Lactoferrin Contributes a Renoprotective Effect in Acute Kidney Injury and Early Renal Fibrosis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Microarray Analysis
2.2. Cell Culture and Lactoferrin Treatment
2.3. Cell Viability Assay
2.4. Western Blot Analysis
2.5. Immunofluorescence Microscopy
2.6. Transfection of siRNA and LTF DNA
2.7. Folic Acid Mouse Model
2.8. Biochemical Measurements
2.9. Histopathological and Immunohistochemical Analysis
2.10. Masson Staining
2.11. Statistical Analysis
3. Results
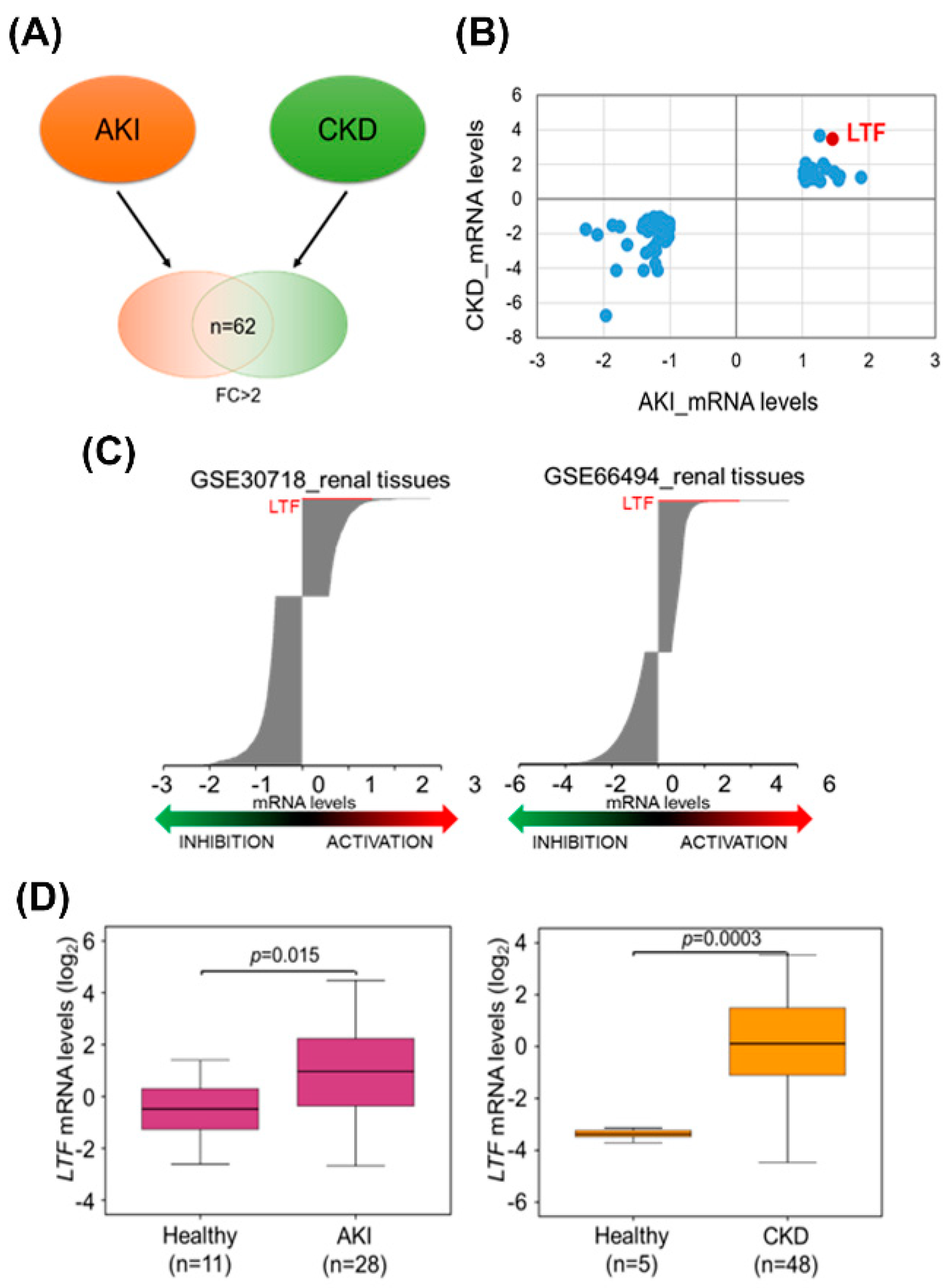
3.1. High Levels of LTF Expression in the Kidney Tissues of AKI and CKD Patients
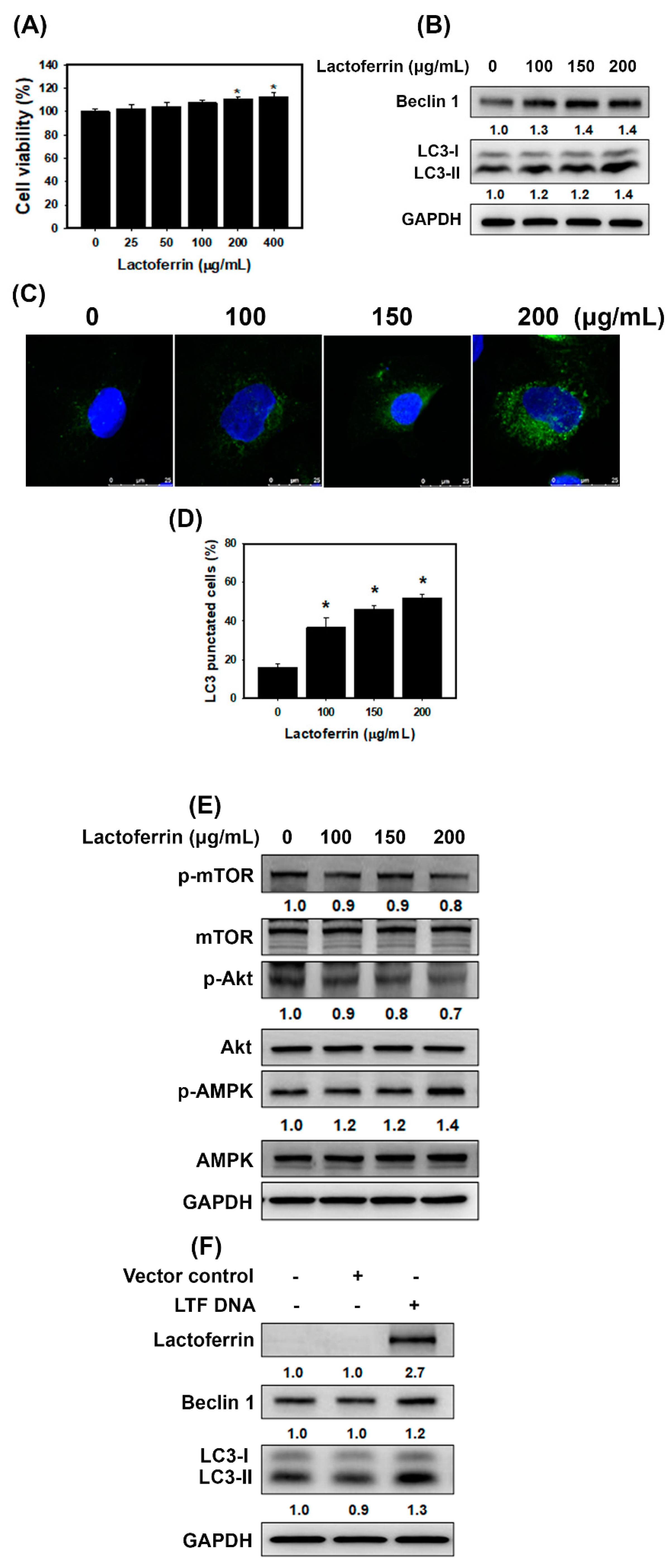
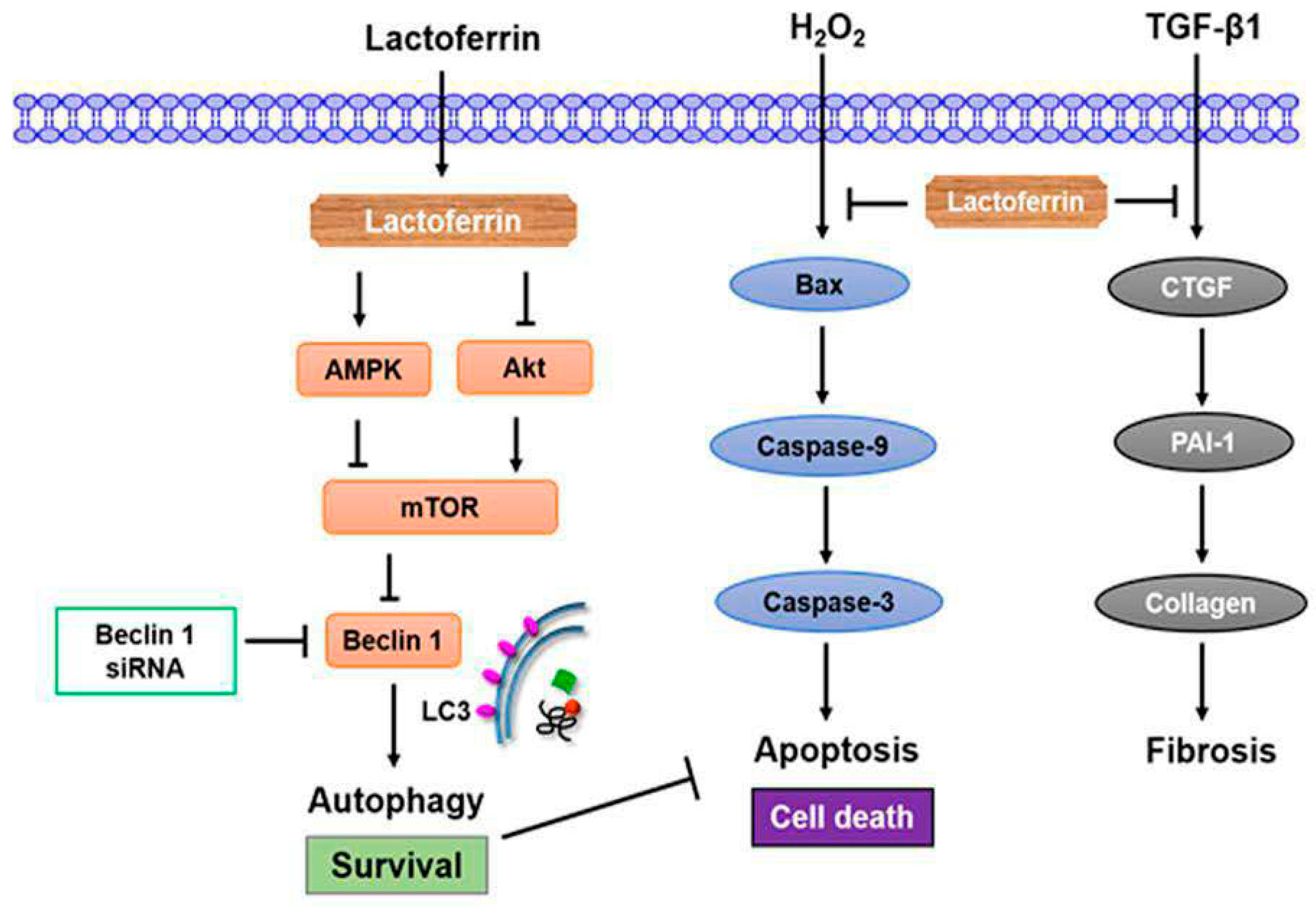
3.2. Lactoferrin Induces Autophagy through the Activation of AMPK and Inhibition of Akt/mTOR Pathway
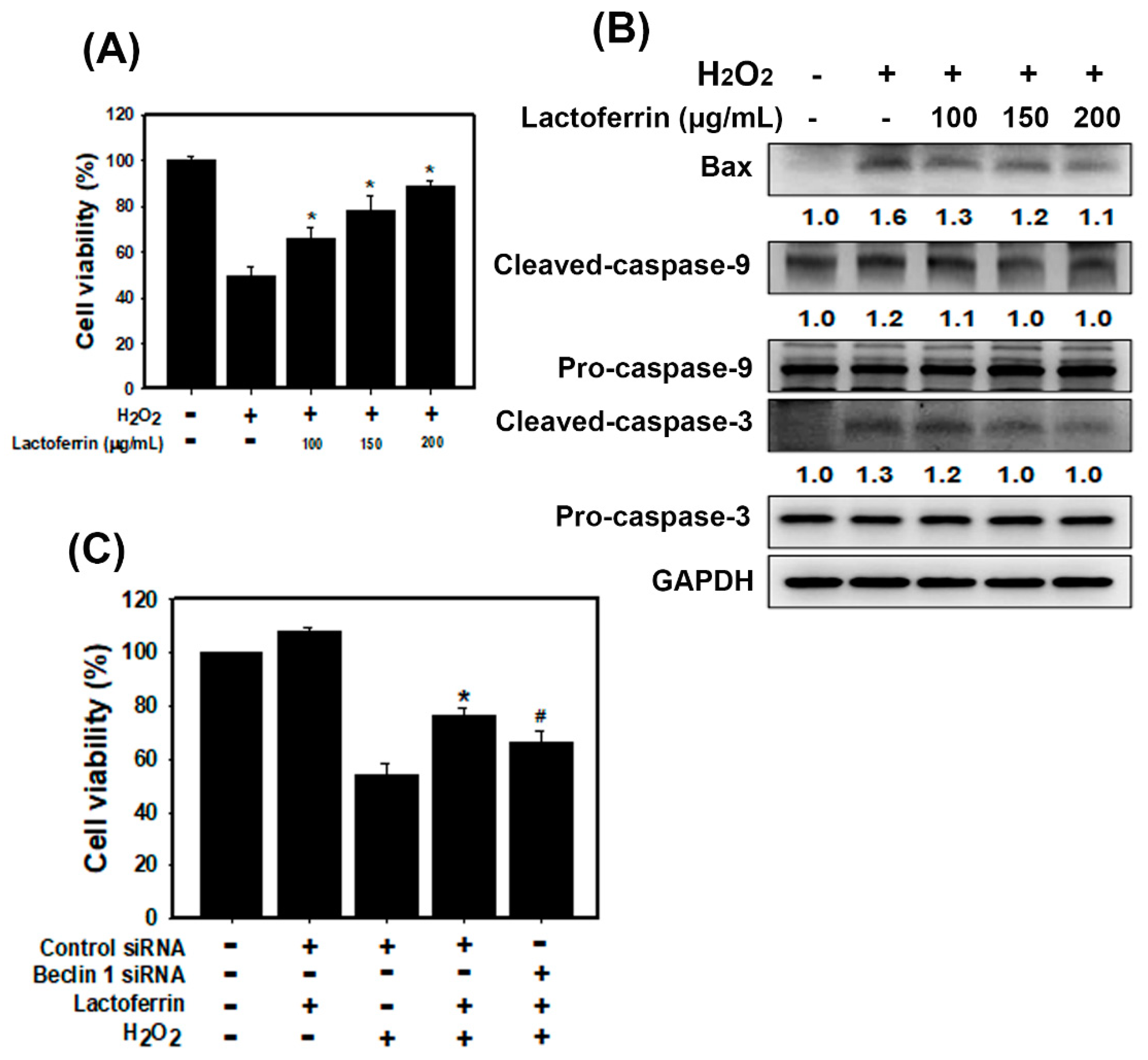
3.3. Lactoferrin Suppresses Oxidative Stress-Induced Cell Death and Apoptosis by Augmenting Autophagy in HK-2 Cells
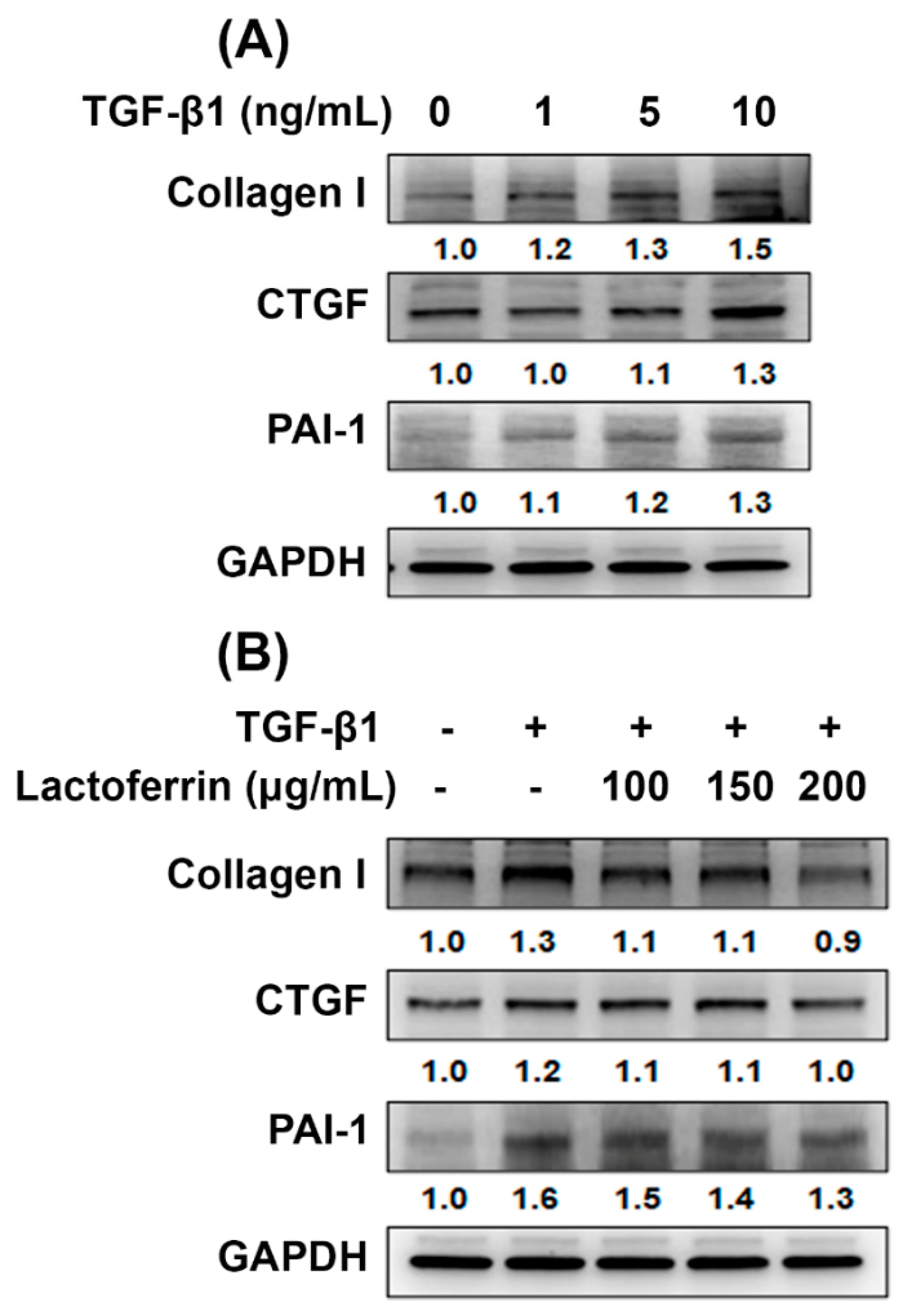
3.4. Lactoferrin Has an Antifibrotic Role in Human Kidney Proximal Tubular Cells
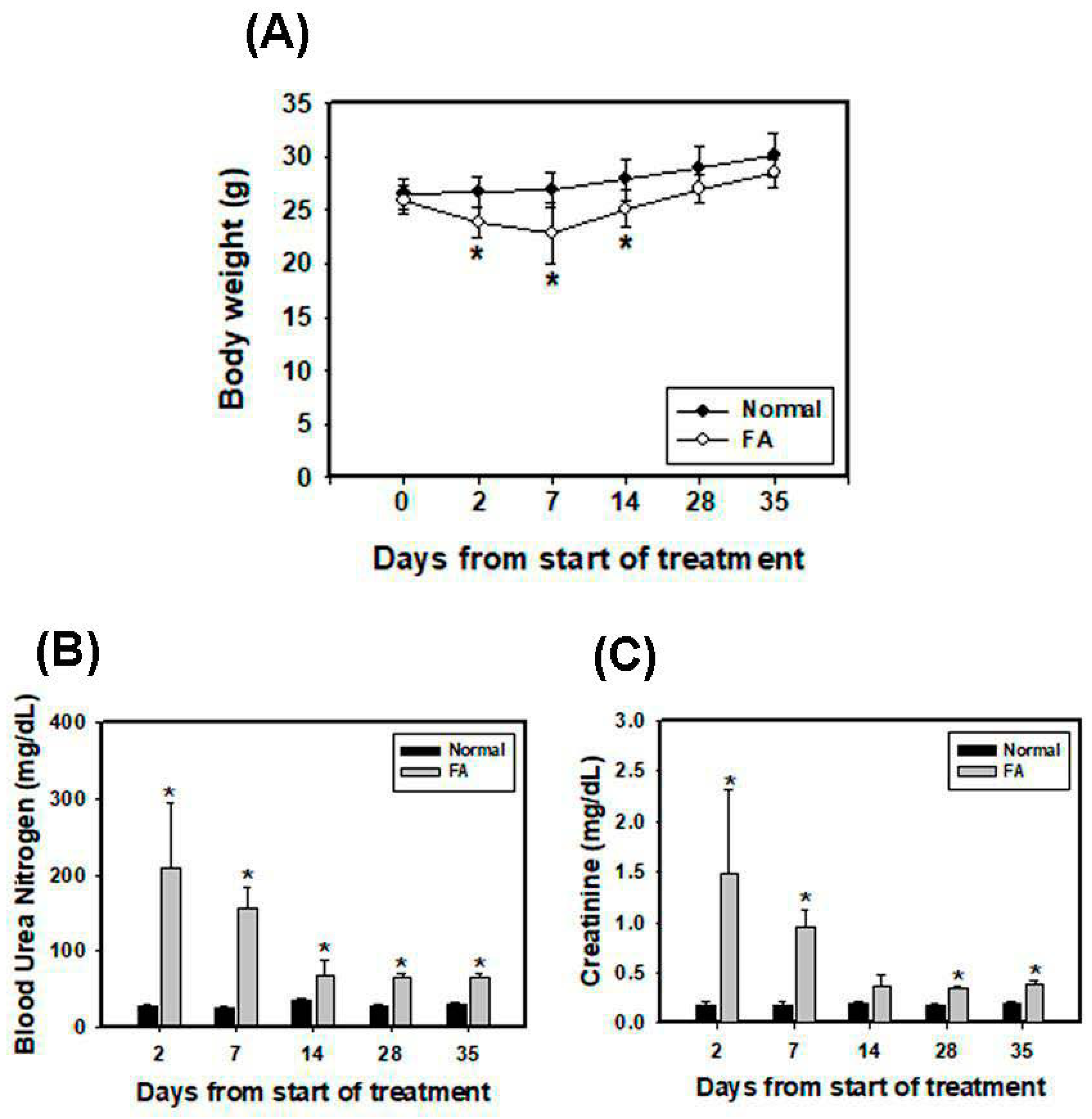
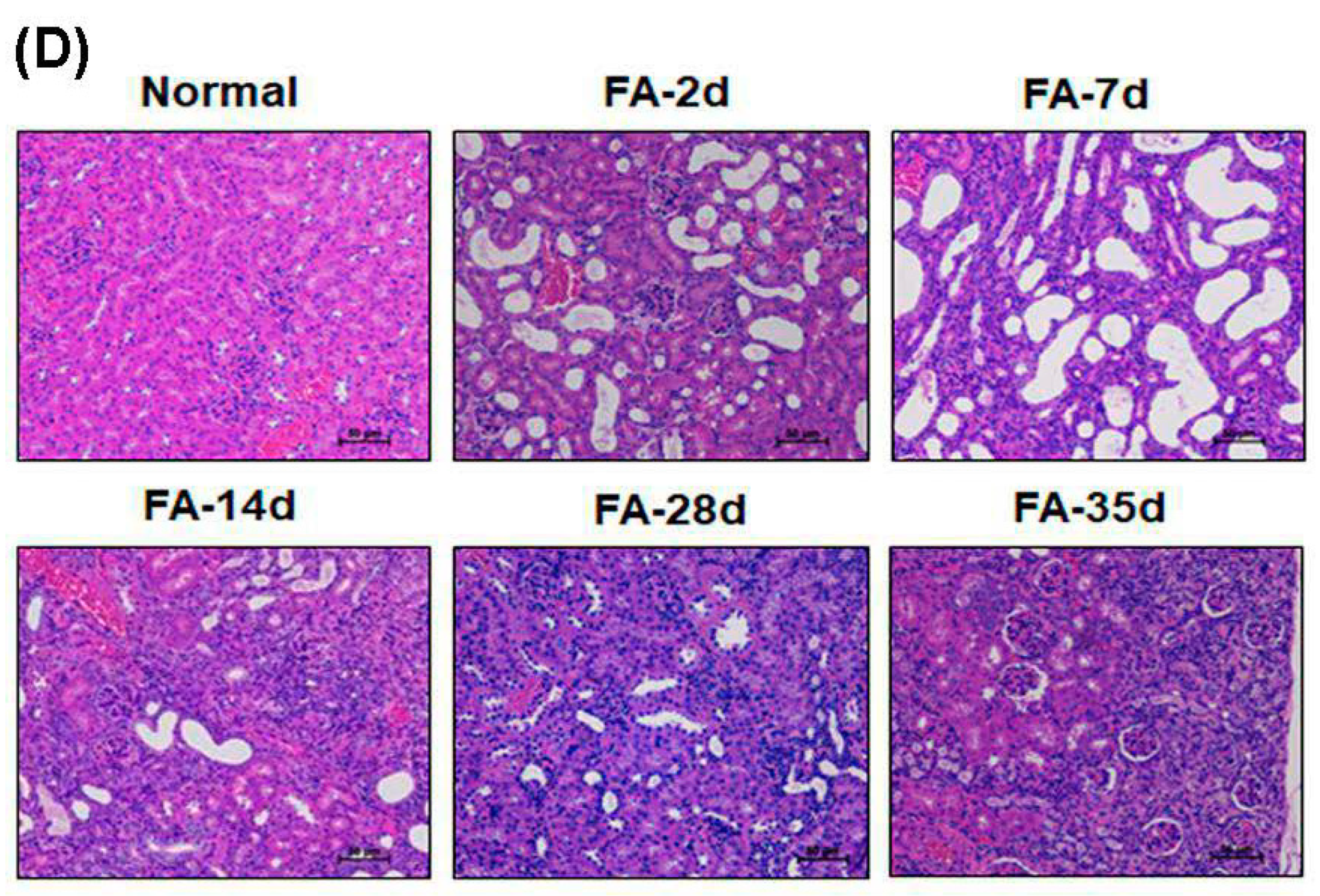
3.5. Folic Acid (FA) Induces AKI and Develops Early Fibrosis in Kidney Tissues
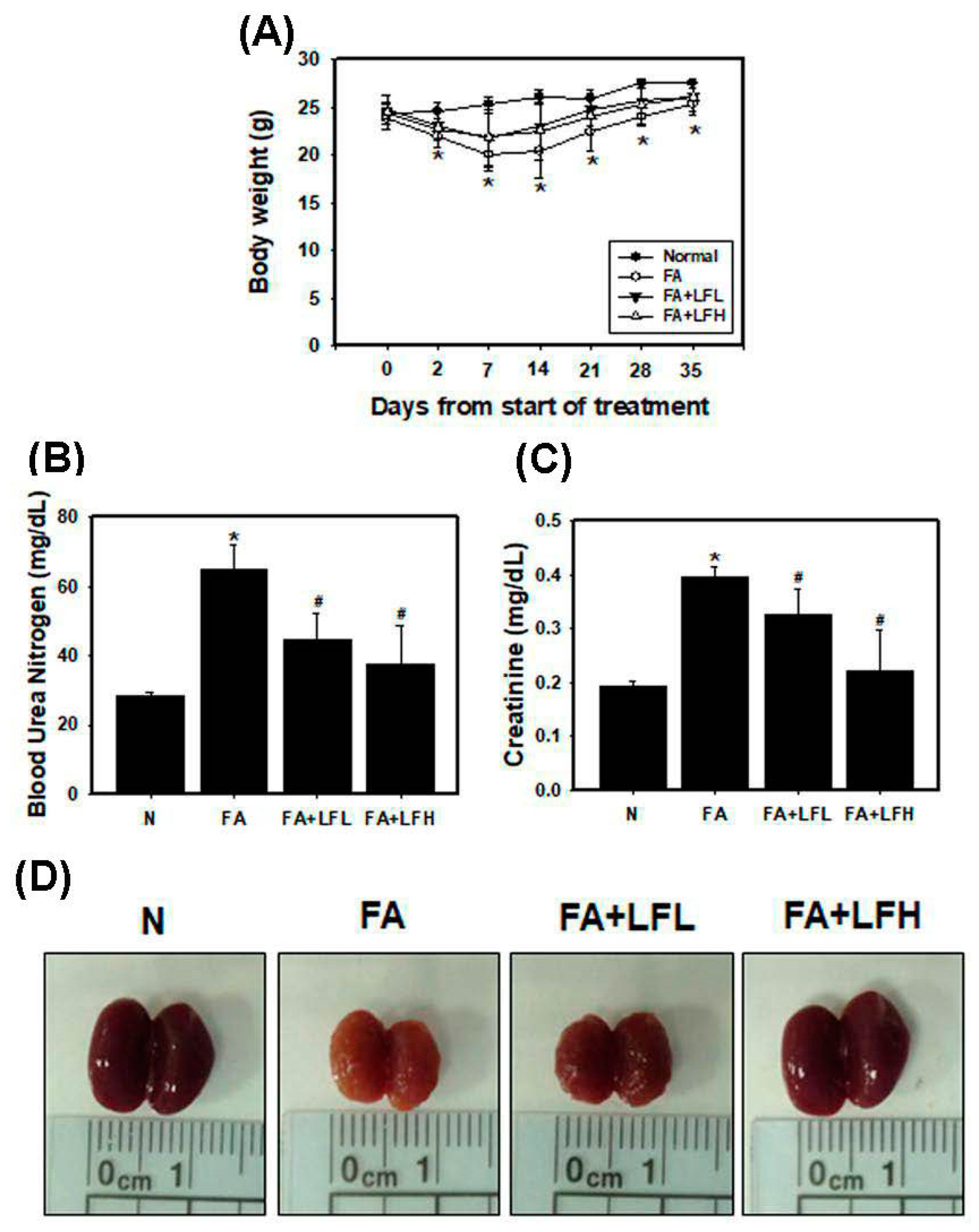
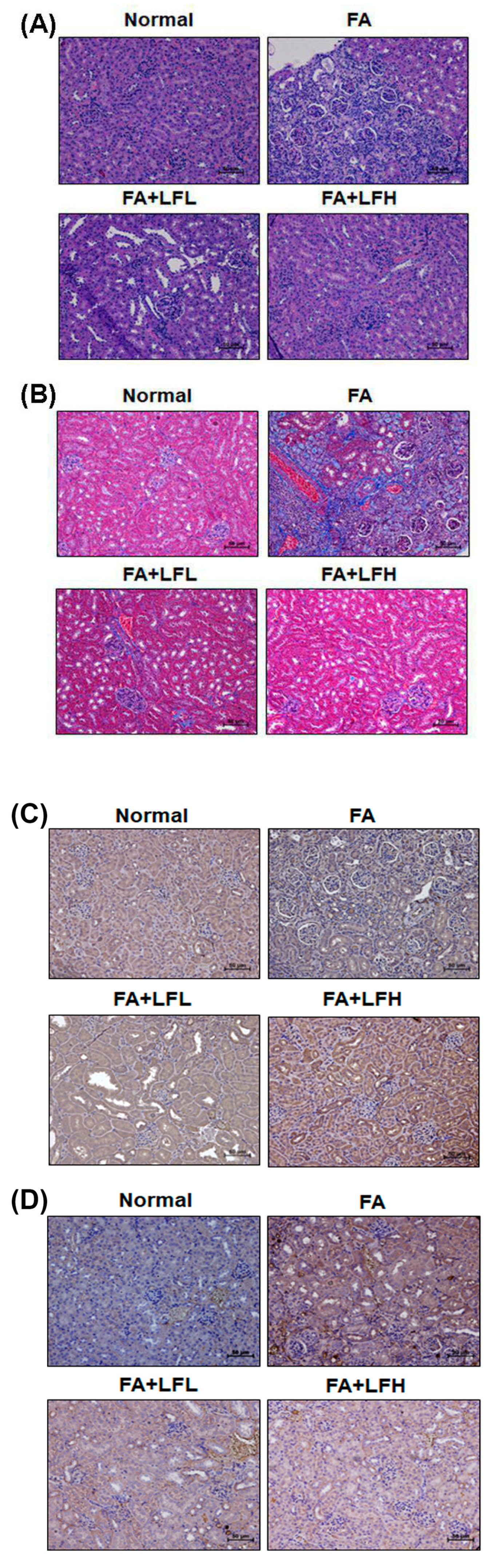
