Ciprofloxacin Loaded Nanostructured Lipid Carriers Incorporated into In-Situ Gels to Improve Management of Bacterial Endophthalmitis

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Quantification of CIP by HPLC
2.2.2. Screening of Lipid Excipients
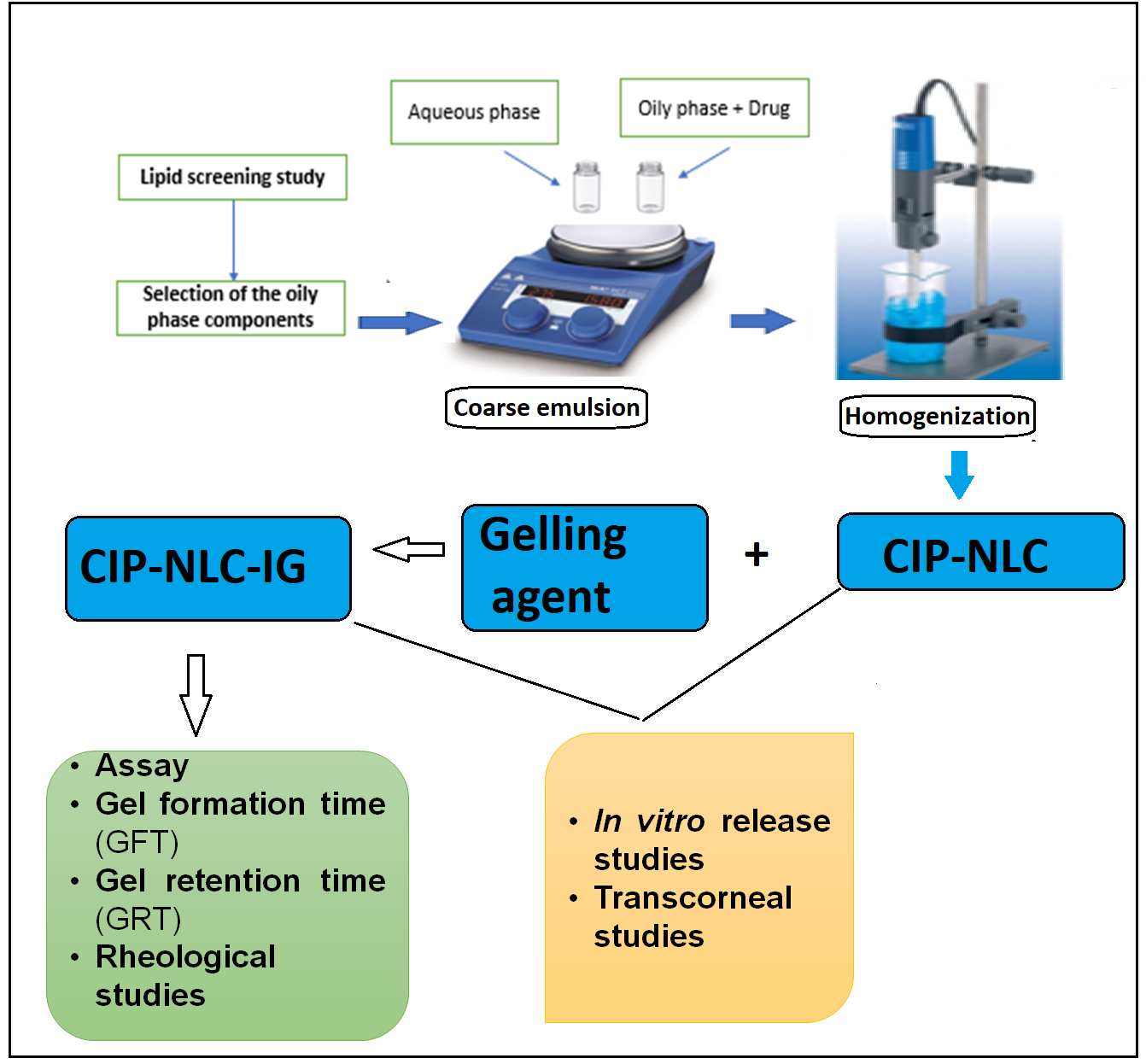
2.2.3. Preparation of CIP-NLCs
2.2.4. Preparation of CIP-NLC-IG
2.2.5. Control Formulation
2.3. Characterization of CIP-NLCs
2.3.1. Measurement of Particle Size (PS), Polydispersity index (PDI) and Zeta Potential (ZP)
2.3.2. Assay (CIP Content)
2.3.3. Entrapment Efficiency (EE)
2.3.4. Stability Studies of CIP-NLCs
2.3.5. Fourier Transform Infrared Spectroscopy (FTIR)
2.4. Characterization of CIP-NLC-IG
2.4.1. Drug Content
2.4.2. Rheological and In Vitro Gelling Characteristics of CIP-NLC-IG
2.4.3. Stability Studies of CIP-NLC-IG
2.4.4. In Vitro Release Studies of CIP-NLC and CIP-NLC-IG
2.4.5. Ex Vivo Transcorneal Permeation Studies
2.4.6. Statistical Analysis
3. Results and Discussion
3.1. Screening of Lipid Excipients
3.2. Physical Characterization of CIP-NLC
3.3. Assay and EE for Optimized CIP-NLC
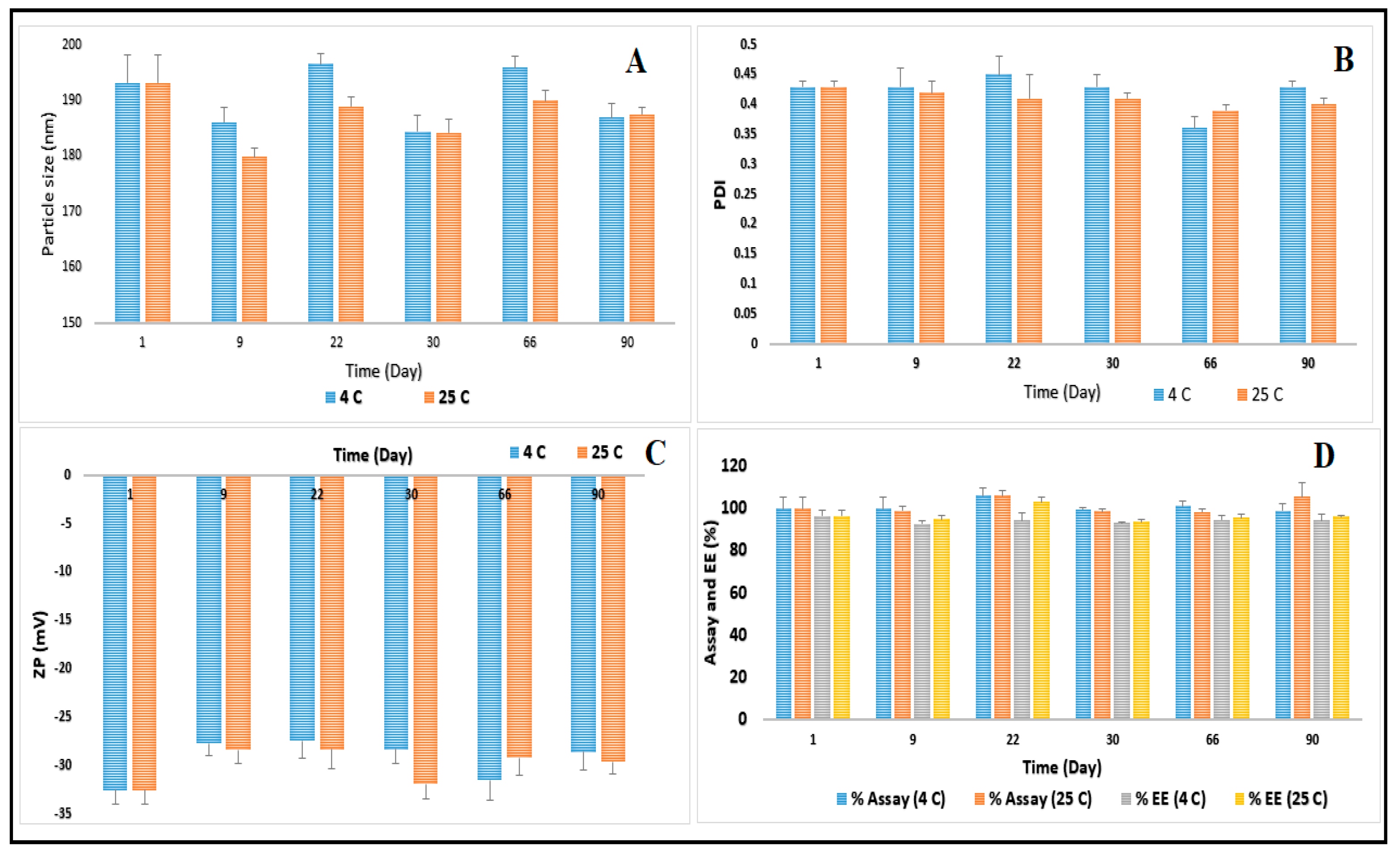
3.4. Stability Studies of CIP-NLC
3.5. Characterization of CIP-NLC-IG
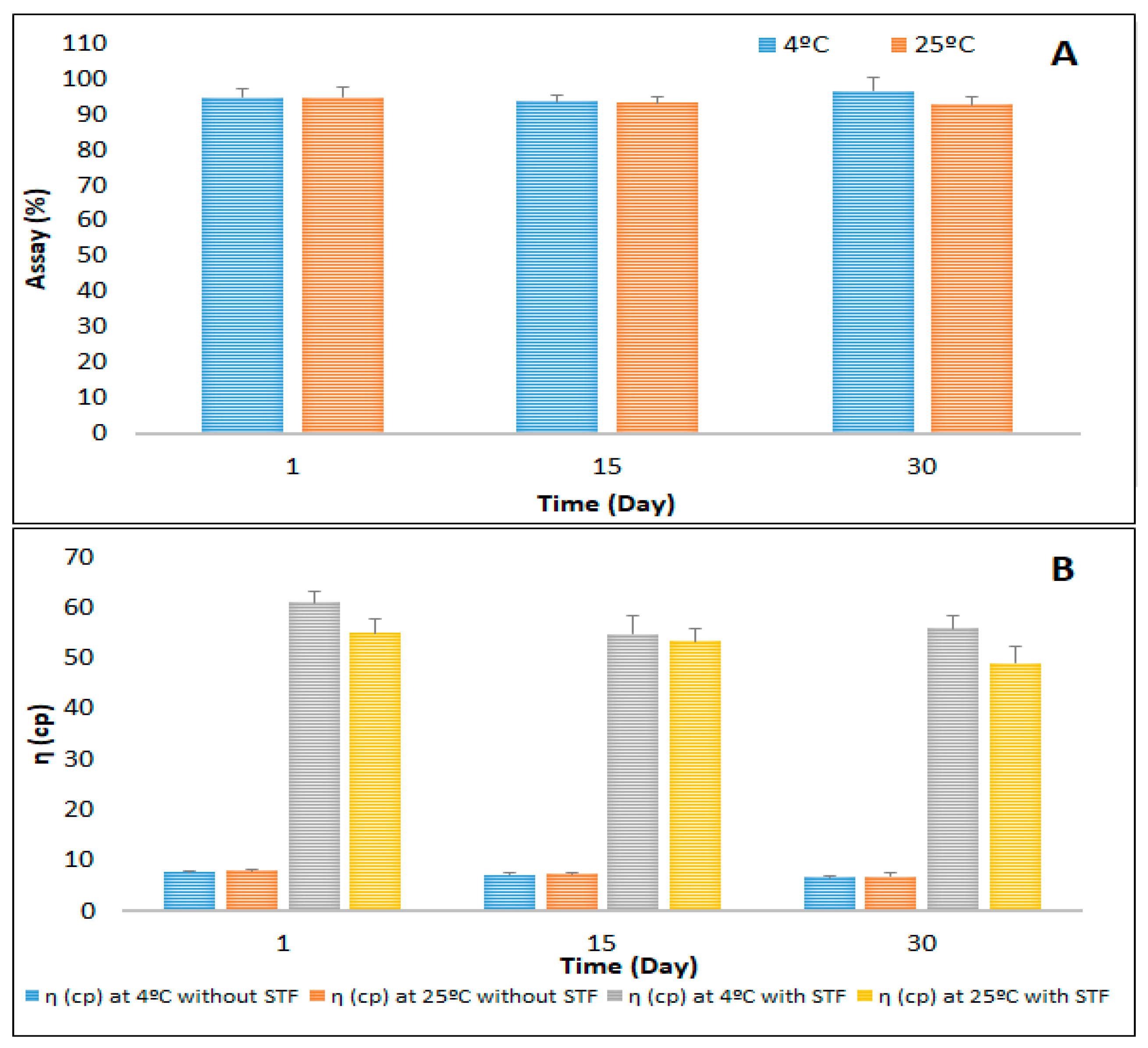
3.6. Stability Studies of the Optimized CIP-NLC-IG
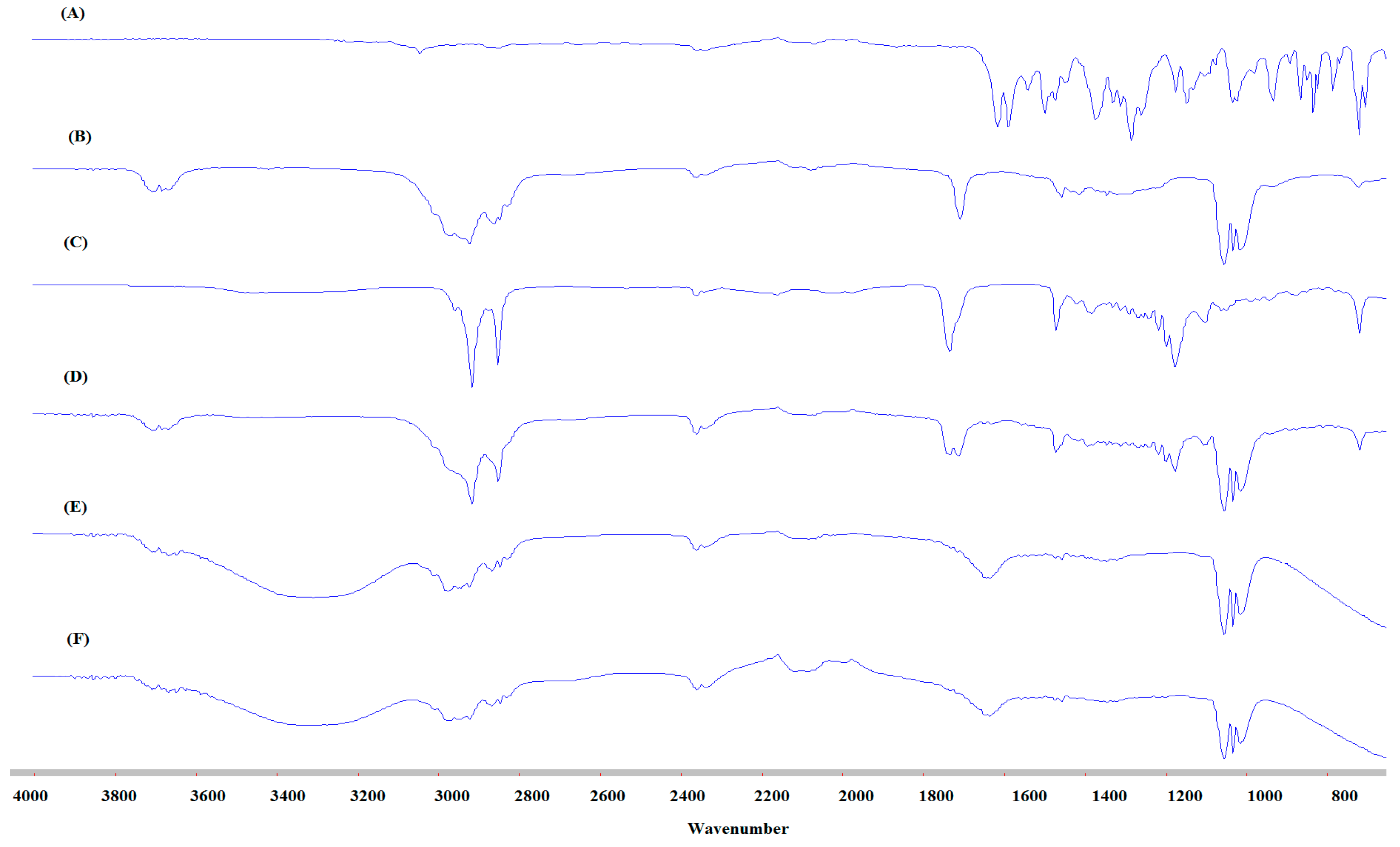
3.7. FTIR Studies
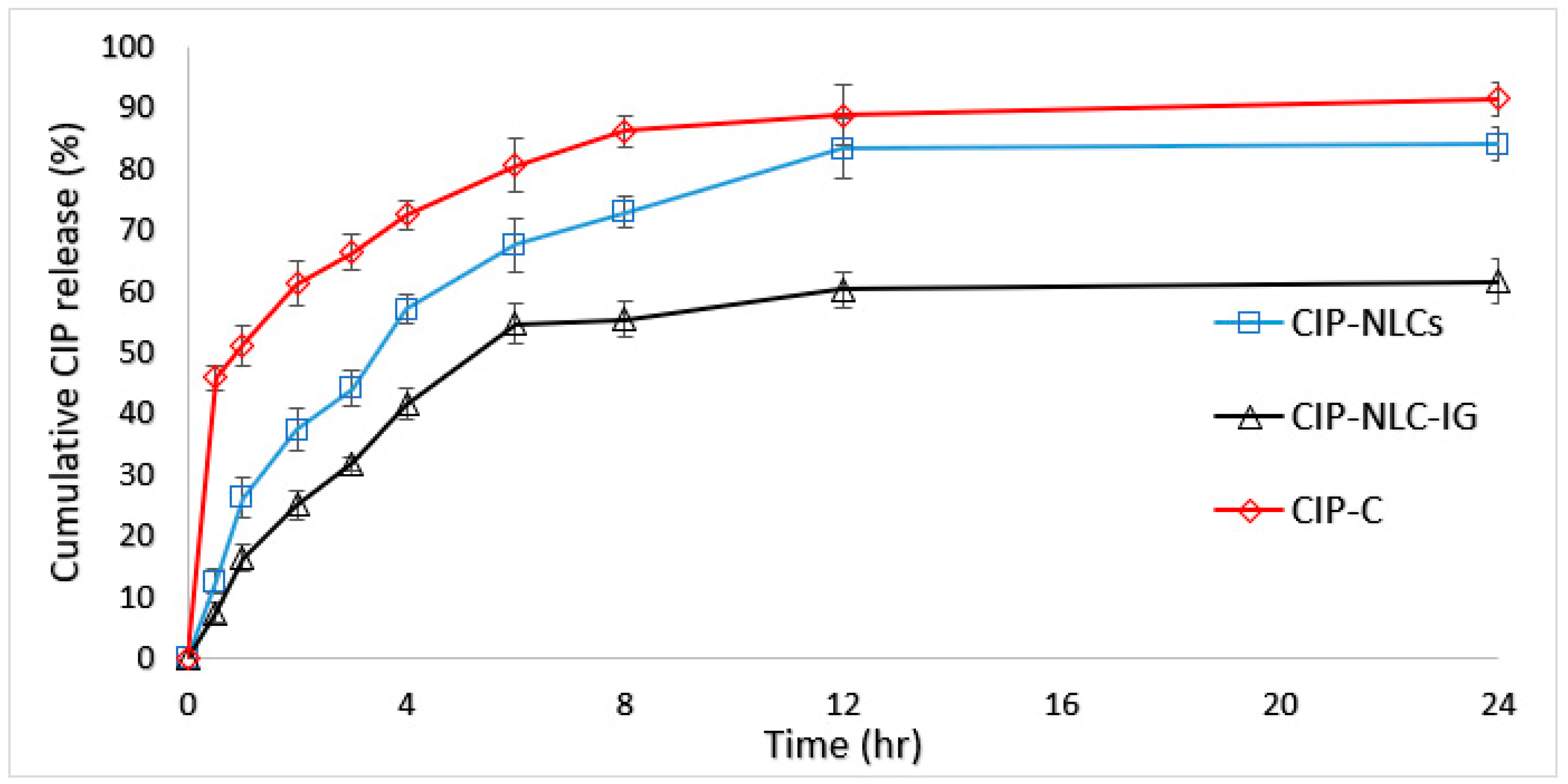
3.8. In Vitro Release Studies
3.9. Ex Vivo Transcorneal Permeation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Durand, M.L. Bacterial and Fungal Endophthalmitis. Clin. Microbiol. Rev. 2017, 30, 597–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertino, J.S. Impact of antibiotic resistance in the management of ocular infections: The role of current and future antibiotics. Clin. Ophthalmol. Auckl. NZ 2009, 3, 507–521. [Google Scholar] [CrossRef] [Green Version]
- Callegan, M.C.; Gilmore, M.S.; Gregory, M.; Ramadan, R.T.; Wiskur, B.J.; Moyer, A.L.; Hunt, J.J.; Novosad, B.D. Bacterial endophthalmitis: Therapeutic challenges and host-pathogen interactions. Prog. Retin. Eye Res. 2007, 26, 189–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simunovic, M.P.; Rush, R.B.; Hunyor, A.P.; Chang, A.A. Endophthalmitis following intravitreal injection versus endophthalmitis following cataract surgery: Clinical features, causative organisms and post-treatment outcomes. Br. J. Ophthalmol. 2012, 96, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Assaad, D.; Wong, D.; Mikhail, M.; Tawfik, S.; Altomare, F.; Berger, A.; Chow, D.; Giavedoni, L. Bacterial endophthalmitis: 10-year review of the culture and sensitivity patterns of bacterial isolates. Can. J. Ophthalmol. 2015, 50, 433–437. [Google Scholar] [CrossRef] [PubMed]
- The Incidence of Endophthalmitis after Cataract Surgery among the U.S. Medicare Population Increased between 1994 and 2001-Science Direct. Available online: https://www.sciencedirect.com/science/article/pii/S0161642005004549 (accessed on 14 February 2020).
