Influence of Aqueous Solubility-Enhancing Excipients on the Microstructural Characteristics of Furosemide-Loaded Electrospun Nanofibers


 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Furosemide-Containing Solutions for Electrospinning
2.3. Preparation of Polymeric Solutions for the Unloaded Electrospun Fibers
2.4. Preparation of Physical Mixtures
2.5. Formulation of Electrospun Nanofibers (NF)
2.6. Characterization of Electrospun Nanofibers
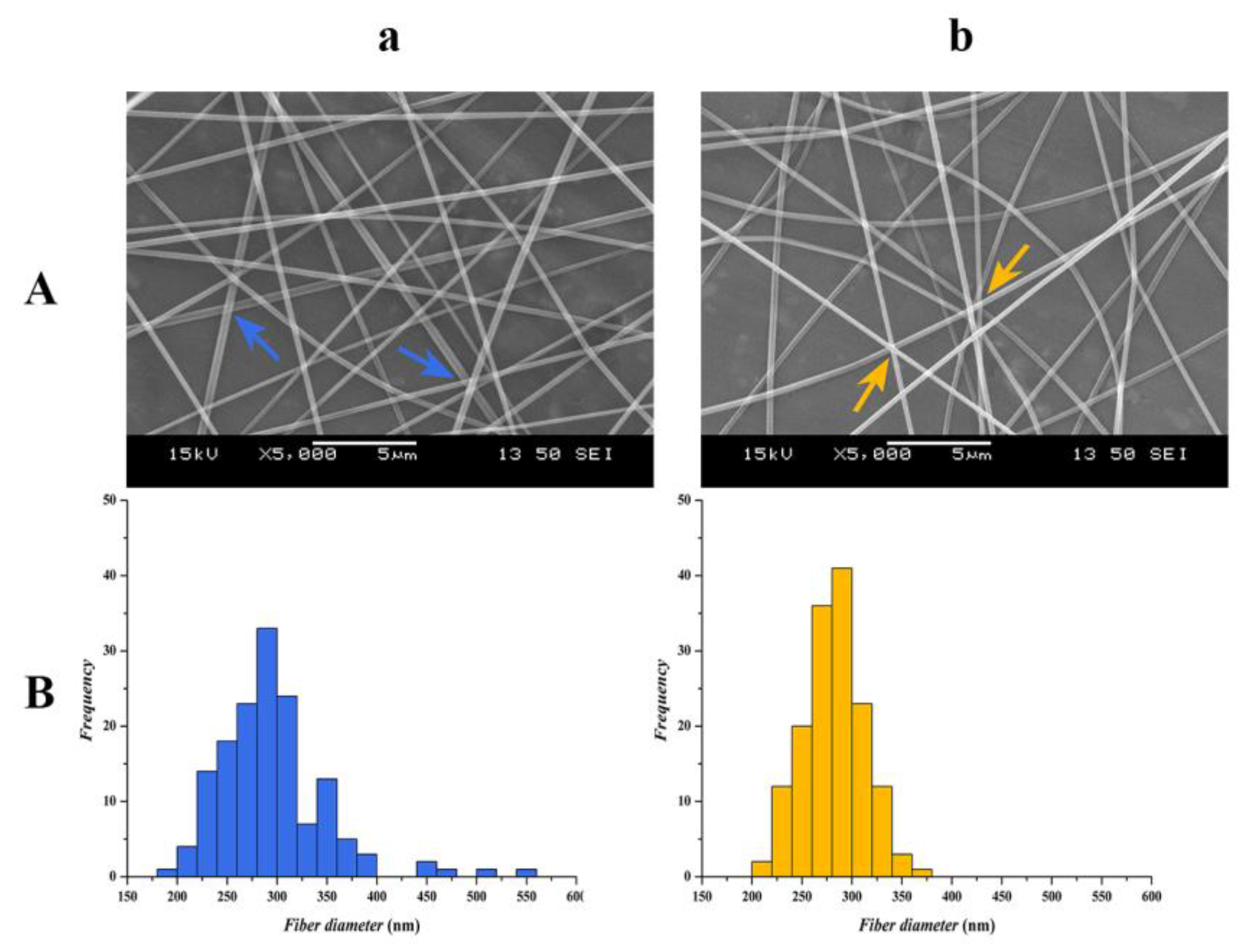
2.6.1. Morphological Study by Scanning Electron Microscopy (SEM)
2.6.2. Statistical Analysis of the Fiber Diameters
2.6.3. Microstructural Evaluation of the Fibers by ATR-FTIR (Attenuated Total Reflectance—Fourier Transform Infrared) Spectroscopy
2.6.4. X-Ray Diffraction (XRD)
2.6.5. Positron Annihilation Lifetime Spectroscopy (PALS)
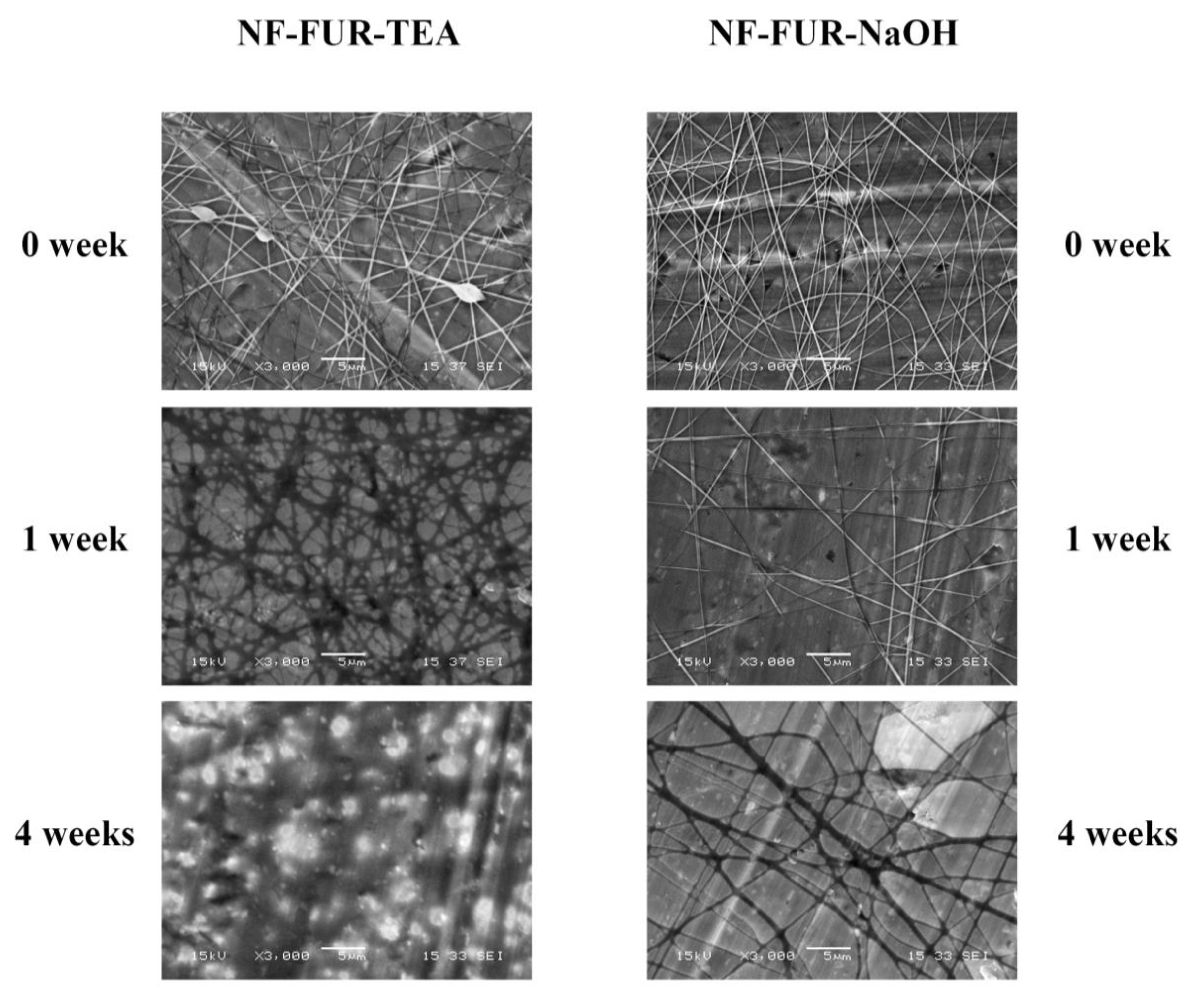
2.6.6. Accelerated Stability Test
2.6.7. Furosemide Content of the Nanofibers
2.6.8. In-Vitro Drug Release Study
3. Results and Discussion
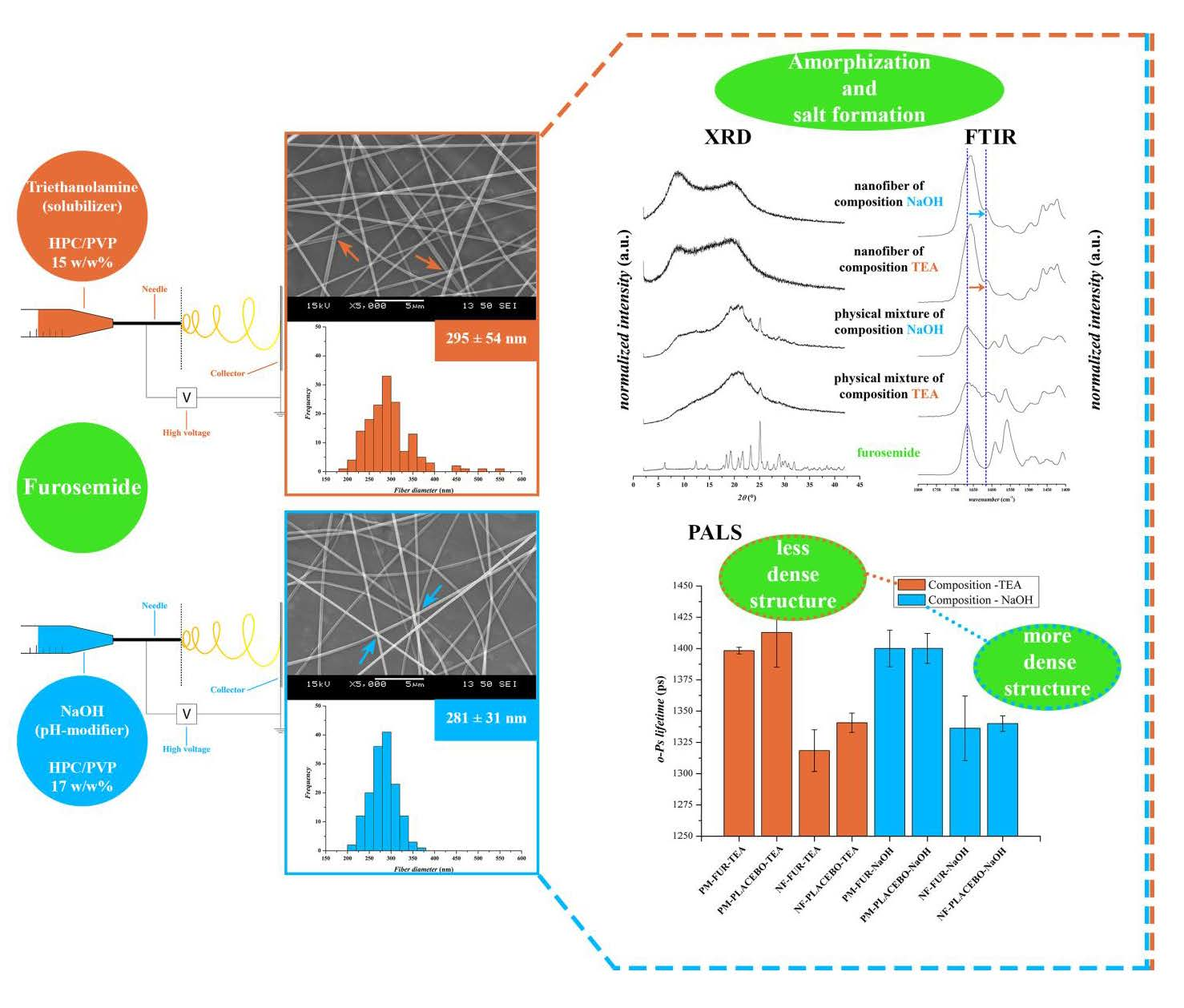
3.1. Morphology Analysis
3.2. Solid-State Characterisation
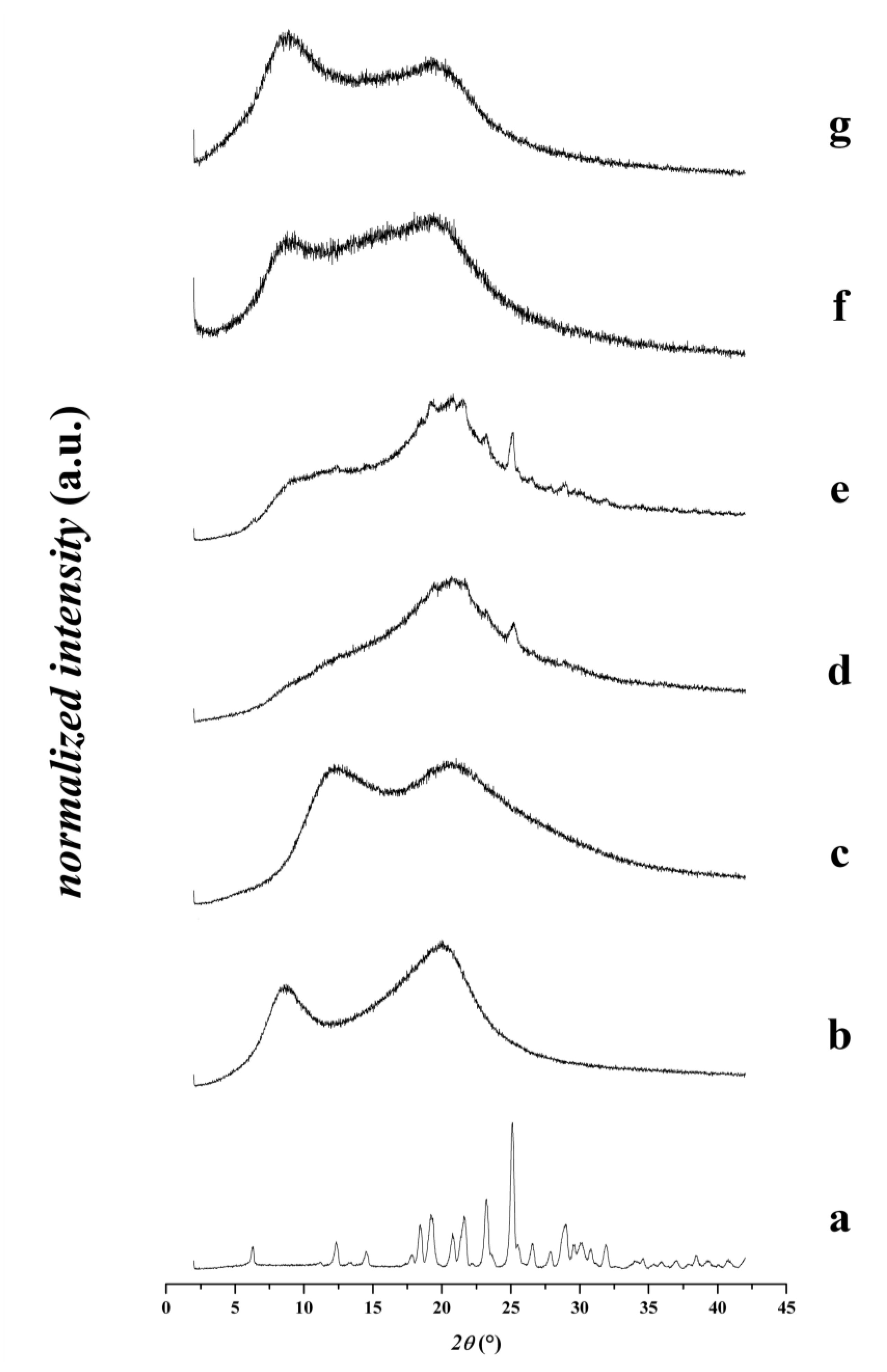
3.2.1. X-Ray Diffraction Patterns of the Fibers
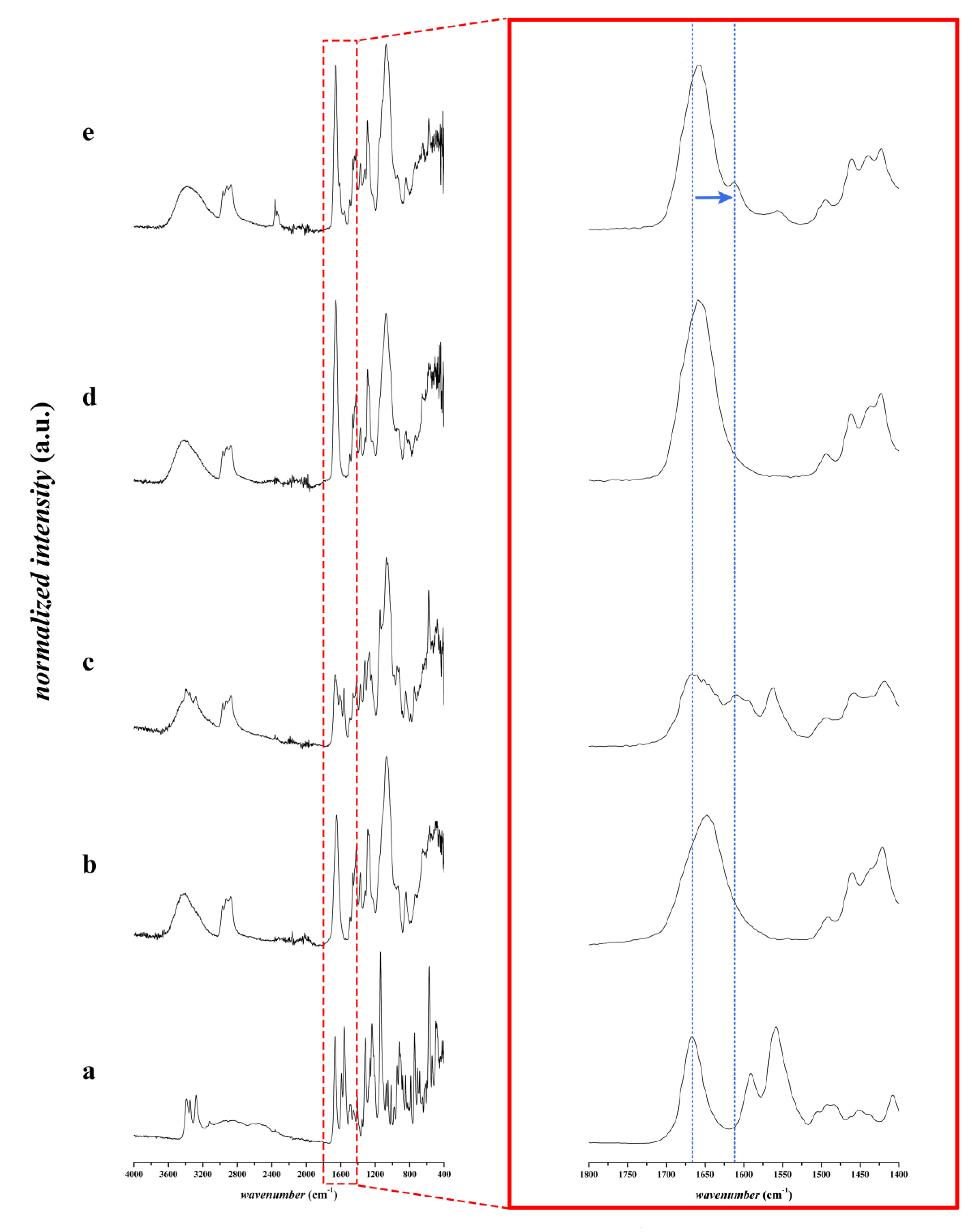
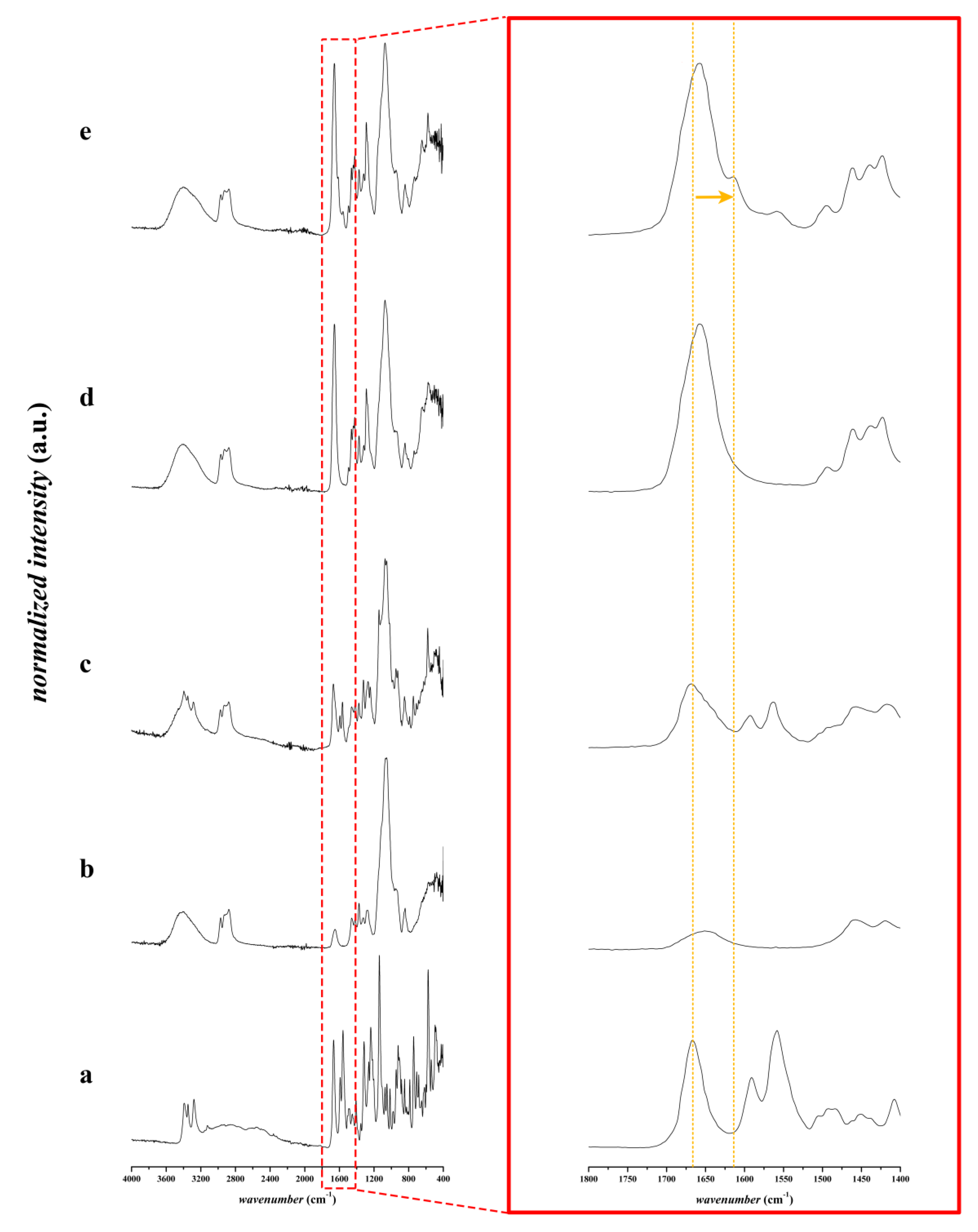
3.2.2. FTIR Spectroscopic Analysis
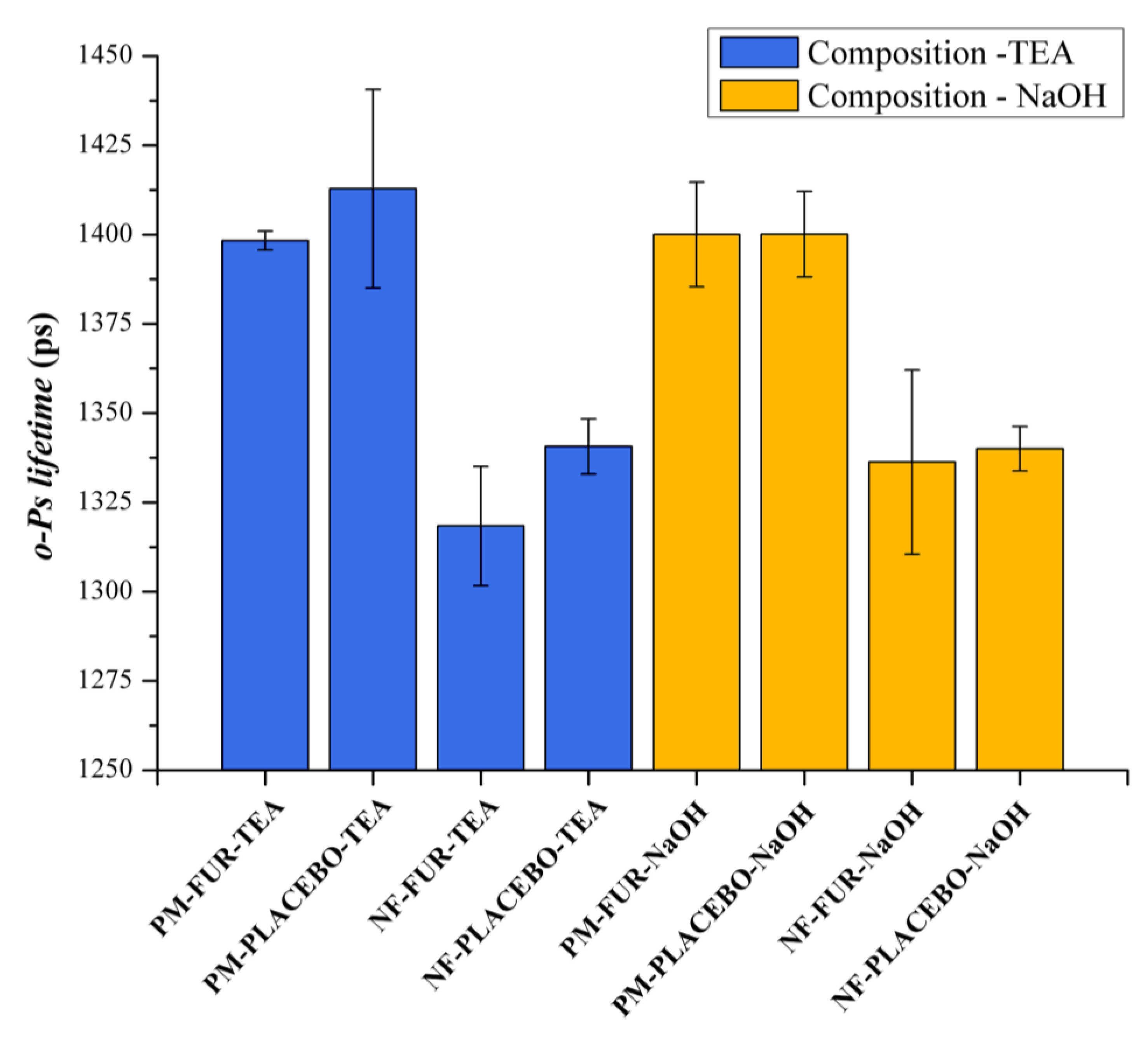
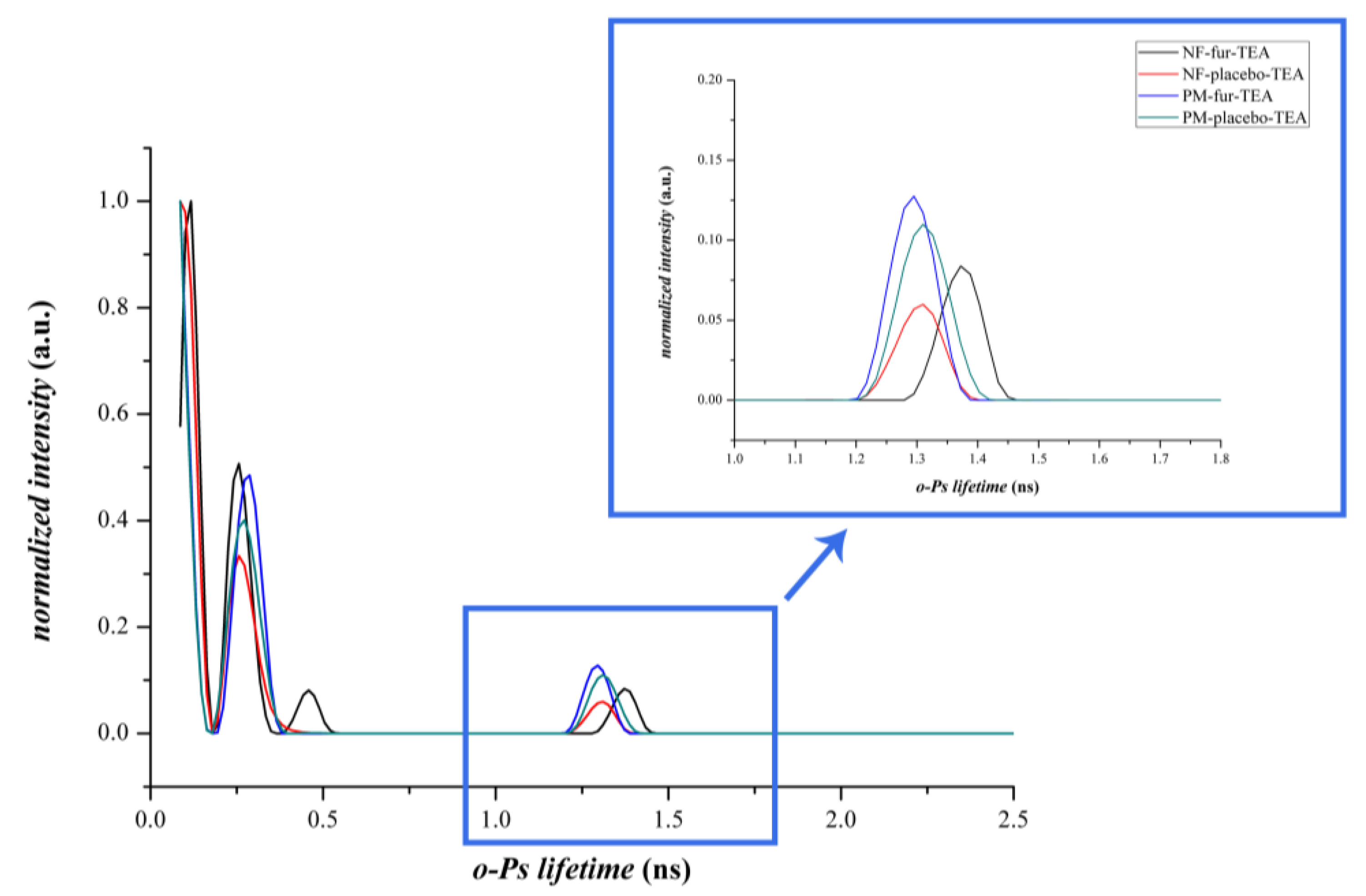
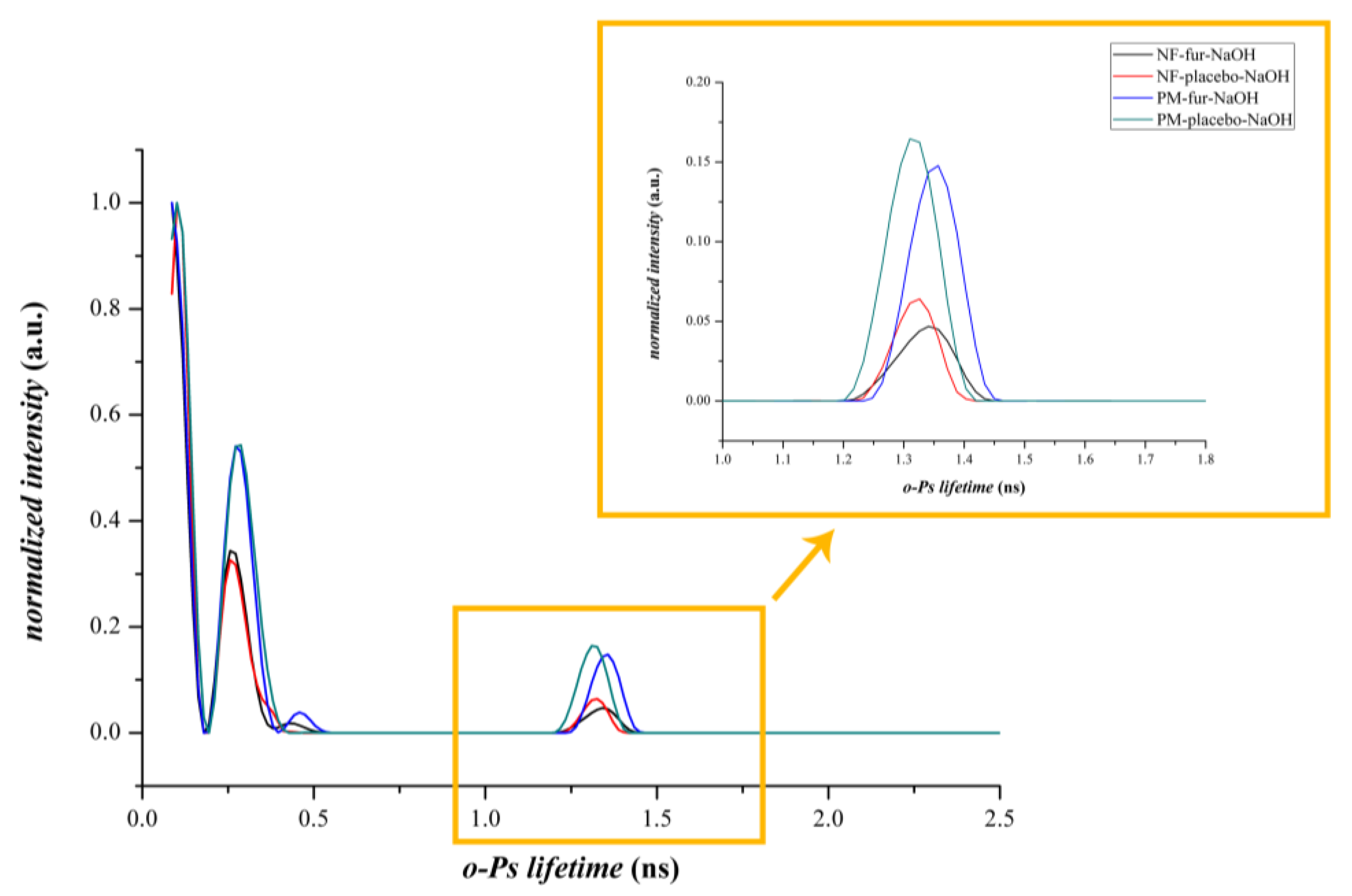
