Control of the Lung Residence Time of Highly Permeable Molecules after Nebulization: Example of the Fluoroquinolones


Abstract
:1. Introduction
2. Control of FQ Lung–Blood Barrier Permeability by Pulmonary Administration of FQ–Metal Complex
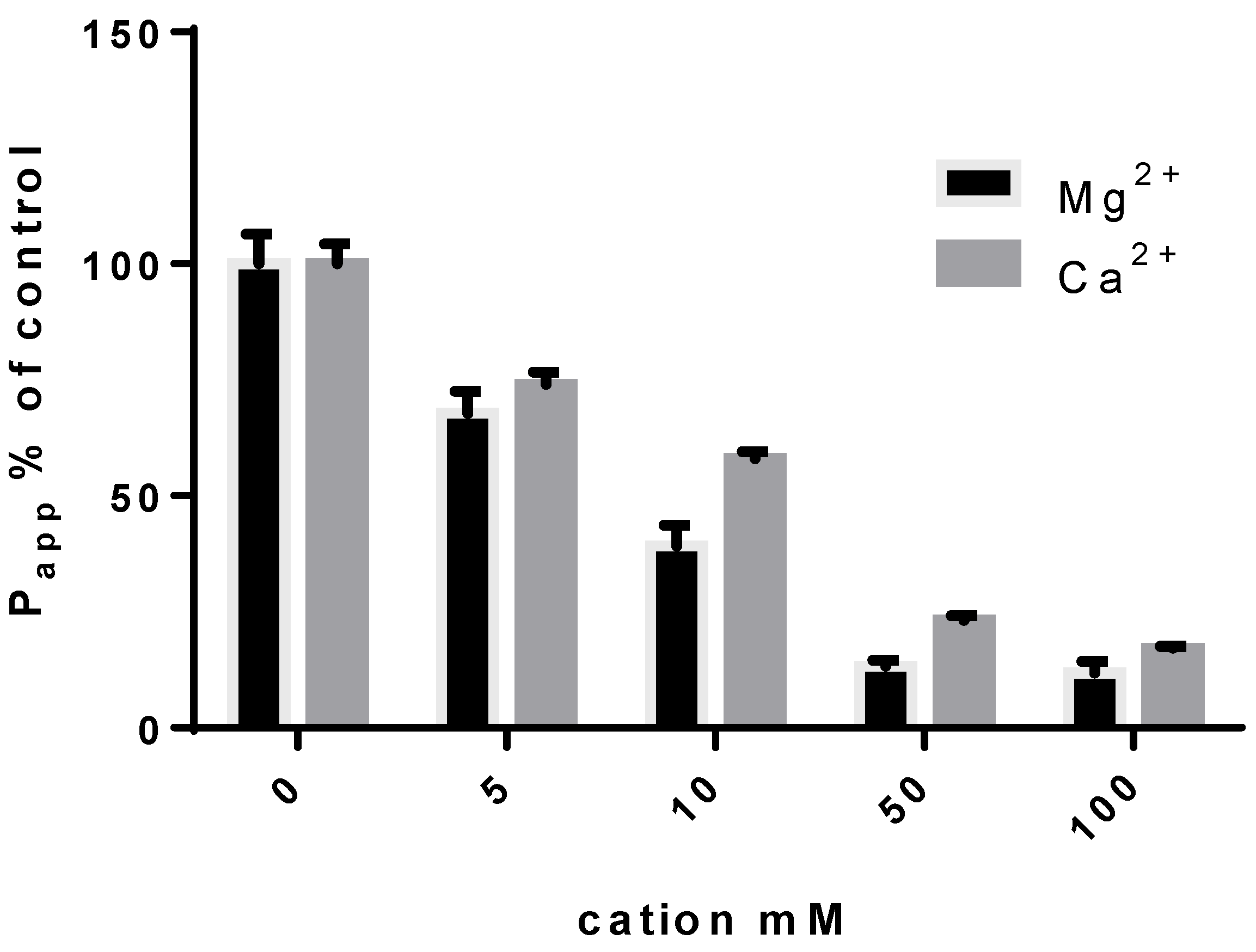
2.1. Metal Cation–FQ Complexes to Control FQs’ Permeability across the Lung–Blood Barrier
2.2. Quinsair®/Aeroquin™: A Solution for Pulmonary Inhalation of the Fluoroquinolone–Metal Complex Marketed
2.2.1. Pharmaceutical Properties—Development
2.2.2. Pharmacokinetics—Clinical Studies
2.2.3. Efficacy
2.2.4. Safety
2.3. Dry Powder of Fluoroquinolone–Metal Complexes for Better Patient Compliance
2.3.1. Pharmaceutical Properties—Development
2.3.2. Pharmacokinetics
2.3.3. Efficacy
2.3.4. Safety
3. Control of the Appearance Rate of FQs in Pulmonary ELF: Controlled Dissolution and Controlled Release
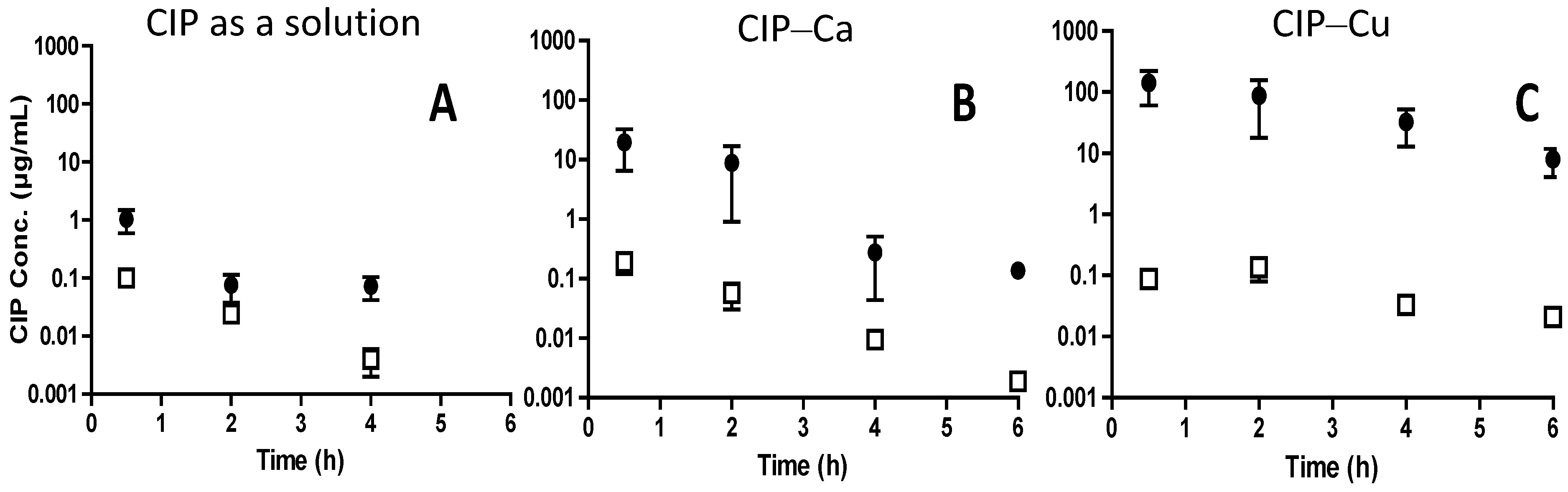
3.1. Control of the Apparent Solubility of CIP in a DPI Formulation to Decrease its Dissolution Rate
3.1.1. Pharmaceutical Properties—Development
3.1.2. Pharmacokinetics—Clinical Studies
3.1.3. Efficacy
3.1.4. Safety
3.2. Control of the FQ Release Rate Using Particulate Systems
3.2.1. Control of the Release Rate Using Polymer Microparticles
3.2.2. Control of the Release Rate of CIP Using Liposomes
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AUC | area under the concentration-time curve |
| BAL | broncho-alveolar lavage |
| BID | twice daily |
| CF | cystic fibrosis |
| CFU | colony forming unity |
| CIP | ciprofloxacin |
| Cmax | maximum concentration |
| COPD | chronic obstructive pulmonary disease |
| DPI | dry powder inhaler |
| ELF | epithelial lining fluid |
| EMA | European medicine agency |
| ESE | emulsification-solvent-evaporation |
| FDA | food and drug administration |
| FQ | fluoroquinolone |
| IT | intratracheal |
| IV | intravenous |
| Ka | association constant |
| Ka(lung) | absorption rate constant from the lung |
| Kel(plasma) | elimination rate constant from the plasma |
| LDH | lactate dehydrogenase |
| LVX | levofloxacin |
| MIC | minimum inhibitory concentration |
| NCFB | non-CF bronchiectasis |
| PA | Pseudomonas aeruginosa |
| Papp | apparent permeability |
| PK | pharmacokinetics |
| PLGA | poly(lactic-co-glycolic acid) |
| QD | once daily |
| TIS | tobramycin solution for inhalation |
References
- Patton, J.S.; Brain, J.D.; Davies, L.A.; Fiegel, J.; Gumbleton, M.; Kim, K.-J.; Sakagami, M.; Vanbever, R.; Ehrhardt, C. The Particle has Landed—Characterizing the Fate of Inhaled Pharmaceuticals. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, S-71–S-87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.T.; Leung, S.S.Y.; Tang, P.; Parumasivam, T.; Loh, Z.H.; Chan, H.-K. Inhaled formulations and pulmonary drug delivery systems for respiratory infections. Adv. Drug Deliv. Rev. 2015, 85, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E.; Mercuri, A.; Wu, S.; Salar-Behzadi, S. Measurements of Deposition, Lung Surface Area and Lung Fluid for Simulation of Inhaled Compounds. Front. Pharm. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.