- Sulkes, D.J.; Scott, I.U.; Flynn, H.W.; Feuer, W.J. Evaluating outpatient versus inpatient costs in endophthalmitis management. Retina 2002, 22, 747–751. [Google Scholar] [CrossRef]
- Clarke, B.; Williamson, T.H.; Gini, G.; Gupta, B. Management of bacterial postoperative endophthalmitis and the role of vitrectomy. Surv. Ophthalmol. 2018, 63, 677–693. [Google Scholar] [CrossRef]
- Group, E.V.S. Results of the Endophthalmitis Vitrectomy Study. A randomized trial of immediate vitrectomy and of intravenous antibiotics for the treatment of postoperative bacterial endophthalmitis. Arch. Ophthalmol. 1995, 113, 1479–1496. [Google Scholar]
- Moshirfar, M.; Feiz, V.; Vitale, A.T.; Wegelin, J.A.; Basavanthappa, S.; Wolsey, D.H. Endophthalmitis after Uncomplicated Cataract Surgery with the Use of Fourth-Generation Fluoroquinolones. Ophthalmology 2007, 114, 686–691. [Google Scholar] [CrossRef]
- Kelkar, A.; Kelkar, J.; Amuaku, W.; Kelkar, U.; Shaikh, A. How to prevent endophthalmitis in cataract surgeries? Indian J. Ophthalmol. 2008, 56, 403–407. [Google Scholar] [CrossRef]
- ESCRS Endophthalmitis Study Group. Prophylaxis of postoperative endophthalmitis following cataract surgery: Results of the ESCRS multicenter study and identification of risk factors. J. Cataract Refract. Surg. 2007, 33, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, R.P.; Dhaliwal, D.K. Ocular bacterial infections: Current and future treatment options. Expert Rev. Anti Infect. Ther. 2005, 3, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Miller, D. Update on the Epidemiology and Antibiotic Resistance of Ocular Infections. Middle East Afr. J. Ophthalmol. 2017, 24, 30–42. [Google Scholar] [CrossRef]
- Davis, R.; Markham, A.; Balfour, J.A. Ciprofloxacin. Drugs 1996, 51, 1019–1074. [Google Scholar] [CrossRef] [PubMed]
- Hyndiuk, R.A.; Eiferman, R.A.; Caldwell, D.R.; Rosenwasser, G.O.; Santos, C.I.; Katz, H.R.; Badrinath, S.S.; Reddy, M.K.; Adenis, J.-P.; Klauss, V.; et al. Comparison of Ciprofloxacin Ophthalmic Solution 0.3% to Fortified Tobramycin-Cefazolin in Treating Bacterial Corneal Ulcers. Ophthalmology 1996, 103, 1854–1863. [Google Scholar] [CrossRef]
- Liu, C.; Ji, J.; Li, S.; Wang, Z.; Tang, L.; Cao, W.; Sun, X. Microbiological Isolates and Antibiotic Susceptibilities: A 10-Year Review of Culture-Proven Endophthalmitis Cases. Curr. Eye Res. 2017, 42, 443–447. [Google Scholar] [CrossRef]
- Friedlaender, M.H. A review of the causes and treatment of bacterial and allergic conjunctivitis. Clin. Ther. 1995, 17, 800–810. [Google Scholar] [CrossRef]
- Balguri, S.P.; Adelli, G.R.; Janga, K.Y.; Bhagav, P.; Majumdar, S. Ocular disposition of ciprofloxacin from topical, PEGylated nanostructured lipid carriers: Effect of molecular weight and density of poly (ethylene) glycol. Int. J. Pharm. 2017, 529, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Deramo, V.A.; Lai, J.C.; Fastenberg, D.M.; Udell, I.J. Acute Endophthalmitis in Eyes Treated Prophylactically with Gatifloxacin and Moxifloxacin. Am. J. Ophthalmol. 2006, 142, 721–725. [Google Scholar] [CrossRef]