3.6. Lactoferrin Is a Therapeutic Intervention in the AKI to CKD Continuum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fang, Y.; Teng, J.; Ding, X. Acute kidney injury in China. Hemodial. Int. 2015, 19, 2–10. [Google Scholar] [CrossRef]
- Horne, K.L.; Packington, R.; Monaghan, J.; Reilly, T.; Selby, N.M. Three-year outcomes after acute kidney injury: Results of a prospective parallel group cohort study. BMJ Open 2017, 7, e015316. [Google Scholar] [CrossRef] [PubMed]
- Coca, S.G.; Singanamala, S.; Parikh, C.R. Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int. 2012, 81, 442–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingorani, S.; Molitoris, B.A.; Himmelfarb, J. Ironing out the pathogenesis of acute kidney injury. Am. J Kidney Dis. 2009, 53, 569–571. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Lin, L.; Tao, X.; Song, Y.; Cui, J.; Wan, J. The role of podocyte damage in the etiology of ischemia-reperfusion acute kidney injury and post-injury fibrosis. BMC Nephrol. 2019, 20, 106. [Google Scholar] [CrossRef] [Green Version]
- Stallons, L.J.; Whitaker, R.M.; Schnellmann, R.G. Suppressed mitochondrial biogenesis in folic acid-induced acute kidney injury and early fibrosis. Toxicol. Lett. 2014, 224, 326–332. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Wei, Q.; Liu, J.; Yi, M.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.; Liu, F.; Venkatachalam, M.A.; et al. AKI on CKD: Heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017, 92, 1071–1083. [Google Scholar] [CrossRef]