3.2.3. Positron Annihilation Lifetime Spectroscopy
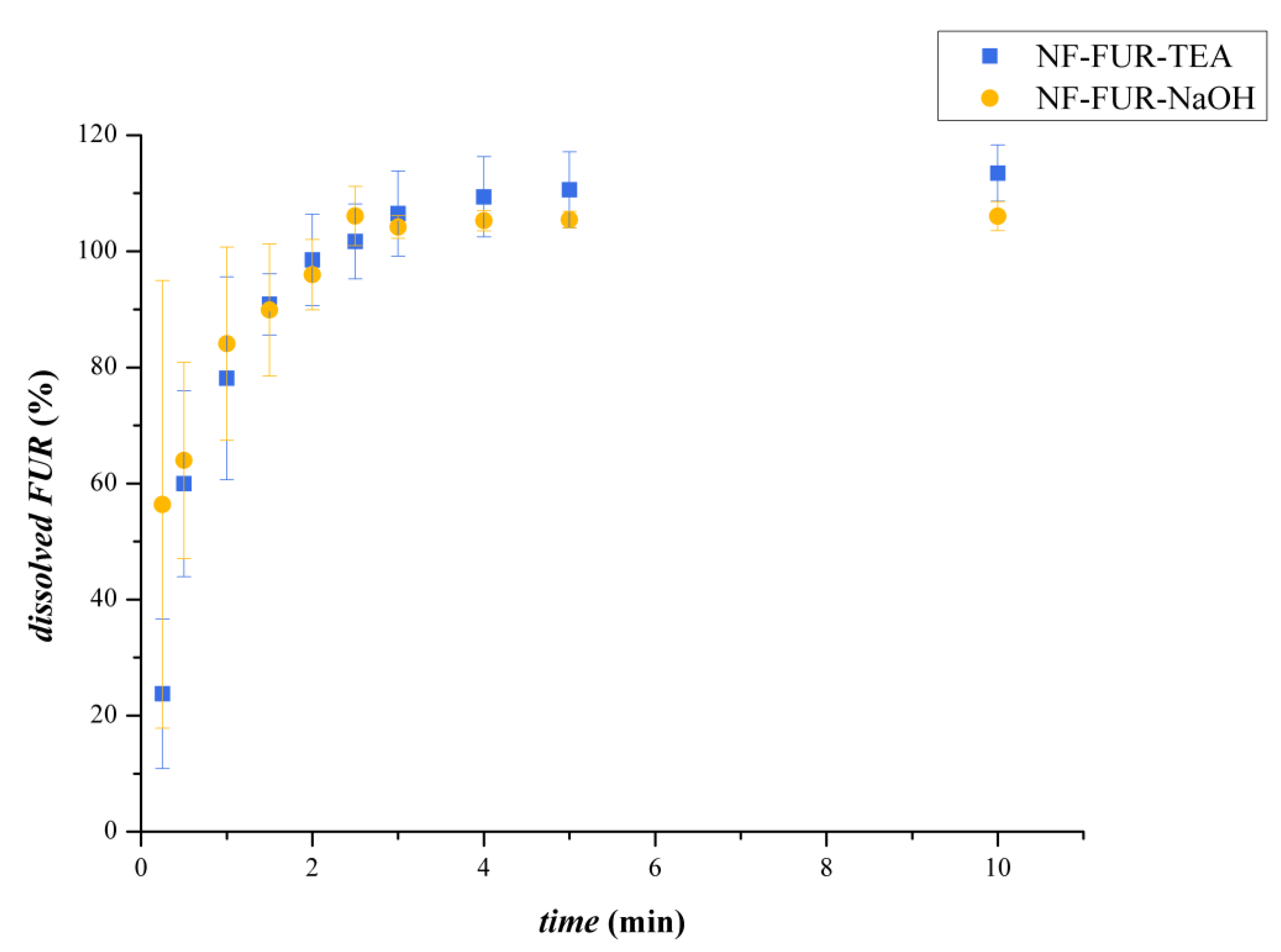
3.2.4. Dissolution Study
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Zordi, N.; Moneghini, M.; Kikic, I.; Grassi, M.; Del Rio Castillo, A.E.; Solinas, D.; Bolger, M.B. Applications of supercritical fluids to enhance the dissolution behaviors of furosemide by generation of microparticles and solid dispersions. Eur. J. Pharm. Biopharm. 2012, 81, 131–141. [Google Scholar] [CrossRef]
- Kortejarvi, H.; Malkki, J.; Shawahna, R.; Scherrmann, J.M.; Urtti, A.; Yliperttula, M. Pharmacokinetic simulations to explore dissolution criteria of bcs i and iii biowaivers with and without mdr-1 efflux transporter. Eur. J. Pharm. Sci. 2014, 61, 18–26. [Google Scholar] [CrossRef]
- Rao Khandavilli, U.B.; Gangavaram, S.; Rajesh Goud, N.; Cherukuvada, S.; Raghavender, S.; Nangia, A.; Manjunatha, S.G.; Nambiar, S.; Pal, S. High solubility crystalline hydrates of na and k furosemide salts. CrystEngComm 2014, 16, 4842–4852. [Google Scholar] [CrossRef]
- Garnero, C.; Chattah, A.K.; Longhi, M. Supramolecular complexes of maltodextrin and furosemide polymorphs: A new approach for delivery systems. Carbohydr. Polym. 2013, 94, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Garnero, C.; Chattah, A.K.; Longhi, M. Improving furosemide polymorphs properties through supramolecular complexes of beta-cyclodextrin. J. Pharm. Biomed. Anal. 