-K.; Nichols, B.L.B.; Edgar, K.J.; Murgia, X.; Loretz, B.; Lehr, C.-M. Challenges and strategies in drug delivery systems for treatment of pulmonary infections. Eur. J. Pharm. Biopharm. 2019, 144, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Hastedt, J.E.; Bäckman, P.; Clark, A.R.; Doub, W.; Hickey, A.; Hochhaus, G.; Kuehl, P.J.; Lehr, C.-M.; Mauser, P.; McConville, J.; et al. Scope and relevance of a pulmonary biopharmaceutical classification system AAPS/FDA/USP Workshop March 16–17th, 2015 in Baltimore, MD. AAPS Open 2016, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.; Gupta, N.; Ahsan, F. Particle engineering to enhance or lessen particle uptake by alveolar macrophages and to influence the therapeutic outcome. Eur. J. Pharm. Biopharm. 2015, 89, 163–174. [Google Scholar] [CrossRef]
- Gontijo, A.V.L.; Grégoire, N.; Lamarche, I.; Gobin, P.; Couet, W.; Marchand, S. Biopharmaceutical Characterization of Nebulized Antimicrobial Agents in Rats: 2. Colistin. Antimicrob. Agents Chemother. 2014, 58, 3950–3956. [Google Scholar] [CrossRef] [Green Version]
- Marchand, S.; Gobin, P.; Brillault, J.; Baptista, S.; Adier, C.; Olivier, J.-C.; Mimoz, O.; Couet, W. Aerosol Therapy with Colistin Methanesulfonate: A Biopharmaceutical Issue Illustrated in Rats. Antimicrob. Agents Chemother. 2010, 54, 3702–3707. [Google Scholar] [CrossRef] [Green Version]
- Marchand, S.; Grégoire, N.; Brillault, J.; Lamarche, I.; Gobin, P.; Couet, W. Biopharmaceutical Characterization of Nebulized Antimicrobial Agents in Rats: 3. Tobramycin. Antimicrob. Agents Chemother. 2015, 59, 6646–6647. [Google Scholar] [CrossRef] [Green Version]
- Marchand, S.; Grégoire, N.; Brillault, J.; Lamarche, I.; Gobin, P.; Couet, W. Biopharmaceutical Characterization of Nebulized Antimicrobial Agents in Rats. 4. Aztreonam. Antimicrob. Agents Chemother. 2016, 60, 3196–3198. [Google Scholar] [CrossRef] [Green Version]
- Gontijo, A.V.L.; Brillault, J.; Grégoire, N.; Lamarche, I.; Gobin, P.; Couet, W.; Marchand, S. Biopharmaceutical Characterization of Nebulized Antimicrobial Agents in Rats: 1. Ciprofloxacin, Moxifloxacin, and Grepafloxacin. Antimicrob. Agents Chemother. 2014, 58, 3942–3949. [Google Scholar] [CrossRef] [Green Version]
- Stass, H.; Weimann, B.; Nagelschmitz, J.; Rolinck-Werninghaus, C.; Staab, D. Tolerability and pharmacokinetic properties of ciprofloxacin dry powder for inhalation in patients with cystic fibrosis: A Phase I, randomized, dose-escalation study. Clin. Ther. 2013, 35, 1571–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stass, H.; Nagelschmitz, J.; Willmann, S.; Delesen, H.; Gupta, A.; Baumann, S. Inhalation of a dry powder ciprofloxacin formulation in healthy subjects: A phase I study. Clin. Drug Investig. 2013, 33, 419–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endermann, R.; Labischinski, H.; Ladel, C.; Petersen, U.; Newton, B. Treatment of Bacterial Diseases of the Respiratory Organs. U.S. Patent 8034817B2, 2011. [Google Scholar]
- Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. Available online: https://www.ncbi.nlm.nih.gov/books/NBK548196/ (accessed on 20 January 2020).