- Bostanciklioglu, M. An update on the interactions between Alzheimer’s disease, autophagy and inflammation. Gene 2019, 705, 157–166. [Google Scholar] [CrossRef]
- Camuzard, O.; Santucci-Darmanin, S.; Carle, G.F.; Pierrefite-Carle, V. Role of autophagy in osteosarcoma. J. Bone Oncol. 2019, 16, 100235. [Google Scholar] [CrossRef]
- Jiang, S.; Sun, J.; Mohammadtursun, N.; Hu, Z.; Li, Q.; Zhao, Z.; Zhang, H.; Dong, J. Dual role of autophagy/mitophagy in chronic obstructive pulmonary disease. Pulm. Pharmacol. Ther. 2019. [Google Scholar] [CrossRef]
- Li, L.; Kang, H.; Zhang, Q.; D’Agati, V.D.; Al-Awqati, Q.; Lin, F. FoxO3 activation in hypoxic tubules prevents chronic kidney disease. J. Clin. Invest. 2019, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, X.; Wang, Y.; Li, H.; Fan, J.; Shen, J.; Yu, X.; Zhou, Y.; Mao, H. ATG5-mediated autophagy suppresses NF-kappaB signaling to limit epithelial inflammatory response to kidney injury. Cell Death Dis. 2019, 10, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Peng, X.; Wang, Y.; Cao, S.; Xiong, L.; Fan, J.; Wang, Y.; Zhuang, S.; Yu, X.; Mao, H. Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G2/M arrest and renal fibrosis. Autophagy 2016, 12, 1472–1486. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.F.; Lee, Y.H.; Hsu, Y.H.; Chen, Y.J.; Lin, Y.F.; Cheng, F.Y.; Chiu, H.W. Resveratrol-loaded nanoparticles conjugated with kidney injury molecule-1 as a drug delivery system for potential use in chronic kidney disease. Nanomedicine 2017, 12, 2741–2756. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, L.; Meng, L.; Cao, G.; Wu, Y. Sirtuin 6 overexpression relieves sepsis-induced acute kidney injury by promoting autophagy. Cell Cycle 2019, 18, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Levay, P.F.; Viljoen, M. Lactoferrin: A general review. Haematologica 1995, 80, 252–267. [Google Scholar]
- Berlutti, F.; Pantanella, F.; Natalizi, T.; Frioni, A.; Paesano, R.; Polimeni, A.; Valenti, P. Antiviral properties of lactoferrin--a natural immunity molecule. Molecules 2011, 16, 6992–7018. [Google Scholar] [CrossRef] [Green Version]
- Velusamy, S.K.; Poojary, R.; Ardeshna, R.; Alabdulmohsen, W.; Fine, D.H.; Velliyagounder, K. Protective effects of human lactoferrin during Aggregatibacter actinomycetemcomitans-induced bacteremia in lactoferrin-deficient mice. Antimicrob. Agents Chemother. 