2014, 95, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Melocchi, A.; Loreti, G.; Del Curto, M.D.; Maroni, A.; Gazzaniga, A.; Zema, L. Evaluation of hot-melt extrusion and injection molding for continuous manufacturing of immediate-release tablets. J. Pharm. Sci. 2015, 104, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Gong, C.; Gou, M.; Fu, S.; Guo, Q.; Shi, S.; Luo, F.; Guo, G.; Qiu, L.; Qian, Z. Biodegradable poly(epsilon-caprolactone)-poly(ethylene glycol) copolymers as drug delivery system. Int. J. Pharm. 2009, 381, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, M.; Gosselin, P.; Mateescu, M.A. Carboxymethyl starch mucoadhesive microspheres as gastroretentive dosage form. Int. J. Pharm. 2015, 496, 497–508. [Google Scholar] [CrossRef]
- Lvov, Y.; Abdullayev, E. Functional polymer–clay nanotube composites with sustained release of chemical agents. Prog. Polym. Sci. 2013, 38, 1690–1719. [Google Scholar] [CrossRef]
- Sebe, I.; Szabo, P.; Kallai-Szabo, B.; Zelko, R. Incorporating small molecules or biologics into nanofibers for optimized drug release: A review. Int. J. Pharm. 2015, 494, 516–530. [Google Scholar] [CrossRef]
- Kovács, A.; Démuth, B.; Meskó, A.; Zelkó, R. Preformulation studies of furosemide-loaded electrospun nanofibrous systems for buccal administration. Polymers 2017, 9, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.G.; Li, J.J.; Williams, G.R.; Zhao, M. Electrospun