- Gaspar, M.C.; Grégoire, N.; Sousa, J.J.; Pais, A.A.; Lamarche, I.; Gobin, P.; Olivier, J.-C.; Marchand, S.; Couet, W. Pulmonary pharmacokinetics of levofloxacin in rats after aerosolization of immediate-release chitosan or sustained-release PLGA microspheres. Eur. J. Pharm. Sci. 2016, 93, 184–191. [Google Scholar] [CrossRef]
- Serisier, D.J.; Bilton, D.; De Soyza, A.; Thompson, P.J.; Kolbe, J.; Greville, H.W.; Cipolla, D.; Bruinenberg, P.; Gonda, I. ORBIT-2 investigators Inhaled, dual release liposomal ciprofloxacin in non-cystic fibrosis bronchiectasis (ORBIT-2): A randomised, double-blind, placebo-controlled trial. Thorax 2013, 68, 812–817. [Google Scholar] [CrossRef] [Green Version]
- Nurbaeti, S.N.; Brillault, J.; Tewes, F.; Olivier, J.-C. Sustained-release microparticle dry powders of chloramphenicol palmitate or thiamphenicol palmitate prodrugs for lung delivery as aerosols. Eur. J. Pharm. Sci. 2019, 105028. [Google Scholar] [CrossRef]
- Cipolla, D.; Blanchard, J.; Gonda, I. Development of liposomal ciprofloxacin to treat lung infections. Pharmaceutics 2016, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- McShane, P.J.; Weers, J.G.; Tarara, T.E.; Haynes, A.; Durbha, P.; Miller, D.P.; Mundry, T.; Operschall, E.; Elborn, J.S. Ciprofloxacin Dry Powder for Inhalation (ciprofloxacin DPI): Technical design and features of an efficient drug–device combination. Pulm. Pharmacol. Ther. 2018, 50, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Türeli, N.G.; Torge, A.; Juntke, J.; Schwarz, B.C.; Schneider-Daum, N.; Türeli, A.E.; Lehr, C.-M.; Schneider, M. Ciprofloxacin-loaded PLGA nanoparticles against cystic fibrosis P. aeruginosa lung infections. Eur. J. Pharm. Biopharm. 2017, 117, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Lamy, B.; Tewes, F.; Serrano, D.R.; Lamarche, I.; Gobin, P.; Couet, W.; Healy, A.M.; Marchand, S. New aerosol formulation to control ciprofloxacin pulmonary concentration. J. Control. Release 2018, 271, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Tewes, F.; Brillault, J.; Lamy, B.; O’Connell, P.; Olivier, J.-C.; Couet, W.; Healy, A.M. Ciprofloxacin-Loaded Inorganic–Organic Composite Microparticles To Treat Bacterial Lung Infection. Mol. Pharm. 2015, 13, 100–112. [Google Scholar] [CrossRef]
- Lamy, B.; Remedios Serrano, D.; O’Connell, P.; Couet, W.; Marchand, S.; Healy, A.M.; Tewes, F. Use of leucine to improve aerodynamic properties of ciprofloxacin-loaded maltose microparticles for inhalation. Eur. J. Pharm. Res. 2019, 1, 2–11. [Google Scholar] [CrossRef]
- Griffith, D.C.; Dudley, M.N.; Surber, M.W.; Bostian, K.A.; Rodny, O. Aerosol Fluoroquinolone Formulations for Improved Pharmacokinetics. U.S. Patent 8815838B2, 2014. [Google Scholar]
- Uivarosi, V. Metal complexes of quinolone antibiotics and their applications: An update. Molecules 2013, 18, 11153–11197. [Google Scholar] [CrossRef] [PubMed]
- Frost, R.W.; Lasseter, K.C.; Noe, A.J.; Shamblen, E.C.; Lettieri, J.T. Effects of aluminum hydroxide and calcium carbonate antacids on the bioavailability of ciprofloxacin. Antimicrob. Agents Chemother. 1992, 36, 830–832. [Google Scholar] [CrossRef] [Green Version]
- Kara, M.; Hasinoff, B.; McKay, D.; Campbell, N. Clinical and chemical interactions between iron preparations and ciprofloxacin. Br. J. Clin. Pharmacol. 1991, 31, 257–261. [Google Scholar] [CrossRef]
- Wallis, S.C.; Charles, B.G.; Gahan, L.R.; Filippich, L.J.; Bredhauer, M.G.; Duckworth, P.A. Interaction of norfloxacin with divalent and trivalent pharmaceutical cations. In vitro complexation and in vivo pharmacokinetic studies in the dog. J. Pharm. Sci. 1996, 85, 803–809. [Google Scholar] [CrossRef]
