N-Alkylisatin-Loaded Liposomes Target the Urokinase Plasminogen Activator System in Breast Cancer


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Liposome Preparation and Characterization
2.2. Cell Lines and uPA and uPAR Expression
2.3. Assessment of Cellular Uptake and Localization of Liposomes
2.4. 3D multicellular Tumor Spheroid Cytotoxicity Assays
2.5. Pharmacokinetics and Biodistribution of N-AI PAI-2 Liposomes in Mice
2.6. Toxicology of N-AI Liposomes in Mice
2.7. Data Analysis
3. Results
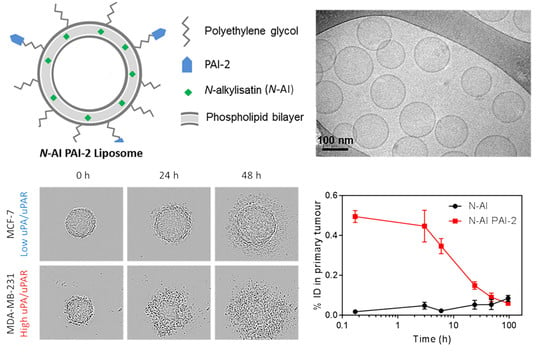
3.1. Preparation and Characterization of Liposomes
3.2. PAI-2 Liposomes Are Taken up by Cells through RME-Dependent and Non-Dependent Mechanisms
3.3. Cytotoxicity of N-AI PAI-2 Liposomes against Breast Cancer Cells
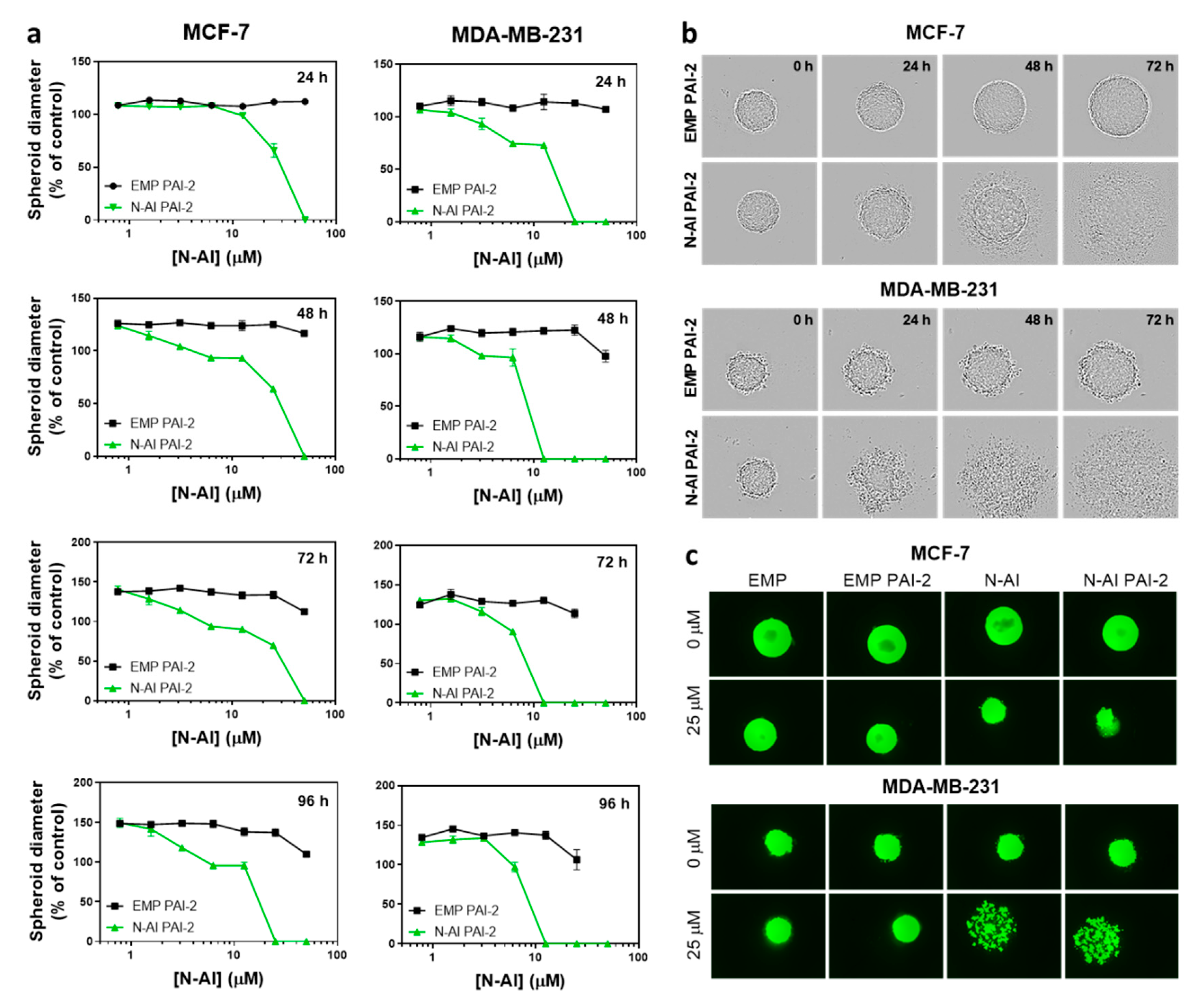
3.4. Cytotoxicity of N-AI PAI-2 Liposomes against Breast Cancer Spheroids
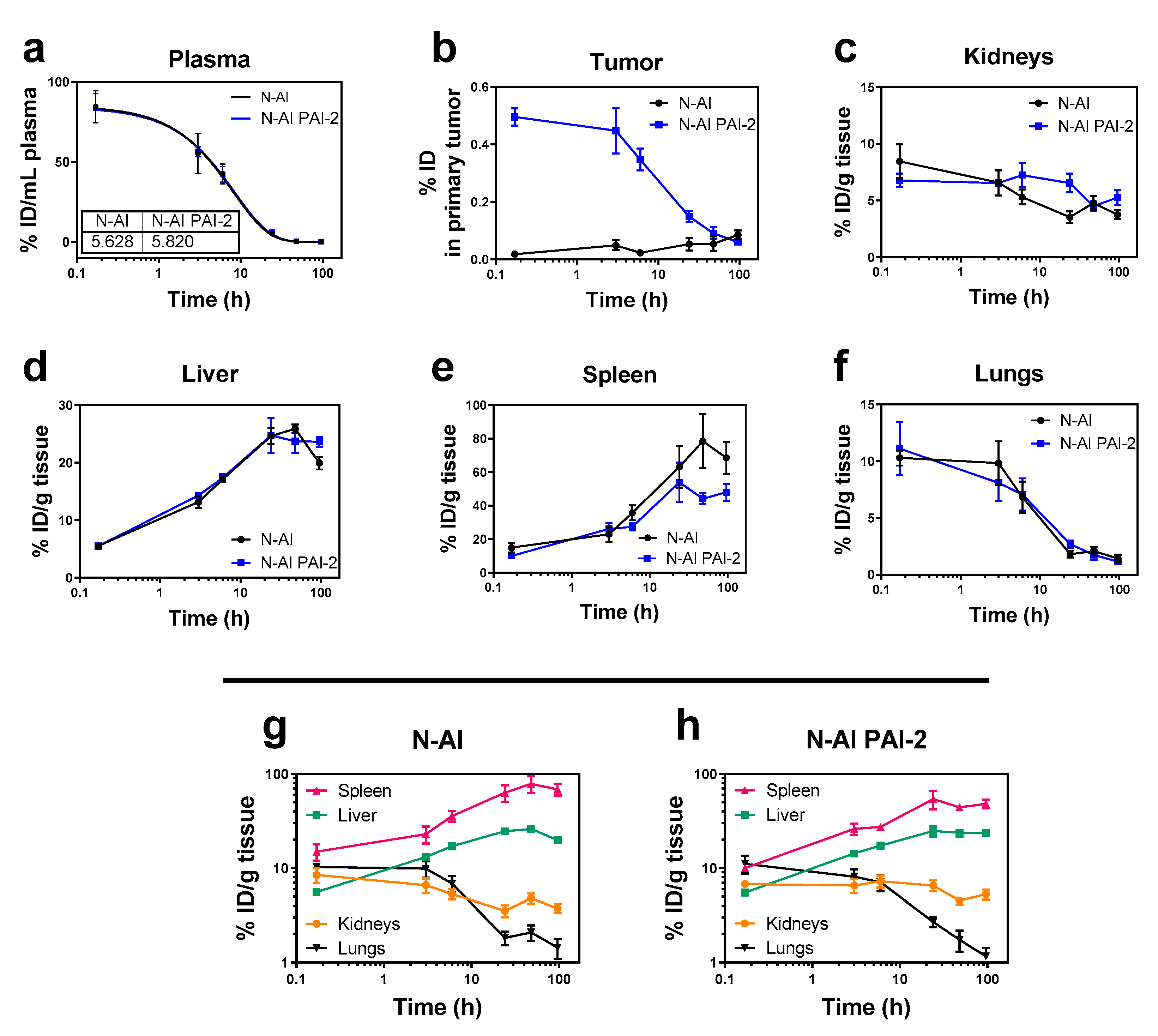
3.5. Pharmacokinetics and Biodistribution of N-AI PAI-2 Liposomes
3.6. Maximum Tolerated Dose of N-AI-Loaded Liposomes in Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Ward, E.M.; DeSantis, C.E.; Lin, C.C.; Kramer, J.L.; Jemal, A.; Kohler, B.; Brawley, O.W.; Gansler, T. Cancer statistics: Breast cancer in situ. CA Cancer J. Clin. 2015, 65, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulou, I.; Mylona, E.; Kapranou, A.; Mavrommatis, J.; Markaki, S.; Zoumbouli, C.; Keramopoulos, A.; Nakopoulou, L. The prognostic value of the topographic distribution of uPAR expression in invasive breast carcinomas. Cancer Lett. 2007, 246, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Indira Chandran, V.; Eppenberger-Castori, S.; Venkatesh, T.; Vine, K.L.; Ranson, M. HER2 and uPAR cooperativity contribute to metastatic phenotype of HER2-positive breast cancer. Oncoscience 2015, 2, 207–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranson, M.; Andronicos, N.M. Plasminogen binding and cancer: Promises and pitfalls. Front. Biosci. J. Virtual Libr. 2003, 8, s294–s304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, L.; Fritsche, H.; Mennel, R.; Norton, L.; Ravdin, P.; Taube, S.; Somerfield, M.R.; Hayes, D.F.; Bast, R.C. American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 5287–5312. [Google Scholar] [CrossRef] [Green Version]