amorphous solid dispersions of poorly water-soluble drugs: A review. J. Control Release 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelipenko, J.; Kocbek, P.; Kristl, J. Critical attributes of nanofibers: Preparation, drug loading, and tissue regeneration. Int. J. Pharm. 2015, 484, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Kapahi, H.; Khan, N.; Bhardwaj, A.; Mishra, N. Implication of nanofibers in oral drug delivery. Curr. Pharm. Des. 2015, 21, 2021–2036. [Google Scholar] [CrossRef]
- Sebe, I.; Petzke, M.; Zelkó, R.; Szabó, B. Nano- és mikroszálas rendszerek előállítása és gyógyszerészeti alkalmazásuk lehetőségei i. Acta Pharmaceutica Hungarica 2013, 83, 1–9. [Google Scholar]
- Yu, D.G.; Wang, M.; Li, X.; Liu, X.; Zhu, L.M.; Annie Bligh, S.W. Multifluid electrospinning for the generation of complex nanostructures. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, e1601. [Google Scholar] [CrossRef]
- Zhao, K.; Wang, W.; Yang, Y.; Wang, K.; Yu, D.-G. From taylor cone to solid nanofiber in tri-axial electrospinning: Size relationships. Results Phys. 2019, 15, 102770. [Google Scholar] [CrossRef]
- Kazsoki, A.; Szabo, P.; Domjan, A.; Balazs, A.; Bozo, T.; Kellermayer, M.; Farkas, A.; Balogh-Weiser, D.; Pinke, B.; Darcsi, A.; et al. Microstructural distinction of electrospun nanofibrous drug delivery systems formulated with different excipients. Mol. Pharm. 2018, 15, 4214–4225. [Google Scholar] [CrossRef]