- Simon, Ž.; Katja, B.; Darko, U.; Marjan, V.; Albin, K. Metal cation–fluoroquinolone complexes do not permeate through the intestinal absorption barrier. J. Pharm. Biomed. Anal. 2010, 53, 655–659. [Google Scholar] [CrossRef]
- Brillault, J.; Tewes, F.; Couet, W.; Olivier, J.C. In vitro biopharmaceutical evaluation of ciprofloxacin/metal cation complexes for pulmonary administration. Eur. J. Pharm. Sci. 2017, 97, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Surber, M.W.; Bostian, K.A.; Dudley, M.N.; Lomovskaya, O.; Griffith, D.C. Aerosolized Fluoroquinolones and Uses Thereof. U.S. Patent 8524735B2, 2013. [Google Scholar]
- Seedher, N.; Agarwal, P. Effect of metal ions on some pharmacologically relevant interactions involving fluoroquinolone antibiotics. Drug Metab. Drug Interact. 2010, 25, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.H.M.; Chiu, F.C.K.; Li, R.C. Mechanistic Investigation of the Reduction in Antimicrobial Activity of Ciprofloxacin by Metal Cations. Pharm. Res. 1997, 14, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, D.; Cuthbertson, L.; Doherty, C.; Campana, S.; Ravenni, N.; Taccetti, G.; Govan, J.R.W. Early Pseudomonas aeruginosa infection in individuals with cystic fibrosis: Is susceptibility testing justified? J. Antimicrob. Chemother. 2010, 65, 2373–2375. [Google Scholar] [CrossRef] [PubMed]
- Sibum, I.; Hagedoorn, P.; de Boer, A.H.; Frijlink, H.W.; Grasmeijer, F. Challenges for pulmonary delivery of high powder doses. Int. J. Pharm. 2018, 548, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Stuart Elborn, J.; Geller, D.E.; Conrad, D.; Aaron, S.D.; Smyth, A.R.; Fischer, R.; Kerem, E.; Bell, S.C.; Loutit, J.S.; Dudley, M.N.; et al. A phase 3, open-label, randomized trial to evaluate the safety and efficacy of levofloxacin inhalation solution (APT-1026) versus tobramycin inhalation solution in stable cystic fibrosis patients. J. Cyst. Fibros. 2015, 14, 507–514. [Google Scholar] [CrossRef]
- Geller, D.E.; Flume, P.A.; Griffith, D.C.; Morgan, E.; White, D.; Loutit, J.S.; Dudley, M.N. Pharmacokinetics and Safety of MP-376 (Levofloxacin Inhalation Solution) in Cystic Fibrosis Subjects. Antimicrob. Agents Chemother. 2011, 55, 2636–2640. [Google Scholar] [CrossRef] [Green Version]
- Loutit, J.S.; Morgan, E.E.; Dudley, M.N.; Griffith, D.C.; Lomovskaya, O. Use of aerosolized levofloxacin for treating cystic fibrosis. U.S. Patent 9700564B2, 2017. [Google Scholar]
- Geller, D.E.; Flume, P.A.; Staab, D.; Fischer, R.; Loutit, J.S.; Conrad, D.J. Levofloxacin Inhalation Solution (MP-376) in Patients with Cystic Fibrosis with Pseudomonas aeruginosa. Am. J. Respir. Crit. Care Med. 2011, 183, 1510–1516. [Google Scholar] [CrossRef]
- Flume, P.A.; VanDevanter, D.R.; Morgan, E.E.; Dudley, M.N.; Loutit, J.S.; Bell, S.C.; Kerem, E.; Fischer, R.; Smyth, A.R.; Aaron, S.D.; et al. A phase 3, multi-center, multinational, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of levofloxacin inhalation solution (APT-1026) in stable cystic fibrosis patients. J. Cyst. Fibros. 2016, 15, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Stockmann, C.; Sherwin, C.M.T.; Ampofo, K.; Spigarelli, M.G. Development of levofloxacin inhalation solution to treat Pseudomonas aeruginosa in patients with cystic fibrosis. Adv. Respir. Dis. 2014, 8, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Dudley, M.; Griffith, D.; Rodny, O. Methods of Treating a Pulmonary Bacterial Infection Using Fluoroquinolones. U.S. Patent 20120035166A1, 2012. [Google Scholar]
- EMA. European Medicines Agency: EMA/CHMP/676680/2014—Assessment Report Quinsair; European Medicines Agency: Amsterdam, The Netherlands, 2014.