- Duffy, M.J.; McGowan, P.M.; Harbeck, N.; Thomssen, C.; Schmitt, M. uPA and PAI-1 as biomarkers in breast cancer: Validated for clinical use in level-of-evidence-1 studies. Breast Cancer Res. 2014, 16, 428. [Google Scholar] [CrossRef] [Green Version]
- Urban, P.; Vuaroqueaux, V.; Labuhn, M.; Delorenzi, M.; Wirapati, P.; Wight, E.; Senn, H.J.; Benz, C.; Eppenberger, U.; Eppenberger-Castori, S. Increased expression of urokinase-type plasminogen activator mRNA determines adverse prognosis in ErbB2-positive primary breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 4245–4253. [Google Scholar] [CrossRef]
- Bianchi, E.; Cohen, R.L.; Thor, A.T.; Todd, R.F.; Mizukami, I.F.; Lawrence, D.A.; Ljung, B.M.; Shuman, M.A.; Smith, H.S. The urokinase receptor is expressed in invasive breast cancer but not in normal breast tissue. Cancer Res. 1994, 54, 861–866. [Google Scholar]
- Brungs, D.; Chen, J.; Aghmesheh, M.; Vine, K.L.; Becker, T.M.; Carolan, M.G.; Ranson, M. The urokinase plasminogen activation system in gastroesophageal cancer: A systematic review and meta-analysis. Oncotarget 2017, 8, 23099–23109. [Google Scholar] [CrossRef] [Green Version]
- Harris, N.L.E.; Vennin, C.; Conway, J.R.W.; Vine, K.L.; Pinese, M.; Cowley, M.J.; Shearer, R.F.; Lucas, M.C.; Herrmann, D.; Allam, A.H.; et al. SerpinB2 regulates stromal remodelling and local invasion in pancreatic cancer. Oncogene 2017, 36, 4288–4298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.C.; Mall, R.; Braselmann, H.; Feuchtinger, A.; Molatore, S.; Lindner, K.; Walch, A.; Gross, E.; Schmitt, M.; Falkenberg, N.; et al. uPAR enhances malignant potential of triple-negative breast cancer by directly interacting with uPA and IGF1R. BMC Cancer 2016, 16, 615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubele, M.; Huber, M.C.; Falkenberg, N.; Gross, E.; Braselmann, H.; Walch, A.K.; Schmitt, M. uPA receptor and its interaction partners: Impact as potential therapeutic targets in triple-negative breast cancer. J. Clin. Oncol. 2015, 33, 150. [Google Scholar] [CrossRef]
