Maini, L.; Chierotti, M.R.; et al. The thermodynamically stable form of solid barbituric acid: The enol tautomer. Angew. Chem. Int. Ed. 2011, 50, 7924–7926. [Google Scholar] [CrossRef]
- Paul, M.E.; da Silva, T.H.; King, M.D. True polymorphic phase transition or dynamic crystal disorder? an investigation into the unusual phase behavior of barbituric acid dihydrate. Cryst. Growth Des. 2019, 19, 4745–4753. [Google Scholar] [CrossRef]
- Braga, D.; Cadoni, M.; Grepioni, F.; Maini, L.; Rubini, K. Gas–solid reactions between the different polymorphic modifications of barbituric acid and amines. CrystEngComm 2006, 8, 756–763. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal | HIMO | TBA/HIMO | BA/HIMO |
|---|---|---|---|
| Empirical formula | C5H8N2O2 | C5H8N2O2+ C4H3N2O2S | C5H8N2O2+ C4H3N2O3 |
| Formula weight | 128.14 | 272.28 | 256.23 |
| Temperature | 293 K | 293 K | 173 K |
| Crystal system | Triclinic | Triclinic | Triclinic |
| Space group | P-1 | P-1 | P-1 |
| a (Å) | 7.5947(4) | 10.7777(2) | 5.4971(4) |
| b (Å) | 8.5233(5) | 11.0568(3) | 7.6116(5) |
| c (Å) | 9.7643(4) | 11.9869(2) | 13.9014(11) |
| α (°) | 85.772(4) | 97.968(2) | 93.952(6) |
| β (°) | 78.587(4) | 97.799(2) | 98.627(7) |
| γ(°) | 83.727(5) | 118.738(3) | 101.157(6) |
| Volume (Å)3 | 614.97(6) | 1206.26(5) | 561.32(8) |
| Z | 4 | 4 | 2 |
| Z’ | 2 | 2 | 1 |
| Density (g·cm−3) | 1.384 | 1.499 | 1.510 |
| Θ Range (°) | 4.63 to 75.46 | 3.79 to 75.25 | 3.23 to 74.96 |
| Index ranges | −9 ≤ h ≤ 9, −10 ≤ k ≤ 10, −12 ≤ l ≤ 12 | −13 ≤ h ≤ 13, −13 ≤ k ≤ 13, −13 ≤ l ≤ 13 | −6 ≤ h ≤ 6, −9 ≤ k ≤ 9, −17≤ l ≤ 17 |
| Nref | 2489 | 4726 | 2268 |
| R (reflections) | 0.0494 (2300) | 0.0535 (4485) | 0.0899 (1825) |
| wR2 (reflections) | 0.1570 (2489) | 0.1745 (4726) | 0.2963 (2268) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wróblewska, A.; Śniechowska, J.; Kaźmierski, S.; Wielgus, E.; Bujacz, G.D.; Mlostoń, G.; Chworos, A.; Suwara, J.; Potrzebowski, M.J. Application of 1-Hydroxy-4,5-Dimethyl-Imidazole 3-Oxide as Coformer in Formation of Pharmaceutical Cocrystals. Pharmaceutics 2020, 12, 359. https://doi.org/10.3390/pharmaceutics12040359
Wróblewska A, Śniechowska J, Kaźmierski S, Wielgus E, Bujacz GD, Mlostoń G, Chworos A, Suwara J, Potrzebowski MJ. Application of 1-Hydroxy-4,5-Dimethyl-Imidazole 3-Oxide as Coformer in Formation of Pharmaceutical Cocrystals. Pharmaceutics. 2020; 12(4):359. https://doi.org/10.3390/pharmaceutics12040359
Chicago/Turabian StyleWróblewska, Aneta, Justyna Śniechowska, Sławomir Kaźmierski, Ewelina Wielgus, Grzegorz D. Bujacz, Grzegorz Mlostoń, Arkadiusz Chworos, Justyna Suwara, and Marek J. Potrzebowski. 2020. "Application of 1-Hydroxy-4,5-Dimethyl-Imidazole 3-Oxide as Coformer in Formation of Pharmaceutical Cocrystals" Pharmaceutics 12, no. 4: 359. https://doi.org/10.3390/pharmaceutics12040359
APA StyleWróblewska, A., Śniechowska, J., Kaźmierski, S., Wielgus, E., Bujacz, G. D., Mlostoń, G., Chworos, A., Suwara, J., & Potrzebowski, M. J. (2020). Application of 1-Hydroxy-4,5-Dimethyl-Imidazole 3-Oxide as Coformer in Formation of Pharmaceutical Cocrystals. Pharmaceutics, 12(4), 359. https://doi.org/10.3390/pharmaceutics12040359