- Sabet, M.; Miller, C.E.; Nolan, T.G.; Senekeo-Effenberger, K.; Dudley, M.N.; Griffith, D.C. Efficacy of Aerosol MP-376, a Levofloxacin Inhalation Solution, in Models of Mouse Lung Infection Due to Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2009, 53, 3923–3928. [Google Scholar] [CrossRef] [Green Version]
- King, P.; Lomovskaya, O.; Griffith, D.C.; Burns, J.L.; Dudley, M.N. In Vitro Pharmacodynamics of Levofloxacin and Other Aerosolized Antibiotics under Multiple Conditions Relevant to Chronic Pulmonary Infection in Cystic Fibrosis. Antimicrob. Agents Chemother. 2010, 54, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, B.G.S.; Awad, R.; Marchand, S.; Couet, W.; Tewes, F. In vitro evaluation of Pseudomonas aeruginosa chronic lung infection models: Are agar and calcium-alginate beads interchangeable? Eur. J. Pharm. Biopharm. 2019, 143, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Conrad, D.; Flume, P.; Sindel, L.; Andrews, S.; Morgan, L.; Loutit, J.; Geller, D.E. Phase 2b Study Of Inhaled MP-376 (Aeroquin, Levofloxacin Inhalation Solution) In Stable Cystic Fibrosis (CF) Patients With Chronic Pseudomonas Aeruginosa (PA) Lung Infection. In A102. Advances in Cystic Fibrosis; American Thoracic Society: New York, NY, USA, 2010; p. A2339. [Google Scholar]
- NHS England. Clinical Commissioning Policy: Levofloxacin Nebuliser Solution for Chronic Pseudomonas Lung Infection in Cystic Fibrosis (Adults); NHS England: London, UK, 2018. [Google Scholar]
- Elborn, J.S.; Vataire, A.-L.; Fukushima, A.; Aballea, S.; Khemiri, A.; Moore, C.; Medic, G.; Hemels, M.E. Comparison of inhaled antibiotics for the treatment of chronic Pseudomonas aeruginosa lung infection in patients with cystic fibrosis: Systematic literature review and network meta-analysis. Clin. Ther. 2016, 38, 2204–2226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weers, J. Inhaled antimicrobial therapy—Barriers to effective treatment. Adv. Drug Deliv. Rev. 2015, 85, 24–43. [Google Scholar] [CrossRef]
- Tiddens, H.A.W.M.; Bos, A.C.; Mouton, J.W.; Devadason, S.; Janssens, H.M. Inhaled antibiotics: Dry or wet? Eur. Respir. J. 2014, 44, 1308–1318. [Google Scholar] [CrossRef] [Green Version]
- Tewes, F.; Gobbo, O.L.; Ehrhardt, C.; Healy, A.M. Amorphous Calcium Carbonate Based-Microparticles for Peptide Pulmonary Delivery. ACS Appl. Mater. Interfaces 2016, 8, 1164–1175. [Google Scholar] [CrossRef]
- Tewes, F.; Brillault, J.; Smyth, H. Inhalable Microparticles Loaded with A Fluoroquinolone/Metal Cation Complex for the Treatment of Respiratory Diseases. Patent WO2018104759A1, 2018. [Google Scholar]
- Lamy, B. Development and Biopharmaceutical Evaluation of Fluoroquinolone-loaded Microparticles for Inhalation. Ph.D. Thesis, University of Poitiers, Poitiers, France, 2018. [Google Scholar]
- Kukut Hatipoglu, M.; Hickey, A.J.; Garcia-Contreras, L. Pharmacokinetics and pharmacodynamics of high doses of inhaled dry powder drugs. Int. J. Pharm. 2018, 549, 306–316. [Google Scholar] [CrossRef]
- Bjarnsholt, T.; Jensen, P.Ø.; Fiandaca, M.J.; Pedersen, J.; Hansen, C.R.; Andersen, C.B.; Pressler, T.; Givskov, M.; Høiby, N. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatric Pulmonol. 2009, 44, 547–558. [Google Scholar] [CrossRef]
- Müller, L.; Murgia, X.; Siebenbürger, L.; Börger, C.; Schwarzkopf, K.; Sewald, K.; Häussler, S.; Braun, A.; Lehr, C.-M.; Hittinger, M. Human airway mucus alters susceptibility of Pseudomonas aeruginosa biofilms to tobramycin, but not colistin. J. Antimicrob. Chemother. 2018, 73, 2762–2769. [Google Scholar] [CrossRef]
- Bahamondez-Canas, T.F.; Zhang, H.; Tewes, F.; Leal, J.; Smyth, H.D. PEGylation of tobramycin improves mucus penetration and antimicrobial activity against Pseudomonas aeruginosa biofilms in vitro. Mol. Pharm. 2018, 15, 1643–1652. [Google Scholar] [CrossRef]
- Cao, B.; Christophersen, L.; Kolpen, M.; Jensen, P.Ø.; Sneppen, K.; Høiby, N.; Moser, C.; Sams, T. Diffusion retardation by binding of tobramycin in an alginate biofilm model. PLoS ONE 2016, 11, e0153616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tewes, F.; Bahamondez-Canas, T.F.; Smyth, H.D.C. Efficacy of Ciprofloxacin and Its Copper Complex against Pseudomonas aeruginosa Biofilms. Aaps Pharmscitech. 2019. [Google Scholar] [CrossRef] [PubMed]
- Bahamondez-Canas, T. Drug Delivery Strategies to Treat Pseudomonas aeruginosa Biofilm Infections. Ph.D. Thesis, University of Texas, Austin, TX, USA, 2018. [Google Scholar]
- Kukavica-Ibrulj, I.; Levesque, R.C. Animal models of chronic lung infection with Pseudomonas aeruginosa: Useful tools for cystic fibrosis studies. Lab. Anim. 2008, 42, 389–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bragonzi, A.; Worlitzsch, D.; Pier, G.B.; Timpert, P.; Ulrich, M.; Hentzer, M.; Andersen, J.B.; Givskov, M.; Conese, M.; Döring, G. Nonmucoid Pseudomonas aeruginosa expresses alginate in the lungs of patients with cystic fibrosis and in a mouse model. J. Infect. Dis. 2005, 192, 410–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Growcott, E.J.; Coulthard, A.; Amison, R.; Hardaker, E.L.; Saxena, V.; Malt, L.; Jones, P.; Grevot, A.; Poll, C.; Osborne, C. Characterisation of a refined rat model of respiratory infection with Pseudomonas aeruginosa and the effect of ciprofloxacin. J. Cyst. Fibros. 2011, 10, 166–174. [Google Scholar] [CrossRef] [Green Version]