- Benz, M.S.; Scott, I.U.; Flynn, H.W.; Miller, D. In Vitro Susceptibilities to Antimicrobials of Pathogens Isolated from the Vitreous Cavity of Patients with Endophthalmitis. Invest. Ophthalmol. Vis. Sci. 2002, 43, 4428. [Google Scholar]
- Patel, A.; Cholkar, K.; Agrahari, V.; Mitra, A.K. Ocular drug delivery systems: An overview. World J. Pharmacol. 2013, 2, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Adelli, G.R.; Balguri, S.P.; Punyamurthula, N.; Bhagav, P.; Majumdar, S. Development and evaluation of prolonged release topical indomethacin formulations for ocular inflammation. Invest. Ophthalmol. Vis. Sci. 2014, 55, 463. [Google Scholar]
- Balguri, S.P.; Adelli, G.R.; Majumdar, S. Topical ophthalmic lipid nanoparticle formulations (SLN, NLC) of indomethacin for delivery to the posterior segment ocular tissues. Eur. J. Pharm. Biopharm. 2016, 109, 224–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janga, K.Y.; Tatke, A.; Dudhipala, N.; Balguri, S.P.; Ibrahim, M.M.; Maria, D.N.; Jablonski, M.M.; Majumdar, S. Gellan Gum Based Sol-to-Gel Transforming System of Natamycin Transfersomes Improves Topical Ocular Delivery. J. Pharmacol. Exp. Ther. 2019, 370, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Patil, H.; Feng, X.; Ye, X.; Majumdar, S.; Repka, M.A. Continuous Production of Fenofibrate Solid Lipid Nanoparticles by Hot-Melt Extrusion Technology: A Systematic Study Based on a Quality by Design Approach. AAPS J. 2015, 17, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Tatke, A.; Dudhipala, N.; Janga, K.Y.; Soneta, B.; Avula, B.; Majumdar, S. Melt-Cast Films Significantly Enhance Triamcinolone Acetonide Delivery to the Deeper Ocular Tissues. Pharmaceutics 2019, 11, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, A.; Lakhani, P.; Taskar, P.; Wu, K.-W.; Sweeney, C.; Avula, B.; Wang, Y.-H.; Khan, I.A.; Majumdar, S. Formulation Development, Optimization, and In Vitro–In Vivo Characterization of Natamycin-Loaded PEGylated Nano-Lipid Carriers for Ocular Applications. J. Pharm. Sci. 2018, 107, 2160–2171. [Google Scholar] [CrossRef]
- Tatke, A.; Dudhipala, N.; Janga, K.Y.; Balguri, S.P.; Avula, B.; Jablonski, M.M.; Majumdar, S. In Situ Gel of Triamcinolone Acetonide-Loaded Solid Lipid Nanoparticles for Improved Topical Ocular Delivery: Tear Kinetics and Ocular Disposition Studies. Nanomaterials 2019, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Yoon, G.; Park, J.W.; Yoon, I.-S. Solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs): Recent advances in drug delivery. J. Pharm. Investig. 2013, 43, 353–362. [Google Scholar] [CrossRef]
- Dudhipala, N. A comprehensive review on solid lipid nanoparticles as delivery vehicle for enhanced pharmacokinetic and pharmacodynamic activity of poorly soluble drugs. Int. J. Pharm. Sci. Nanotechol. 2019, 12, 4421–4440. [Google Scholar]
