Identification and Pharmaceutical Characterization of a New Itraconazole Terephthalic Acid Cocrystal

 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
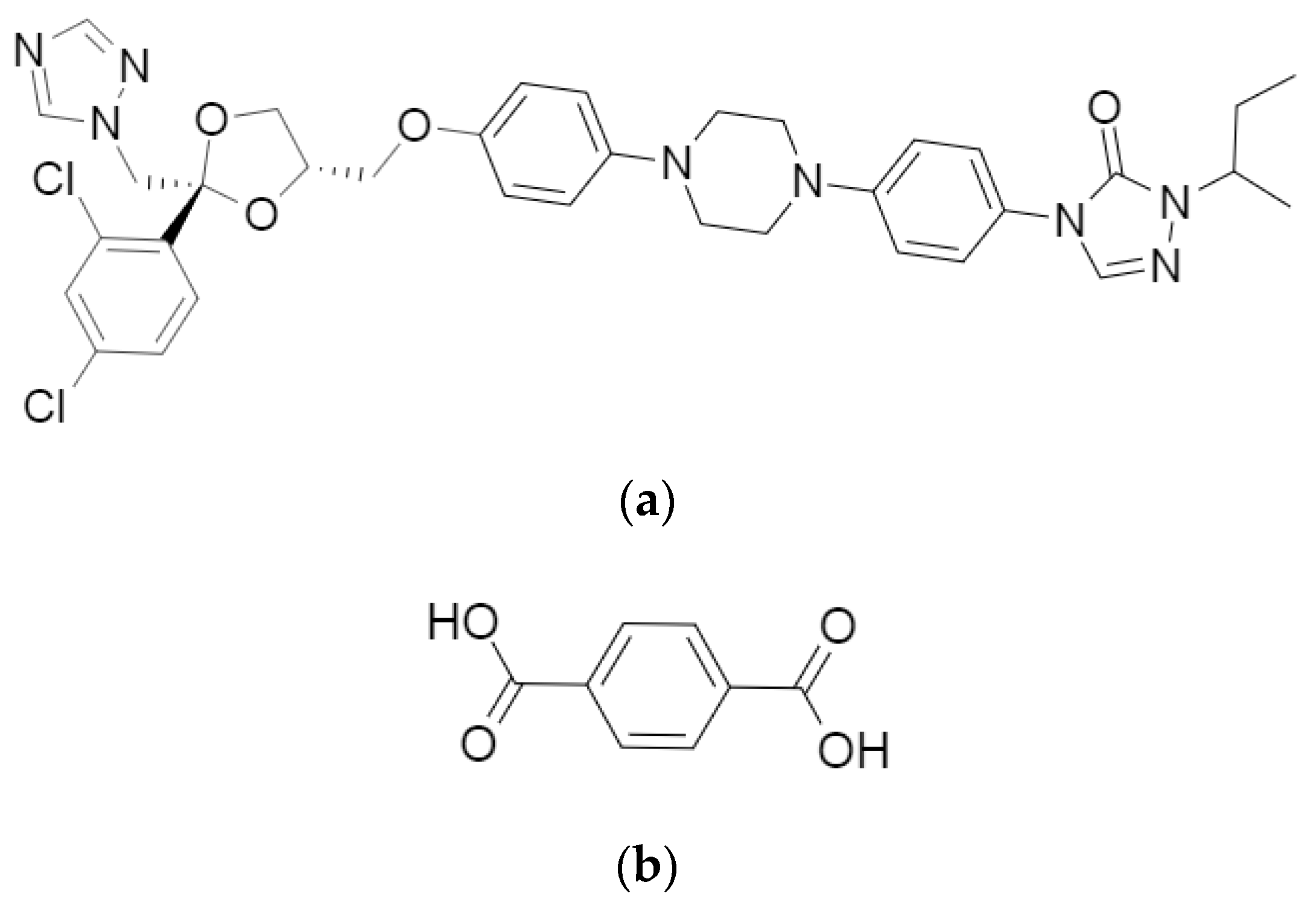
2.1. Materials
2.2. Methods
2.2.1. Neat Grinding (NG) and Liquid-Assisted Grinding (LG)
2.2.2. Cocrystallisation by Slurrying
2.2.3. Cocrystallisation by Ball Milling (BM)
2.2.4. Cocrystallisation by Slow Evaporation
2.2.5. Freeze Drying of Itraconazole (ITR)
2.2.6. Differential Scanning Calorimetry (DSC)