- Grillon, A.; Schramm, F.; Kleinberg, M.; Jehl, F. Comparative Activity of Ciprofloxacin, Levofloxacin and Moxifloxacin against Klebsiella pneumoniae, Pseudomonas aeruginosa and Stenotrophomonas maltophilia Assessed by Minimum Inhibitory Concentrations and Time-Kill Studies. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Janssen, R.; Wouters, E.F.M.; Janssens, W.; Daamen, W.; Hagedoorn, P.; De Wit, H.A.J.M.; Serré, J.; Gayan-Ramirez, G.; Franssen, F.; Reynaert, N.L.; et al. Copper-heparin inhalation therapy to repair emphysema: A scientific rationale. Int. J. Chronic Obstr. Pulm. Dis. 2019, 14, 2587–2602. [Google Scholar] [CrossRef] [Green Version]
- Mutti, A.; Corradi, M.; Goldoni, M.; Vettori, M.V.; Bernard, A.; Apostoli, P. Exhaled metallic elements and serum pneumoproteins in asymptomatic smokers and patients with COPD or asthma. Chest 2006, 129, 1288–1297. [Google Scholar] [CrossRef] [Green Version]
- Wehbe, M.; Leung, A.W.; Abrams, M.J.; Orvig, C.; Bally, M.B. A Perspective–can copper complexes be developed as a novel class of therapeutics? Dalton Trans. 2017, 46, 10758–10773. [Google Scholar] [CrossRef]
- Ovet, H.; Oztay, F. The copper chelator tetrathiomolybdate regressed bleomycin-induced pulmonary fibrosis in mice, by reducing lysyl oxidase expressions. Biol. Trace Elem. Res. 2014, 162, 189–199. [Google Scholar] [CrossRef]
- Stockmann, C.; Roberts, J.K.; Yellepeddi, V.K.; Sherwin, C.M.T. Clinical Pharmacokinetics of Inhaled Antimicrobials. Clin. Pharm. 2015, 54, 473–492. [Google Scholar] [CrossRef] [PubMed]
- Velaga, S.P.; Djuris, J.; Cvijic, S.; Rozou, S.; Russo, P.; Colombo, G.; Rossi, A. Dry powder inhalers: An overview of the in vitro dissolution methodologies and their correlation with the biopharmaceutical aspects of the drug products. Eur. J. Pharm. Sci. 2018, 113, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Quon, B.S.; Goss, C.H.; Ramsey, B.W. Inhaled Antibiotics for Lower Airway Infections. Ann. Am. Thorac. Soc. 2014, 11, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Weers, J.G.; Tarara, T. Pulmonary Delivery of a Fluoroquinolone. Patent CA2724009A1, 2009. [Google Scholar]
- Comer, J.E.A. 5.16—Ionization Constants and Ionization Profiles. In Comprehensive Medicinal Chemistry II; Taylor, J.B., Triggle, D.J., Eds.; Elsevier: Oxford, UK, 2007; pp. 357–397. ISBN 978-0-08-045044-5. [Google Scholar]
- Yu, X.; Zipp, G.L.; Davidson III, G.W.R. The Effect of Temperature and pH on the Solubility of Quinolone Compounds: Estimation of Heat of Fusion. Pharm. Res. 1994, 11, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Blokhina, S.V.; Sharapova, A.V.; Ol’khovich, M.V.; Volkova, T.V.; Perlovich, G.L. Solubility, lipophilicity and membrane permeability of some fluoroquinolone antimicrobials. Eur. J. Pharm. Sci. 2016, 93, 29–37. [Google Scholar] [CrossRef]
- Roca Jalil, M.E.; Baschini, M.; Sapag, K. Influence of pH and antibiotic solubility on the removal of ciprofloxacin from aqueous media using montmorillonite. Appl. Clay Sci. 2015, 114, 69–76. [Google Scholar] [CrossRef]
- Tarara, T.E.; Weers, J.G. Pharmaceutical Formulation with an Insoluble Active Agent for Pulmonary Administration. Patent EP1589947B1, 2016. [Google Scholar]
- Weers, J.; Tarara, T. The PulmoSphereTM platform for pulmonary drug delivery. Ther. Deliv. 2014, 5, 277–295. [Google Scholar] [CrossRef]
- Geller, D.E.; Weers, J.; Heuerding, S. Development of an Inhaled Dry-Powder Formulation of Tobramycin Using PulmoSphereTM Technology. J. Aerosol Med. Pulm. Drug Deliv. 2011, 24, 175–182. [Google Scholar] [CrossRef]
- Buttini, F.; Balducci, A.G.; Colombo, G.; Sonvico, F.; Montanari, S.; Pisi, G.; Rossi, A.; Colombo, P.; Bettini, R. Dose administration maneuvers and patient care in tobramycin dry powder inhalation therapy. Int. J. Pharm. 2018, 548, 182–191. [Google Scholar] [CrossRef]
- Stass, H.; Nagelschmitz, J.; Watz, H.; Kirsten, A.M. Safety and pharmacokinetics of two dose strengths of ciprofloxacin dry powder for inhalation (DPI) in patients with moderate to severe COPD. Eur. Respir. J. 2012, 40, 2817. [Google Scholar]
- BAYER. Ciprofloxacin DPI (BAY q3939)—Briefing Document for FDA Advisory Committee Meeting; FDA: White Oak, MD, USA, 2017; p. 146.
- Wilson, R.; Welte, T.; Polverino, E.; De Soyza, A.; Greville, H.; O’Donnell, A.; Alder, J.; Reimnitz, P.; Hampel, B. Ciprofloxacin dry powder for inhalation in non-cystic fibrosis bronchiectasis: A phase II randomised study. Eur. Respir. J. 2013, 41, 1107–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksamit, T.; Bandel, T.-J.; Criollo, M.; De Soyza, A.; Elborn, J.S.; Operschall, E.; Polverino, E.; Roth, K.; Winthrop, K.L.; Wilson, R. The RESPIRE trials: Two phase III, randomized, multicentre, placebo-controlled trials of Ciprofloxacin Dry Powder for Inhalation (Ciprofloxacin DPI) in non-cystic fibrosis bronchiectasis. Contemp. Clin. Trials 2017, 58, 78–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksamit, T.; De Soyza, A.; Bandel, T.-J.; Criollo, M.; Elborn, J.S.; Operschall, E.; Polverino, E.; Roth, K.; Winthrop, K.L.; Wilson, R. RESPIRE 2: A phase III placebo-controlled randomised trial of ciprofloxacin dry powder for inhalation in non-cystic fibrosis bronchiectasis. Eur. Respir. J. 2018, 51, 1702053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chotirmall, S.H.; Chalmers, J.D. RESPIRE: Breathing new life into bronchiectasis. Eur. Respir. J. 2018. [Google Scholar] [CrossRef]
- FDA. Ciprofloxacin Dry Powder for Inhalation (DPI)—Meeting of the Antimicrobial Drugs Advisory Committee (AMDAC); FDA: White Oak, MD, USA, 2017; p. 51.