- Lakhani, P.; Patil, A.; Majumdar, S. Recent advances in topical nano drug-delivery systems for the anterior ocular segment. Ther. Deliv. 2018, 9, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.C.; Yang, H. Hydrogel-based ocular drug delivery systems: Emerging fabrication strategies, applications, and bench-to-bedside manufacturing considerations. J. Controlled Release 2019, 306, 29–39. [Google Scholar] [CrossRef]
- Üstündağ Okur, N.; Yozgatlı, V.; Okur, M.E.; Yoltaş, A.; Siafaka, P.I. Improving therapeutic efficacy of voriconazole against fungal keratitis: Thermo-sensitive in situ gels as ophthalmic drug carriers. J. Drug Deliv. Sci. Technol. 2019, 49, 323–333. [Google Scholar] [CrossRef]
- Hosny, K.M. Ciprofloxacin as Ocular Liposomal Hydrogel. AAPS PharmSciTech 2010, 11, 241–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, A.; Lakhani, P.; Taskar, P.; Avula, B.; Majumdar, S. Carboxyvinyl Polymer and Guar-Borate Gelling System Containing Natamycin Loaded PEGylated Nanolipid Carriers Exhibit Improved Ocular Pharmacokinetic Parameters. J. Ocul. Pharmacol. Ther. 2020. [Google Scholar] [CrossRef]
- Janga, K.Y.; Tatke, A.; Balguri, S.P.; Lamichanne, S.P.; Ibrahim, M.M.; Maria, D.N.; Jablonski, M.M.; Majumdar, S. Ion sensitive in situ hydrogels of natamycin bilosomes for enhanced and prolonged ocular pharmacotherapy: In vitro permeability, cytotoxicity and in vivo evaluation. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1039–1050. [Google Scholar] [CrossRef] [Green Version]
- Imre, S.; Dogaru, M.T.; Vari, C.E.; Muntean, T.; Kelemen, L. Validation of an HPLC method for the determination of ciprofloxacin in human plasma. J. Pharm. Biomed. Anal. 2003, 33, 125–130. [Google Scholar] [CrossRef]
- Dudhipala, N.; Veerabrahma, K. Candesartan cilexetil loaded solid lipid nanoparticles for oral delivery: Characterization, pharmacokinetic and pharmacodynamic evaluation. Drug Deliv. 2016, 23, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Dudhipala, N.; Gorre, T. Neuroprotective Effect of Ropinirole Lipid Nanoparticles Enriched Hydrogel for Parkinson’s Disease: In Vitro, Ex Vivo, Pharmacokinetic and Pharmacodynamic Evaluation. Pharmaceutics 2020, 12, 448. [Google Scholar] [CrossRef]
- Poonia, N.; Kharb, R.; Lather, V.; Pandita, D. Nanostructured lipid carriers: Versatile oral delivery vehicle. Future Sci. OA 2016, 2. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Iqbal, S.; Zhao, Z. Preparation of Ergosterol-Loaded Nanostructured Lipid Carriers for Enhancing Oral Bioavailability and Antidiabetic Nephropathy Effects. AAPS PharmSciTech 2020, 21, 64. [Google Scholar] [CrossRef]
- Tirumalesh, C.; Suram, D.; Dudhipala, N.; Banala, N. Enhanced pharmacokinetic activity of Zotepine via nanostructured lipid carrier system in Wistar rats for oral application. Pharm. Nanotechnol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhang, Y.; Li, D.; Chen, Y.; Sun, J.; Kong, F. Transferrin-modified nanostructured lipid carriers as multifunctional nanomedicine for codelivery of DNA and doxorubicin. Int. J. Nanomed. 2014, 9, 4107–4116. [Google Scholar] [CrossRef] [Green Version]