- Al-Mahmood, S.; Sapiezynski, J.; Garbuzenko, O.B.; Minko, T. Metastatic and triple-negative breast cancer: Challenges and treatment options. Drug Deliv. Transl. Res. 2018, 8, 1483–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keam, B.; Im, S.A.; Kim, H.J.; Oh, D.Y.; Kim, J.H.; Lee, S.H.; Chie, E.K.; Han, W.; Kim, D.W.; Moon, W.K.; et al. Prognostic impact of clinicopathologic parameters in stage II/III breast cancer treated with neoadjuvant docetaxel and doxorubicin chemotherapy: Paradoxical features of the triple negative breast cancer. BMC Cancer 2007, 7, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazar, A.P.; Ahn, R.W.; O’Halloran, T.V. Development of Novel Therapeutics Targeting the Urokinase Plasminogen Activator Receptor (uPAR) and Their Translation toward the Clinic. Curr. Pharm. Des. 2011, 17, 1970–1978. [Google Scholar] [CrossRef] [Green Version]
- Matthews, H. Synthesis and Biological Evaluation of Plasminogen Activation Inhibitors As Antitumour/Antimetastasis Agents. Ph.D. Thesis, University of Wollongong, Wollongong, Australia, 2011. [Google Scholar]
- Al-Ejeh, F.; Croucher, D.; Ranson, M. Kinetic analysis of plasminogen activator inhibitor type-2: Urokinase complex formation and subsequent internalisation by carcinoma cell lines. Exp. Cell Res. 2004, 297, 259–271. [Google Scholar] [CrossRef]
- Croucher, D.; Saunders, D.N.; Ranson, M. The urokinase/PAI-2 complex: A new high affinity ligand for the endocytosis receptor low density lipoprotein receptor-related protein. J. Biol. Chem. 2006, 281, 10206–10213. [Google Scholar] [CrossRef] [Green Version]
- Cochran, B.J.; Croucher, D.R.; Lobov, S.; Saunders, D.N.; Ranson, M. Dependence on endocytic receptor binding via a minimal binding motif underlies the differential prognostic profiles of SerpinE1 and SerpinB2 in cancer. J. Biol. Chem. 2011, 286, 24467–24475. [Google Scholar] [CrossRef] [Green Version]