- Ernst, J.; Klinger-Strobel, M.; Arnold, K.; Thamm, J.; Hartung, A.; Pletz, M.W.; Makarewicz, O.; Fischer, D. Polyester-based particles to overcome the obstacles of mucus and biofilms in the lung for tobramycin application under static and dynamic fluidic conditions. Eur. J. Pharm. Biopharm. 2018, 131, 120–129. [Google Scholar] [CrossRef]
- Sah, E.; Sah, H. Recent Trends in Preparation of Poly(lactide-co-glycolide) Nanoparticles by Mixing Polymeric Organic Solution with Antisolvent. J. Nanomater. 2015, 2015, 794601. [Google Scholar] [CrossRef] [Green Version]
- Thomas, N.; Thorn, C.; Richter, K.; Thierry, B.; Prestidge, C. Efficacy of Poly-Lactic-Co-Glycolic Acid Micro- and Nanoparticles of Ciprofloxacin Against Bacterial Biofilms. J. Pharm. Sci. 2016, 105, 3115–3122. [Google Scholar] [CrossRef]
- Shen, J.; Burgess, D.J. Accelerated in-vitro release testing methods for extended-release parenteral dosage forms. J. Pharm. Pharmacol. 2012, 64, 986–996. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.-I.; Na, H.-S.; Seo, D.-H.; Kim, D.-G.; Lee, H.-C.; Jang, M.-K.; Na, S.-K.; Roh, S.-H.; Kim, S.-I.; Nah, J.-W. Ciprofloxacin-encapsulated poly(dl-lactide-co-glycolide) nanoparticles and its antibacterial activity. Int. J. Pharm. 2008, 352, 317–323. [Google Scholar] [CrossRef]
- Cheow, W.S.; Chang, M.W.; Hadinoto, K. Antibacterial efficacy of inhalable antibiotic-encapsulated biodegradable polymeric nanoparticles against E. coli biofilm cells. J. Biomed. Nanotechnol. 2010, 6, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Cheow, W.S.; Chang, M.W.; Hadinoto, K. Antibacterial Efficacy of Inhalable Levofloxacin-Loaded Polymeric Nanoparticles Against E. coli Biofilm Cells: The Effect of Antibiotic Release Profile. Pharm. Res. 2010, 27, 1597–1609. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Yuan, Z.; Zhang, W.; Wei, D.; Hu, N. Preparation, in vitro release and antibacterial activity evaluation of rifampicin and moxifloxacin-loaded poly(D,L-lactide-co-glycolide) microspheres. Artif. Cells Nanomed. Biotechnol. 2019, 47, 790–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torshabi, M.; Nojehdehian, H.; Tabatabaei, F.S. In vitro behavior of poly-lactic-co-glycolic acid microspheres containing minocycline, metronidazole, and ciprofloxacin. J. Investig. Clin. Dent. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Amikacin Liposome Inhalation Suspension: A Review in Mycobacterium avium Complex Lung Disease. Drugs 2019, 79, 555–562. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Leifer, F.; Rose, S.; Chun, D.Y.; Thaisz, J.; Herr, T.; Nashed, M.; Joseph, J.; Perkins, W.R.; DiPetrillo, K. Amikacin Liposome Inhalation Suspension (ALIS) Penetrates Non-tuberculous Mycobacterial Biofilms and Enhances Amikacin Uptake Into Macrophages. Front. Microbiol. 2018, 9, 915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haworth, C.S.; Bilton, D.; Chalmers, J.D.; Davis, A.M.; Froehlich, J.; Gonda, I.; Thompson, B.; Wanner, A.; O’Donnell, A.E. Inhaled liposomal ciprofloxacin in patients with non-cystic fibrosis bronchiectasis and chronic lung infection with Pseudomonas aeruginosa (ORBIT-3 and ORBIT-4): Two phase 3, randomised controlled trials. Lancet Respir. Med. 2019, 7, 213–226. [Google Scholar] [CrossRef] [Green Version]
- Cipolla, D.; Shekunov, B.; Blanchard, J.; Hickey, A. Lipid-based carriers for pulmonary products: Preclinical development and case studies in humans. Adv. Drug Deliv. Rev. 2014, 75, 53–80. [Google Scholar] [CrossRef]
- Weers, J. Comparison of Phospholipid-Based Particles for Sustained Release of Ciprofloxacin Following Pulmonary Administration to Bronchiectasis Patients. Pulm. Ther. 2019, 5, 127–150. [Google Scholar] [CrossRef] [Green Version]
- Chorepsima, S.; Kechagias, K.S.; Kalimeris, G.; Triarides, N.A.; Falagas, M.E. Spotlight on inhaled ciprofloxacin and its potential in the treatment of non-cystic fibrosis bronchiectasis. Drug Des. Dev. 2018, 12, 4059–4066. [Google Scholar] [CrossRef] [Green Version]
- Hamblin, K.A.; Wong, J.P.; Blanchard, J.D.; Atkins, H.S. The potential of liposome-encapsulated ciprofloxacin as a tularemia therapy. Front. Cell Infect. Microbiol. 2014, 4, 79. [Google Scholar] [CrossRef] [Green Version]
- Norville, I.H.; Hatch, G.J.; Bewley, K.R.; Atkinson, D.J.; Hamblin, K.A.; Blanchard, J.D.; Armstrong, S.J.; Pitman, J.K.; Rayner, E.; Hall, G.; et al. Efficacy of liposome-encapsulated ciprofloxacin in a murine model of Q fever. Antimicrob. Agents Chemother. 2014, 58, 5510–5518. [Google Scholar] [CrossRef] [Green Version]