- Khames, A.; Khaleel, M.A.; El-Badawy, M.F.; El-Nezhawy, A.O.H. Natamycin solid lipid nanoparticles–sustained ocular delivery system of higher corneal penetration against deep fungal keratitis: Preparation and optimization. Int. J. Nanomed. 2019, 14, 2515–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javadzadeh, Y.; Ahadi, F.; Davaran, S.; Mohammadi, G.; Sabzevari, A.; Adibkia, K. Preparation and physicochemical characterization of naproxen–PLGA nanoparticles. Colloids Surf. B Biointerfaces 2010, 81, 498–502. [Google Scholar] [CrossRef]
- Ghadiri, M.; Fatemi, S.; Vatanara, A.; Doroud, D.; Najafabadi, A.R.; Darabi, M.; Rahimi, A.A. Loading hydrophilic drug in solid lipid media as nanoparticles: Statistical modeling of entrapment efficiency and particle size. Int. J. Pharm. 2012, 424, 128–137. [Google Scholar] [CrossRef]
- Araújo, J.; Gonzalez-Mira, E.; Egea, M.A.; Garcia, M.L.; Souto, E.B. Optimization and physicochemical characterization of a triamcinolone acetonide-loaded NLC for ocular antiangiogenic applications. Int. J. Pharm. 2010, 393, 167–175. [Google Scholar] [CrossRef]
- Foster, K.A.; Yazdanian, M.; Audus, K.L. Microparticulate uptake mechanisms of in-vitro cell culture models of the respiratory epithelium. J. Pharm. Pharmacol. 2001, 53, 57–66. [Google Scholar] [CrossRef]
- Desai, M.P.; Labhasetwar, V.; Walter, E.; Levy, R.J.; Amidon, G.L. The Mechanism of Uptake of Biodegradable Microparticles in Caco-2 Cells Is Size Dependent. Pharm. Res. 1997, 14, 1568–1573. [Google Scholar] [CrossRef]
- Kulkarni, S.A.; Feng, S.-S. Effects of Particle Size and Surface Modification on Cellular Uptake and Biodistribution of Polymeric Nanoparticles for Drug Delivery. Pharm. Res. 2013, 30, 2512–2522. [Google Scholar] [CrossRef]
- Dudhipala, N.; Veerabrahma, K. Improved anti-hyperlipidemic activity of Rosuvastatin Calcium via lipid nanoparticles: Pharmacokinetic and pharmacodynamic evaluation. Eur. J. Pharm. Biopharm. 2017, 110, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Butreddy, A.; Dudhipala, N.; Veerabrahma, K. Development of olmesartan medoxomil lipid-based nanoparticles and nanosuspension: Preparation, characterization and comparative pharmacokinetic evaluation. Artif. Cells Nanomed. Biotechnol. 2018, 46, 126–137. [Google Scholar] [CrossRef] [Green Version]
- zur Mühlen, A.; Schwarz, C.; Mehnert, W. Solid lipid nanoparticles (SLN) for controlled drug delivery–Drug release and release mechanism. Eur. J. Pharm. Biopharm. 1998, 45, 149–155. [Google Scholar] [CrossRef]
- Dudhipala, N.; Janga, K.Y.; Gorre, T. Comparative study of nisoldipine-loaded nanostructured lipid carriers and solid lipid nanoparticles for oral delivery: Preparation, characterization, permeation and pharmacokinetic evaluation. Artif. Cells Nanomed. Biotechnol. 2018, 46, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Sood, A.; Mehta, V.; Malairaman, U. Formulation and physicochemical evaluation of nanostructured lipid carrier for codelivery of clotrimazole and ciprofloxacin. Asian J. Pharm. Clin. Res. 2016, 356–360. [Google Scholar]