2014, 58, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Tsubota, A.; Yoshikawa, T.; Nariai, K.; Mitsunaga, M.; Yumoto, Y.; Fukushima, K.; Hoshina, S.; Fujise, K. Bovine lactoferrin potently inhibits liver mitochondrial 8-OHdG levels and retrieves hepatic OGG1 activities in Long-Evans Cinnamon rats. J. Hepatol. 2008, 48, 486–493. [Google Scholar] [CrossRef]
- Hegazy, R.; Salama, A.; Mansour, D.; Hassan, A. Renoprotective Effect of Lactoferrin against Chromium-Induced Acute Kidney Injury in Rats: Involvement of IL-18 and IGF-1 Inhibition. PLoS ONE 2016, 11, e0151486. [Google Scholar] [CrossRef] [Green Version]
- Abrink, M.; Larsson, E.; Gobl, A.; Hellman, L. Expression of lactoferrin in the kidney: Implications for innate immunity and iron metabolism. Kidney Int. 2000, 57, 2004–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bort, A.; Sanchez, B.G.; Mateos-Gomez, P.A.; Diaz-Laviada, I.; Rodriguez-Henche, N. Capsaicin Targets Lipogenesis in HepG2 Cells Through AMPK Activation, AKT Inhibition and PPARs Regulation. Int. J. Mol. Sci. 2019, 20, 1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldkorn, T.; Balaban, N.; Shannon, M.; Chea, V.; Matsukuma, K.; Gilchrist, D.; Wang, H.; Chan, C. H2O2 acts on cellular membranes to generate ceramide signaling and initiate apoptosis in tracheobronchial epithelial cells. J. Cell Sci. 1998, 111, 3209–3220. [Google Scholar] [PubMed]