- Stutchbury, T.K.; Al-Ejeh, F.; Stillfried, G.E.; Croucher, D.R.; Andrews, J.; Irving, D.; Links, M.; Ranson, M. Preclinical evaluation of 213Bi-labeled plasminogen activator inhibitor type 2 in an orthotopic murine xenogenic model of human breast carcinoma. Mol. Cancer Ther. 2007, 6, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Vine, K.L.; Locke, J.M.; Ranson, M.; Pyne, S.G.; Bremner, J.B. An Investigation into the Cytotoxicity and Mode of Action of Some Novel N-Alkyl-Substituted Isatins. J. Med. Chem. 2007, 50, 5109–5117. [Google Scholar] [CrossRef] [PubMed]
- Vine, K.L.; Belfiore, L.; Jones, L.; Locke, J.M.; Wade, S.; Minaei, E.; Ranson, M. N-alkylated isatins evade P-gp mediated efflux and retain potency in MDR cancer cell lines. Heliyon 2016, 2, e00060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keenan, B.; Finol-Urdaneta, R.K.; Hope, A.; Bremner, J.B.; Kavallaris, M.; Lucena-Agell, D.; Oliva, M.A.; Diaz, J.F.; Vine, K.L. N-alkylisatin-based microtubule destabilizers bind to the colchicine site on tubulin and retain efficacy in drug resistant acute lymphoblastic leukemia cell lines with less in vitro neurotoxicity. Cancer Cell Int. 2020, 20, 170. [Google Scholar] [CrossRef] [PubMed]
- Vine, K.L.; Chandran, V.I.; Locke, J.M.; Matesic, L.; Lee, J.; Skropeta, D.; Bremner, J.B.; Ranson, M. Targeting Urokinase and the Transferrin Receptor with Novel, Anti-Mitotic N-Alkylisatin Cytotoxin Conjugates Causes Selective Cancer Cell Death and Reduces Tumor Growth. Curr. Cancer Drug Targets 2012, 12, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, N.; Andrade, F.; Segovia, N.; Ferrer-Tasies, L.; Sala, S.; Veciana, J.; Ventosa, N. Lipid-based nanovesicles for nanomedicine. Chem. Soc. Rev. 2016, 45, 6520–6545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubernator, J. Active methods of drug loading into liposomes: Recent strategies for stable drug entrapment and increased in vivo activity. Expert Opin. Drug Deliv. 2011, 8, 565–580. [Google Scholar] [CrossRef]