- Balogh, A.; Farkas, B.; Farago, K.; Farkas, A.; Wagner, I.; Van Assche, I.; Verreck, G.; Nagy, Z.K.; Marosi, G. Melt-blown and electrospun drug-loaded polymer fiber mats for dissolution enhancement: A comparative study. J. Pharm. Sci. 2015, 104, 1767–1776. [Google Scholar] [CrossRef] [Green Version]
- Vasconcelos, T.; Marques, S.; das Neves, J.; Sarmento, B. Amorphous solid dispersions: Rational selection of a manufacturing process. Adv. Drug Deliv. Rev. 2016, 100, 85–101. [Google Scholar] [CrossRef]
- Panda, B.; Parihar, A.S.; Mallick, S. Effect of plasticizer on drug crystallinity of hydroxypropyl methylcellulose matrix film. Int. J. Biol. Macromol. 2014, 67, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, S.P.; Dugar, R.P. Application of surfactants in solid dispersion technology for improving solubility of poorly water soluble drugs. J. Drug Deliv. Sci. Technol. 2017, 41, 68–77. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O′Reilly, N.J. Polymeric amorphous solid dispersions: A review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class ii drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.; Kwak, S.Y. Effect of nanoscale confinement on molecular mobility and drug release properties of cellulose acetate/sulindac nanofibers. J. Appl. Polym. Sci. 2019, 136, 47863. [Google Scholar] [CrossRef]