- Eddy, A.A.; Neilson, E.G. Chronic kidney disease progression. J. Am. Soc. Nephrol. 2006, 17, 2964–2966. [Google Scholar] [CrossRef] [Green Version]
- Bottinger, E.P. TGF-beta in renal injury and disease. Semin. Nephrol. 2007, 27, 309–320. [Google Scholar] [CrossRef]
- Grotendorst, G.R.; Okochi, H.; Hayashi, N. A novel transforming growth factor beta response element controls the expression of the connective tissue growth factor gene. Cell Growth Differ. 1996, 7, 469–480. [Google Scholar]
- Eddy, A.A.; Fogo, A.B. Plasminogen activator inhibitor-1 in chronic kidney disease: Evidence and mechanisms of action. J. Am. Soc. Nephrol. 2006, 17, 2999–3012. [Google Scholar] [CrossRef] [Green Version]
- Thadhani, R.; Pascual, M.; Bonventre, J.V. Acute renal failure. New Engl. J. Med. 1996, 334, 1448–1460. [Google Scholar] [CrossRef]
- Long, D.A.; Price, K.L.; Ioffe, E.; Gannon, C.M.; Gnudi, L.; White, K.E.; Yancopoulos, G.D.; Rudge, J.S.; Woolf, A.S. Angiopoietin-1 therapy enhances fibrosis and inflammation following folic acid-induced acute renal injury. Kidney Int. 2008, 74, 300–309. [Google Scholar] [CrossRef]
- Fink, M.; Henry, M.; Tange, J.D. Experimental folic acid nephropathy. Pathology 1987, 19, 143–149. [Google Scholar] [CrossRef]