- Uddin, M.S.; Mamun, A.A.; Kabir, M.T.; Setu, J.R.; Zaman, S.; Begum, Y.; Amran, M.S. Quality Control Tests for Ophthalmic Pharmaceuticals: Pharmacopoeial Standards and Specifications. J. Adv. Med. Pharm. Sci. 2017, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Morsi, N.; Ibrahim, M.; Refai, H.; El Sorogy, H. Nanoemulsion-based electrolyte triggered in situ gel for ocular delivery of acetazolamide. Eur. J. Pharm. Sci. 2017, 104, 302–314. [Google Scholar] [CrossRef]
- Lee, D.H.; Condrate, R.A. FTIR spectral characterization of thin film coatings of oleic acid on glasses: I. Coatings on glasses from ethyl alcohol. J. Mater. Sci. 1999, 34, 139–146. [Google Scholar]
- Yang, K.; Peng, H.; Wen, Y.; Li, N. Re-examination of characteristic FTIR spectrum of secondary layer in bilayer oleic acid-coated Fe3O4 nanoparticles. Appl. Surf. Sci. 2010, 256, 3093–3097. [Google Scholar] [CrossRef]
- Deore, R.; Kavitha, K.; Tamizhmani, T. Preparation and Evaluation of Sustained Release Matrix Tablets of Tramadol Hydrochloride Using Glyceryl Palmitostearate. Trop. J. Pharm. Res. 2010, 9. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, S.; Chakraborti, C.K.; Mishra, S.C. Qualitative analysis of controlled release ciprofloxacin/carbopol 934 mucoadhesive suspension. J. Adv. Pharm. Technol. Res. 2011, 2, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Palem, C.R.; Gannu, R.; Doodipala, N.; Yamsani, V.V.; Yamsani, M.R. Transmucosal.delivery of domperidone from bilayered buccal patches: In vitro, ex vivo and in vivo characterization. Arch. Pharm. Res. 2011, 34, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Baig, M.S.; Ahad, A.; Aslam, M.; Imam, S.S.; Aqil, M.; Ali, A. Application of Box–Behnken design for preparation of levofloxacin-loaded stearic acid solid lipid nanoparticles for ocular delivery: Optimization, in vitro release, ocular tolerance, and antibacterial activity. Int. J. Biol. Macromol. 2016, 85, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Narendar, D.; Someshwar, K.; Arjun, N.; Madhusudan, R.Y. Quality by design approach.for development and optimization of Quetiapine Fumarate effervescent floating matrix tablets.for improved oral delivery. J. Pharm. Investi. 2016, 46, 253–263. [Google Scholar] [CrossRef]
- Sharma, D.; Maheshwari, D.; Philip, G.; Rana, R.; Bhatia, S.; Singh, M.; Gabrani, R.; Sharma, .S.K.; Ali, J.; Sharma, R.K.; et al. Formulation and Optimization of Polymeric Nanoparticles for.Intranasal Delivery of Lorazepam Using Box-Behnken Design: In Vitro and In Vivo Evaluation. BioMed. Res. Int. 2014, 2014, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Bhaskar, K.; Anbu, J.; Ravichandiran, V.; Venkateswarlu, V.; Rao, Y.M. Lipid nanoparticles for transdermal delivery of flurbiprofen: Formulation, in vitro, ex vivo and in vivo studies. Lipids Health Dis. 2009, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Palem, C.R.; Dudhipala, N.; Battu, S.K.; Goda, S.; Repka, M.A.; Yamsani, M.R. Combined.dosage form of pioglitazone and felodipine as mucoadhesive pellets via hot melt extrusion for.improved buccal delivery with application of quality by design approach. J. Drug Deliv. Sci. Technol. 2015, 30, 209–219. [Google Scholar] [CrossRef]