- Nielsen, L.H.; Gordon, S.; Holm, R.; Selen, A.; Rades, T.; Mullertz, A. Preparation of an amorphous sodium furosemide salt improves solubility and dissolution rate and leads to a faster tmax after oral dosing to rats. Eur. J. Pharm. Biopharm. 2013, 85, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, P.; Eldrup, M.; Mogensen, O.E.; Pedersen, N.J. Program system for analysing positron lifetime spectra and angular correlation curves. Comput. Phys. Commun. 1981, 23, 307–335. [Google Scholar] [CrossRef]
- Shukla, A.; Peter, M.; Hoffmann, L. Analysis of positron lifetime spectra using quantified maximum entropy and a general linear filter. Nucl. Instrum. Methods Phys. Res. Sect. A-Accel. Spectrom. Dect. Assoc. Equ. 1993, 335, 310–317. [Google Scholar] [CrossRef]
- Babu, N.J.; Cherukuvada, S.; Thakuria, R.; Nangia, A. Conformational and synthon polymorphism in furosemide (lasix). Cryst. Growth Des. 2010, 10, 1979–1989. [Google Scholar] [CrossRef]
- Schonbichler, S.A.; Bittner, L.K.; Weiss, A.K.; Griesser, U.J.; Pallua, J.D.; Huck, C.W. Comparison of nir chemical imaging with conventional nir, raman and atr-ir spectroscopy for quantification of furosemide crystal polymorphs in ternary powder mixtures. Eur. J. Pharm. Biopharm. 2013, 84, 616–625. [Google Scholar] [CrossRef]
- Doherty, C.; York, P. Frusemide crystal forms; solid state and physicochemical analyses. Int. J. Pharm. 1988, 47, 141–155. [Google Scholar] [CrossRef]