- Leelahavanichkul, A.; Yan, Q.; Hu, X.; Eisner, C.; Huang, Y.; Chen, R.; Mizel, D.; Zhou, H.; Wright, E.C.; Kopp, J.B.; et al. Angiotensin II overcomes strain-dependent resistance of rapid CKD progression in a new remnant kidney mouse model. Kidney Int. 2010, 78, 1136–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuk, A.; Bonventre, J.V. Recent advances in acute kidney injury and its consequences and impact on chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2019, 28, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Invest. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Kimoto, Y.; Nishinohara, M.; Sugiyama, A.; Haruna, A.; Takeuchi, T. Protective effect of lactoferrin on Cisplatin-induced nephrotoxicity in rats. J. Vet. Med. Sci. 2013, 75, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.F.; Zhang, Y.H.; Wang, S.; Pang, Z.Q.; Fan, Y.G.; Li, J.Y.; Wang, Z.Y.; Guo, C. Lactoferrin ameliorates dopaminergic neurodegeneration and motor deficits in MPTP-treated mice. Redox Biol. 2019, 21, 101090. [Google Scholar] [CrossRef]
- Aizawa, S.; Hoki, M.; Yamamuro, Y. Lactoferrin promotes autophagy via AMP-activated protein kinase activation through low-density lipoprotein receptor-related protein 1. Biochem. Biophys. Res. Commun. 2017, 493, 509–513. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.A.; Wu, V.C.; Wang, C.Y. Autophagy in Chronic Kidney Diseases. Cells 2019, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Choi, M.E. Autophagy in diabetic nephropathy. J. Endocrinol. 