- Uster, P.S.; Allen, T.M.; Daniel, B.E.; Mendez, C.J.; Newman, M.S.; Zhu, G.Z. Insertion of poly(ethylene glycol) derivatized phospholipid into pre-formed liposomes results in prolonged in vivo circulation time. FEBS Lett. 1996, 386, 243–246. [Google Scholar] [CrossRef] [Green Version]
- Messerschmidt, S.K.; Kolbe, A.; Muller, D.; Knoll, M.; Pleiss, J.; Kontermann, R.E. Novel single-chain Fv’ formats for the generation of immunoliposomes by site-directed coupling. Bioconj. Chem. 2008, 19, 362–369. [Google Scholar] [CrossRef]
- Belfiore, L.; Saunders, D.N.; Ranson, M.; Thurecht, K.J.; Storm, G.; Vine, K.L. Towards clinical translation of ligand-functionalized liposomes in targeted cancer therapy: Challenges and opportunities. J. Control. Release 2018, 277, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Liu, F.T.; Rabinovich, G.A. Galectins as modulators of tumour progression. Nat. Rev. Cancer 2005, 5, 29–41. [Google Scholar] [CrossRef]
- Belfiore, L.; Spenkelink, L.M.; Ranson, M.; van Oijen, A.M.; Vine, K.L. Quantification of ligand density and stoichiometry on the surface of liposomes using single-molecule fluorescence imaging. J. Control. Release 2018, 278, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Kessler, R.J.; Fanestil, D.D. Interference by lipids in the determination of protein using bicinchoninic acid. Anal. Biochem. 1986, 159, 138–142. [Google Scholar] [CrossRef]
- Friedrich, J.; Seidel, C.; Ebner, R.; Kunz-schughart, L.A. Spheroid-based drug screen: Considerations and practical approach. Nat. Protoc. 2009, 4, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Caujolle, F.M.; Caujolle, D.H.; Cros, S.B.; Calvet, M.M. Limits of toxic and teratogenic tolerance of dimethyl sulfoxide. Ann. N. Y. Acad. Sci. 1967, 141, 110–126. [Google Scholar] [CrossRef] [PubMed]
- Biological Actions and Medical Applications of Dimethyl Sulfoxide; Annals of the New York Academy of Sciences: New York, NY, USA, 1983; Volume 411, pp. 1–404.