- Palem, C.R.; Dudhipala, N.; Battu, S.K.; Goda, S.; Repka, M.A.; Yamsani, M.R. Development, optimization and in vivo characterization of domperidone controlled release hot.melt extruded films for buccal delivery. Drug Dev. Ind. Pharm. 2016, 42, 473–484. [Google Scholar] [CrossRef]
- Narendar, D.; Kishan, V. Pharmacokinetic and pharmacodynamic studies of nisoldipine-loaded.solid lipid nanoparticles developed by central composite design. Drug Dev. Ind. Pharm. 2015, 41, 1968–1977. [Google Scholar]
- Narendar, D.; Karthikyadav, J. Lipid nanoparticles of zaleplon for improved oral delivery by.Box-Behnken design: Optimization, in vitro and in vivo evaluation. Drug Dev. Ind. Pharm. 2017, 43, 1205–1214. [Google Scholar]
- Gan, L.; Wang, J.; Jiang, M.; Bartlett, H.; Ouyang, D.; Eperjesi, F.; Liu, J.; Gan, Y. Recent advances in topical ophthalmic drug delivery with lipid-based nanocarriers. Drug Discov. Today 2013, 18, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Seyfoddin, A.; Al-Kassas, R. Development of solid lipid nanoparticles and nanostructured lipid carriers for improving ocular delivery of acyclovir. Drug Dev. Ind. Pharm. 2013, 39, 508–519. [Google Scholar] [CrossRef]
- Alany, R.G.; Rades, T.; Nicoll, J.; Tucker, I.G.; Davies, N.M. W/O microemulsions for ocular delivery: Evaluation of ocular irritation and precorneal retention. J. Control. Rel. 2006, 111, 145–152. [Google Scholar] [CrossRef]
- Kou, L.; Sun, J.; Zhai, Y.; He, Z. The endocytosis and intracellular fate of nanomedicines: Implication for rational design. Asian J. Pharm. Sci. 2013, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Morris, E.R.; Nishinari, K.; Rinaudo, M. Gelation of gellan–A review. Food Hydrocoll. 2012, 28, 373–411. [Google Scholar] [CrossRef]
- Yu, S.; Li, Q.; Li, Y.; Wang, H.; Liu, D.; Yang, X.; Pan, W. A novel hydrogel with dual temperature and pH responsiveness based on a nanostructured lipid carrier as an ophthalmic delivery system: Enhanced trans-corneal permeability and bioavailability of nepafenac. New J. Chem. 2017, 41, 3920–3929. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solid/Liquid-Lipid | Lipid | Solubility |
|---|---|---|
| Solid | Precirol® ATO 5 | (+) |
| Compritol® 888 ATO | (−) | |
| DynasanTM 114 | (−) | |
| GeleolTM | (−) | |
| GelucireTM 43/01 | (−) | |
| DynasanTM 116 | (−) | |
| GelucireTM 50/13 | (−) | |
| GelucireTM 44/14 | (−) | |
| Softisan 154 | (−) | |
| Liquid | soybean oil | (−) |
| Captex® 355 EP | (−) | |
| castor oil | (−) | |
| sesame oil | (−) | |
| Maisine® CC | (−) | |
| Miglyol® 829 | (−) | |
| Oleic acid | (+) | |
| Capryol 90TM | (−) | |
| Olive oil | (−) |
| Formulation Composition (%w/v) | CIP-NLC | CIP-NLC-IG |
|---|---|---|
| Ciprofloxacin | 0.1 | 0.1 |
| Precirol® ATO 5 | 3 | 3 |
| Oleic acid | 1 | 1 |
| Tween® 80 | 2 | 2 |
| Poloxamer 188 | 0.25 | 0.25 |
| Glycerin | 2.25 | 2.25 |
| Gellan gum | - | 0.2 |
| Water | Up to 10 mL | Up to 10 mL |