2015, 224, R15–R30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Kim, S.; Lee, S.Y.; Koo, J.K.; Wang, Z.; Choi, M.E. Autophagy regulates TGF-beta expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J. Am. Soc. Nephrol. 2014, 25, 2835–2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, Y.-H.; Chiu, I.-J.; Lin, Y.-F.; Chen, Y.-J.; Lee, Y.-H.; Chiu, H.-W. Lactoferrin Contributes a Renoprotective Effect in Acute Kidney Injury and Early Renal Fibrosis. Pharmaceutics 2020, 12, 434. https://doi.org/10.3390/pharmaceutics12050434
Hsu Y-H, Chiu I-J, Lin Y-F, Chen Y-J, Lee Y-H, Chiu H-W. Lactoferrin Contributes a Renoprotective Effect in Acute Kidney Injury and Early Renal Fibrosis. Pharmaceutics. 2020; 12(5):434. https://doi.org/10.3390/pharmaceutics12050434
Chicago/Turabian StyleHsu, Yung-Ho, I-Jen Chiu, Yuh-Feng Lin, Yi-Jie Chen, Yu-Hsuan Lee, and Hui-Wen Chiu. 2020. "Lactoferrin Contributes a Renoprotective Effect in Acute Kidney Injury and Early Renal Fibrosis" Pharmaceutics 12, no. 5: 434. https://doi.org/10.3390/pharmaceutics12050434
APA StyleHsu, Y. -H., Chiu, I. -J., Lin, Y. -F., Chen, Y. -J., Lee, Y. -H., & Chiu, H. -W. (2020). Lactoferrin Contributes a Renoprotective Effect in Acute Kidney Injury and Early Renal Fibrosis. Pharmaceutics, 12(5), 434. https://doi.org/10.3390/pharmaceutics12050434