- Smith, M.C.; Crist, R.M.; Clogston, J.D.; McNeil, S.E. Zeta potential: A case study of cationic, anionic, and neutral liposomes. Anal. Bioanal. Chem. 2017, 409, 5779–5787. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Doxil(R)—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Croucher, D.R.; Saunders, D.N.; Stillfried, G.E.; Ranson, M. A structural basis for differential cell signalling by PAI-1 and PAI-2 in breast cancer cells. Biochem. J. 2007, 408, 203–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safhi, M.M.; Sivakumar, S.M.; Jabeen, A.; Zakir, F.; Islam, F.; Anwer, T.; Bagul, U.S.; Elmobark, M.E.; Khan, G.; Siddiqui, R.; et al. Chapter 8—Nanoparticle System for Anticancer Drug Delivery: Targeting to Overcome Multidrug Resistance. In Multifunctional Systems for Combined Delivery, Biosensing and Diagnostics; Grumezescu, A.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 159–169. [Google Scholar] [CrossRef]
- Herda, L.M.; Hristov, D.R.; Lo Giudice, M.C.; Polo, E.; Dawson, K.A. Mapping of Molecular Structure of the Nanoscale Surface in Bionanoparticles. J. Am. Chem. Soc. 2017, 139, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Fiandra, L.; Alessio, G.; Mazzucchelli, S.; Nebuloni, M.; De Palma, C.; Kantner, K.; Pelaz, B.; Rotem, R.; Corsi, F.; et al. Tumour homing and therapeutic effect of colloidal nanoparticles depend on the number of attached antibodies. Nat. Commun. 2016, 7, 13818. [Google Scholar] [CrossRef] [PubMed]
- Faria, M.; Björnmalm, M.; Thurecht, K.J.; Kent, S.J.; Parton, R.G.; Kavallaris, M.; Johnston, A.P.R.; Gooding, J.J.; Corrie, S.R.; Boyd, B.J.; et al. Minimum information reporting in bio–nano experimental literature. Nat. Nanotechnol. 2018, 13, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Webb, D.J.; Jo, M.; Gonias, S.L. Endogenously produced urokinase-type plasminogen activator is a major determinant of the basal level of activated ERK/MAP kinase and prevents apoptosis in MDA-MB-231 breast cancer cells. J. Cell Sci. 2001, 114, 3387–3396. [Google Scholar] [PubMed]
- Ducat, E.; Evrard, B.; Peulen, O.; Piel, G. Cellular uptake of liposomes monitored by confocal microscopy and flow cytometry. J. Drug Deliv. Sci. Technol. 2011, 21, 469–477. [Google Scholar] [CrossRef]
- Willis, M.; Forssen, E. Ligand-targeted liposomes. Adv. Drug Deliv. Rev. 1998, 29, 249–271. [Google Scholar]
- Xiao, K.; Li, Y.; Luo, J.; Lee, J.S.; Xiao, W.; Gonik, A.M.; Agarwal, R.G.; Lam, K.S. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials 2011, 32, 3435–3446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cochran, B.J.; Gunawardhana, L.P.; Vine, K.L.; Lee, J.A.; Lobov, S.; Ranson, M. The CD-loop of PAI-2 (SERPINB2) is redundant in the targeting, inhibition and clearance of cell surface uPA activity. BMC Biotechnol. 2009, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- Conese, M.; Blasi, F. Urokinase/urokinase receptor system: Internalization/degradation of urokinase-serpin complexes: Mechanism and regulation. Biol. Chem. Hoppe-Seyler 1995, 376, 143–155. [Google Scholar]