- Tewes, F.; Brillault, J.; Couet, W.; Olivier, J.-C. Formulation of rifampicin–cyclodextrin complexes for lung nebulization. J. Control. Release 2008, 129, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Dutton, B.; Woods, A.; Sadler, R.; Prime, D.; Barlow, D.J.; Forbes, B.; Jones, S.A. Using polar ion-pairs to control drug delivery to the airways of the lungs. Mol. Pharm. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bandara, H.; Herpin, M.J.; Kolacny Jr, D.; Harb, A.; Romanovicz, D.; Smyth, H.D.C. Incorporation of farnesol significantly increases the efficacy of liposomal ciprofloxacin against Pseudomonas aeruginosa biofilms in vitro. Mol. Pharm. 2016, 13, 2760–2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deygen, I.M.; Egorov, A.M.; Kudryashova, E.V. Structure and stability of fluoroquinolone-(2-hydroxypropyl)-β-cyclodextrin complexes as perspective antituberculosis drugs. Mosc. Univ. Chem. Bull. 2016, 71, 1–6. [Google Scholar] [CrossRef]
- Gursahani, H.; Riggs-Sauthier, J.; Pfeiffer, J.; Lechuga-Ballesteros, D.; Fishburn, C.S. Absorption of Polyethylene Glycol (PEG) Polymers: The Effect of PEG Size on Permeability. J. Pharm. Sci. 2009, 98, 2847–2856. [Google Scholar] [CrossRef]
- Larsen, E.M.; Johnson, R.J. Microbial esterases and ester prodrugs: An unlikely marriage for combating antibiotic resistance. Drug Dev. Res. 2019, 80, 33–47. [Google Scholar] [CrossRef] [Green Version]
- Forde, E.; Devocelle, M. Pro-Moieties of Antimicrobial Peptide Prodrugs. Molecules 2015, 20, 1210–1227. [Google Scholar] [CrossRef]
- Wang, Y.; Yuan, Q.; Feng, W.; Pu, W.; Ding, J.; Zhang, H.; Li, X.; Yang, B.; Dai, Q.; Cheng, L.; et al. Targeted delivery of antibiotics to the infected pulmonary tissues using ROS-responsive nanoparticles. J. Nanobiotechnol. 2019, 17, 103. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| CIP–Cations (M−1, 1:1 Complexes) [31] | LVX-cation (M−2, 2:1 Complexes) [32] | |
|---|---|---|
| Ca2+ | 100 | 562 |
| Fe2+ | 122 * | 34,673 |
| Mg2+ | 720 | 4898 |
| Fe3+ | 1551 * | |
| Zn2+ | 2700 | 27,542 |
| Al3+ | 88,000 | |
| Cu2+ | 906,900 |
| ciprofloxacin | Cu2+ > Al3+ > Zn2+ ≈ Fe3+ > Mg2+ > Fe2+ ≈ Ca2+ | [31] |
| Al3+ > Fe3+ ≈ Cu2+ > Zn2+ > Mg2+ | [33] | |
| Al3+ ≈ Fe3+ > Zn2+ ≈ Mn2+ ≈ Mg2+ | [34] | |
| levofloxacin | Fe3+ > Fe2+ > Mg2+ ≈ Ca2+ | * |
| Al3+ > Fe2+ ≈ Zn2+ > Mg2+ > Ca2+ | [32] | |
| Al3+ ≈ Cu2+ > Fe3+ > Mg2+ | [33] | |
| enrofloxacin | Al3+ > Fe3+ ≈ Cu2+ > Mg2+ | [33] |
| sparfloxacin | Al3+ > Cu2+ > Zn2+ > Mg2+ | [33] |
| moxifloxacin | Fe3+ > Mg2+ ≈ Fe2+ ≈ Ca2+ | * |
| pefloxacin | Fe3+ ≈ Mg2+ > Fe2+ ≈ Ca2+ | * |
| Clinical Study | LVX Sputum Versus Serum Cmax | LVX Sputum Versus Serum AUC0-24 |
|---|---|---|
| MPEX-204 | 5000 to 7000-fold higher in sputum than in serum | 300-fold higher in sputum than in serum |
| MPEX-207 | 3500-fold higher in sputum than in serum | 400-fold higher in sputum than in serum |
| MPEX-209 | 1700-fold higher in sputum than in serum | 600-fold higher in sputum than in serum |
| (CIP–Ca)2+ | (CIP–Cu)2+ | CIP solution | |
|---|---|---|---|
| Ka (M−1) (from [31]) | 100 | 906,900 | |
| Concentration (µM) needed to reduce the CIP Papp by 50% (from [31]) | 40 | 0.04 | |
| ELF Cmax (µg/mL) after IT administration to rats (from [22]) | 26.5 ± 17.7 | 142.3 ± 81.5 | 1.0 ± 0.4 |
| CIP AUCELF to AUCplasma ratio after IT administration to rats (from [22]) | 203 | 1069 | 9.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brillault, J.; Tewes, F. Control of the Lung Residence Time of Highly Permeable Molecules after Nebulization: Example of the Fluoroquinolones. Pharmaceutics 2020, 12, 387. https://doi.org/10.3390/pharmaceutics12040387
Brillault J, Tewes F. Control of the Lung Residence Time of Highly Permeable Molecules after Nebulization: Example of the Fluoroquinolones. Pharmaceutics. 2020; 12(4):387. https://doi.org/10.3390/pharmaceutics12040387
Chicago/Turabian StyleBrillault, Julien, and Frédéric Tewes. 2020. "Control of the Lung Residence Time of Highly Permeable Molecules after Nebulization: Example of the Fluoroquinolones" Pharmaceutics 12, no. 4: 387. https://doi.org/10.3390/pharmaceutics12040387
APA StyleBrillault, J., & Tewes, F. (2020). Control of the Lung Residence Time of Highly Permeable Molecules after Nebulization: Example of the Fluoroquinolones. Pharmaceutics, 12(4), 387. https://doi.org/10.3390/pharmaceutics12040387