| Composition (%w/v) | O-NLC-75 | O-NLC-100 | O-NLC-150 | O-NLC-200 |
|---|---|---|---|---|
| Precirol® ATO 5 | 3 | 3 | 3 | 3 |
| Oleic acid | 1 | 1 | 1 | 1 |
| Tween® 80 | 0.75 | 1 | 1.5 | 2 |
| Poloxamer 188 | 0.25 | 0.25 | 0.25 | 0.25 |
| Glycerin | 2.25 | 2.25 | 2.25 | 2.25 |
| Water | Up to 10 mL | Up to 10 mL | Up to 10 mL | Up to 10 mL |
| PS (nm) | 384.4 ± 7.9 | 291.6 ± 10.4 | 211.7 ± 4.5 | 142.3 ± 3.9 |
| PDI | 0.41 ± 0.06 | 0.42 ± 0.08 | 0.39 ± 0.02 | 0.38 ± 0.01 |
| ZP (mV) | −28.5 ± 1.5 | −29.6 ± 0.7 | −31.4 ± 1.3 | −27.1 ± 1.7 |
| Formulation Composition (%w/v) | O-NLC-200 | O-NLC |
|---|---|---|
| Precirol® ATO 5 | 3 | 4.5 |
| Oleic acid | 1 | 1.5 |
| Tween® 80 | 2 | 2 |
| Poloxamer 188 | 0.25 | 0.25 |
| Glycerin | 2.25 | 2.25 |
| Water | Up to 10 mL | Up to 10 mL |
| PS (nm) | 142.3 ± 3.9 | 226.0 ± 11.2 |
| PDI | 0.38 ± 0.01 | 0.60 ± 0.05 |
| ZP (mV) | −27.1 ± 1.7 | −27.8 ± 0.5 |
| Formulation | Gellan Gum (%w/v) | GT | GRT (h) | η (cP) Without STF | η (cP) With STF | Drug Content (%) |
|---|---|---|---|---|---|---|
| CIP-NLC-IG2 | 0.2 | Immediate | >24 | 7.9 ± 0.2 | 55 ± 2.7 | 94.8 ± 2.4 |
| CIP-NLC-IG3 | 0.3 | Immediate | >24 | 25.4 ± 2.3 | 110 ± 3.5 | 83.1 ± 1.8 |
| CIP-NLC-IG4 | 0.4 | Immediate | >24 | 93.4 ± 4.6 | 211.9 ± 8.7 | 77.4 ± 2.0 |
| Model | Equation | R2 Value | ||
|---|---|---|---|---|
| CIP-C | CIP-NLC | CIP-NLC-IG2 | ||
| Zero-order | Q0 − Q = kt | 0.697 | 0.930 | 0.976 |
| First order | ln Q = kt | 0.797 | 0.984 | 0.995 |
| Higuchi’s matrix | Q0 − Q = kt1/2 | 0.927 | 0.986 | 0.969 |
| Korsemeyer–Peppas | log (Q0 − Q) = n log t + log k | 0.99 | 0.995 | 0.998 |
| Formulation | Flux (µg/min/cm2) | Permeability (×10−5 cm/min) | Fold Enhancement with CIP-C | |
|---|---|---|---|---|
| Flux | p | |||
| CIP-C | 0.04 ± 0.01 | 2.3 ± 0.8 | - | - |
| CIP-NLC | 0.16 ± 0.01 # | 8.1 ± 0.4 # | 4 | 3.5 |
| CIP-NLC-IG2 | 0.09 ± 0.01 # | 4.4 ± 0.4 # | 2.2 | 1.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Youssef, A.; Dudhipala, N.; Majumdar, S. Ciprofloxacin Loaded Nanostructured Lipid Carriers Incorporated into In-Situ Gels to Improve Management of Bacterial Endophthalmitis. Pharmaceutics 2020, 12, 572. https://doi.org/10.3390/pharmaceutics12060572
Youssef A, Dudhipala N, Majumdar S. Ciprofloxacin Loaded Nanostructured Lipid Carriers Incorporated into In-Situ Gels to Improve Management of Bacterial Endophthalmitis. Pharmaceutics. 2020; 12(6):572. https://doi.org/10.3390/pharmaceutics12060572
Chicago/Turabian StyleYoussef, Ahmed, Narendar Dudhipala, and Soumyajit Majumdar. 2020. "Ciprofloxacin Loaded Nanostructured Lipid Carriers Incorporated into In-Situ Gels to Improve Management of Bacterial Endophthalmitis" Pharmaceutics 12, no. 6: 572. https://doi.org/10.3390/pharmaceutics12060572
APA StyleYoussef, A., Dudhipala, N., & Majumdar, S. (2020). Ciprofloxacin Loaded Nanostructured Lipid Carriers Incorporated into In-Situ Gels to Improve Management of Bacterial Endophthalmitis. Pharmaceutics, 12(6), 572. https://doi.org/10.3390/pharmaceutics12060572