- Ivascu, A.; Kubbies, M. Diversity of cell-mediated adhesions in breast cancer spheroids. Int. J. Oncol. 2007, 31, 1403–1413. [Google Scholar] [CrossRef] [Green Version]
- Nichols, J.W.; Bae, Y.H. EPR: Evidence and fallacy. J. Control. Release: Off. J. Control. Release Soc. 2014, 190, 451–464. [Google Scholar] [CrossRef]
- Krasnici, S.; Werner, A.; Eichhorn, M.E.; Schmitt-Sody, M.; Pahernik, S.A.; Sauer, B.; Schulze, B.; Teifel, M.; Michaelis, U.; Naujoks, K.; et al. Effect of the surface charge of liposomes on their uptake by angiogenic tumor vessels. Int. J. Cancer 2003, 105, 561–567. [Google Scholar] [CrossRef]
- Gabizon, A.; Isacson, R.; Libson, E.; Kaufman, B.; Uziely, B.; Catane, R.; Ben-Dor, C.G.; Rabello, E.; Cass, Y.; Peretz, T.; et al. Clinical Studies of Liposome-Encapsulated Doxorubicin. Acta Oncol. 1994, 33, 779–786. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Liposome | Diameter (nm) | Polydispersity Index | Peak Intensity %)# | Zeta Potential (mV) | Phospholipid (mM) |
|---|---|---|---|---|---|
| EMP | 137.6 ± 5.6 | 0.067 ± 0.04 | 100 | −3.63 ± 0.80 | 16.44 |
| N-AI | 139.9 ± 3.9 | 0.093 ± 0.02 | 100 | −3.64 ± 0.59 | 16.45 |
| EMP PAI-2 | 139.7 ± 4.9 | 0.109 ± 0.02 | 100 | −4.05 ± 0.53 | 16.67 |
| N-AI PAI-2 | 141.1 ± 5.0 | 0.086 ± 0.03 | 100 | −4.66 ± 0.52 | 16.62 |
| PK Parameter | N-AI | N-AI PAI-2 |
|---|---|---|
| Cmax (% ID/mL) | 84.66 (± 9.79) | 83.76 (± 9.25) |
| Kelim α (fast) min−1 | 0.061 | 0.058 |
| Kelim β (slow) min−1 | 0.002 | 0.002 |
| T1/2 α (fast) min | 11.419 | 12.050 |
| T1/2 β (slow) min | 408.152 | 410.843 |
| Correlation coefficient (R2) | 0.9629 | 0.9836 |
| AUC (% ID/min/mL) | 860.3 (± 66.89) | 873.4 (± 50.79) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belfiore, L.; Saunders, D.N.; Ranson, M.; Vine, K.L. N-Alkylisatin-Loaded Liposomes Target the Urokinase Plasminogen Activator System in Breast Cancer. Pharmaceutics 2020, 12, 641. https://doi.org/10.3390/pharmaceutics12070641
Belfiore L, Saunders DN, Ranson M, Vine KL. N-Alkylisatin-Loaded Liposomes Target the Urokinase Plasminogen Activator System in Breast Cancer. Pharmaceutics. 2020; 12(7):641. https://doi.org/10.3390/pharmaceutics12070641
Chicago/Turabian StyleBelfiore, Lisa, Darren N. Saunders, Marie Ranson, and Kara L. Vine. 2020. "N-Alkylisatin-Loaded Liposomes Target the Urokinase Plasminogen Activator System in Breast Cancer" Pharmaceutics 12, no. 7: 641. https://doi.org/10.3390/pharmaceutics12070641
APA StyleBelfiore, L., Saunders, D. N., Ranson, M., & Vine, K. L. (2020). N-Alkylisatin-Loaded Liposomes Target the Urokinase Plasminogen Activator System in Breast Cancer. Pharmaceutics, 12(7), 641. https://doi.org/10.3390/pharmaceutics12070641