- Abraham Miranda, J.; Garnero, C.; Chattah, A.K.; Santiago de Oliveira, Y.; Ayala, A.P.; Longhi, M.R. Furosemide:Triethanolamine salt as a strategy to improve the biopharmaceutical properties and photostability of the drug. Cryst. Growth Des. 2019, 19, 2060–2068. [Google Scholar] [CrossRef]
- Zelko, R.; Orban, A.; Suvegh, K. Tracking of the physical ageing of amorphous pharmaceutical polymeric excipients by positron annihilation spectroscopy. J. Pharm. Biomed. Anal. 2006, 40, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Duplatre, G.; Ferreira Marques, M.F.; Da Graca Miguel, M. Size of sodium dodecyl sulfate micelles in aqueous solutions as studied by positron annihilation lifetime spectroscopy. J. Phys. Chem. 1996, 100, 16608–16612. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients | Composition-TEA (w/w%) | Composition-NaOH (w/w%) |
|---|---|---|
| Polymer concentration * | 15 | 17 |
| Furosemide concentration | 1 | 1 |
| Triethanolamine concentration | 1.5 | - |
| NaOH (2 M concentration) solution concentration | - | ~2 |
| Purified water | 82.5 | ~80 |
| Sample Name | o-Ps Lifetime (ns) | Height | FWHM | A |
|---|---|---|---|---|
| PM-FUR-TEA | 1.29325 | 0.13188 | 0.08653 | 0.01215 |
| PM-PLACEBO-TEA | 1.3108 | 0.11264 | 0.09452 | 0.01133 |
| NF-FUR-TEA | 1.37286 | 0.08635 | 0.07866 | 0.00723 |
| NF-PLACEBO-TEA | 1.30485 | 0.06128 | 0.0869 | 0.00567 |
| PM-FUR-NaOH | 1.35086 | 0.15345 | 0.09621 | 0.01571 |
| PM-PLACEBO-NaOH | 1.31382 | 0.17043 | 0.09771 | 0.01773 |
| NF-FUR-NaOH | 1.33622 | 0.04768 | 0.10874 | 0.00552 |
| NF-PLACEBO-NaOH | 1.31933 | 0.06599 | 0.08334 | 0.00585 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, A.; Kazsoki, A.; Démuth, B.; Szirányi, B.; Madarász, J.; Süvegh, K.; Zelkó, R. Influence of Aqueous Solubility-Enhancing Excipients on the Microstructural Characteristics of Furosemide-Loaded Electrospun Nanofibers. Pharmaceutics 2020, 12, 385. https://doi.org/10.3390/pharmaceutics12040385
Kovács A, Kazsoki A, Démuth B, Szirányi B, Madarász J, Süvegh K, Zelkó R. Influence of Aqueous Solubility-Enhancing Excipients on the Microstructural Characteristics of Furosemide-Loaded Electrospun Nanofibers. Pharmaceutics. 2020; 12(4):385. https://doi.org/10.3390/pharmaceutics12040385
Chicago/Turabian StyleKovács, Andrea, Adrienn Kazsoki, Balázs Démuth, Bernadett Szirányi, János Madarász, Károly Süvegh, and Romána Zelkó. 2020. "Influence of Aqueous Solubility-Enhancing Excipients on the Microstructural Characteristics of Furosemide-Loaded Electrospun Nanofibers" Pharmaceutics 12, no. 4: 385. https://doi.org/10.3390/pharmaceutics12040385
APA StyleKovács, A., Kazsoki, A., Démuth, B., Szirányi, B., Madarász, J., Süvegh, K., & Zelkó, R. (2020). Influence of Aqueous Solubility-Enhancing Excipients on the Microstructural Characteristics of Furosemide-Loaded Electrospun Nanofibers. Pharmaceutics, 12(4), 385. https://doi.org/10.3390/pharmaceutics12040385