2.2.7. Powder X-ray Diffraction (PXRD)
2.2.8. Single Crystal X-ray Analysis
2.2.9. Fourier-Transform Infrared Spectroscopy (FTIR) and Raman Spectroscopy
2.2.10. Morphological Analysis
2.2.11. Contact Angle Measurements
2.2.12. Intrinsic Dissolution Rate (IDR) Study
2.2.13. Dissolution Analysis
2.2.13.1. Powder Dissolution of ITR Systems Mixed with Lactose
2.2.13.2. Powder Dissolution of ITR Systems Mixed with Other Excipients
2.2.14. Statistical Analysis
3. Results and Discussion
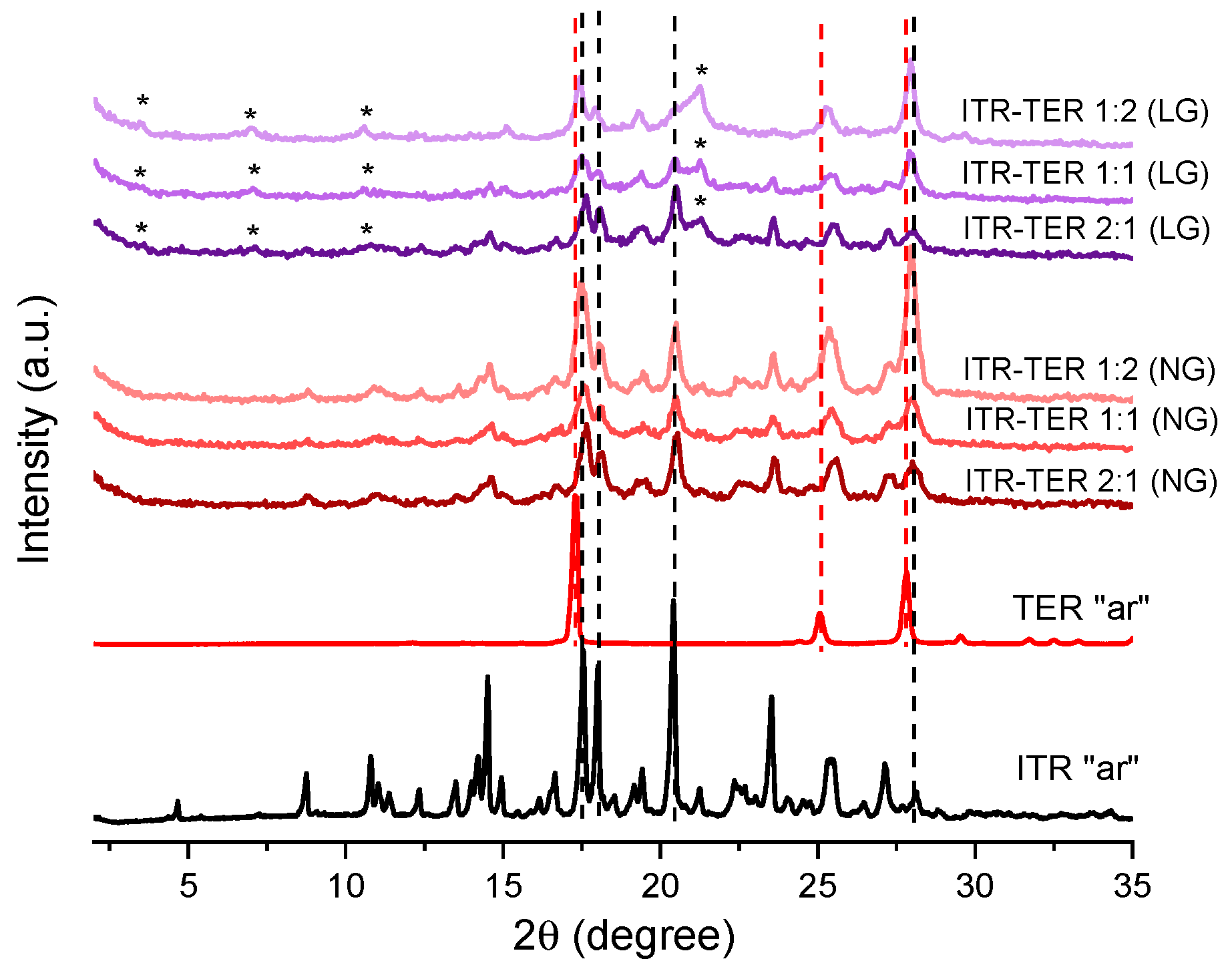
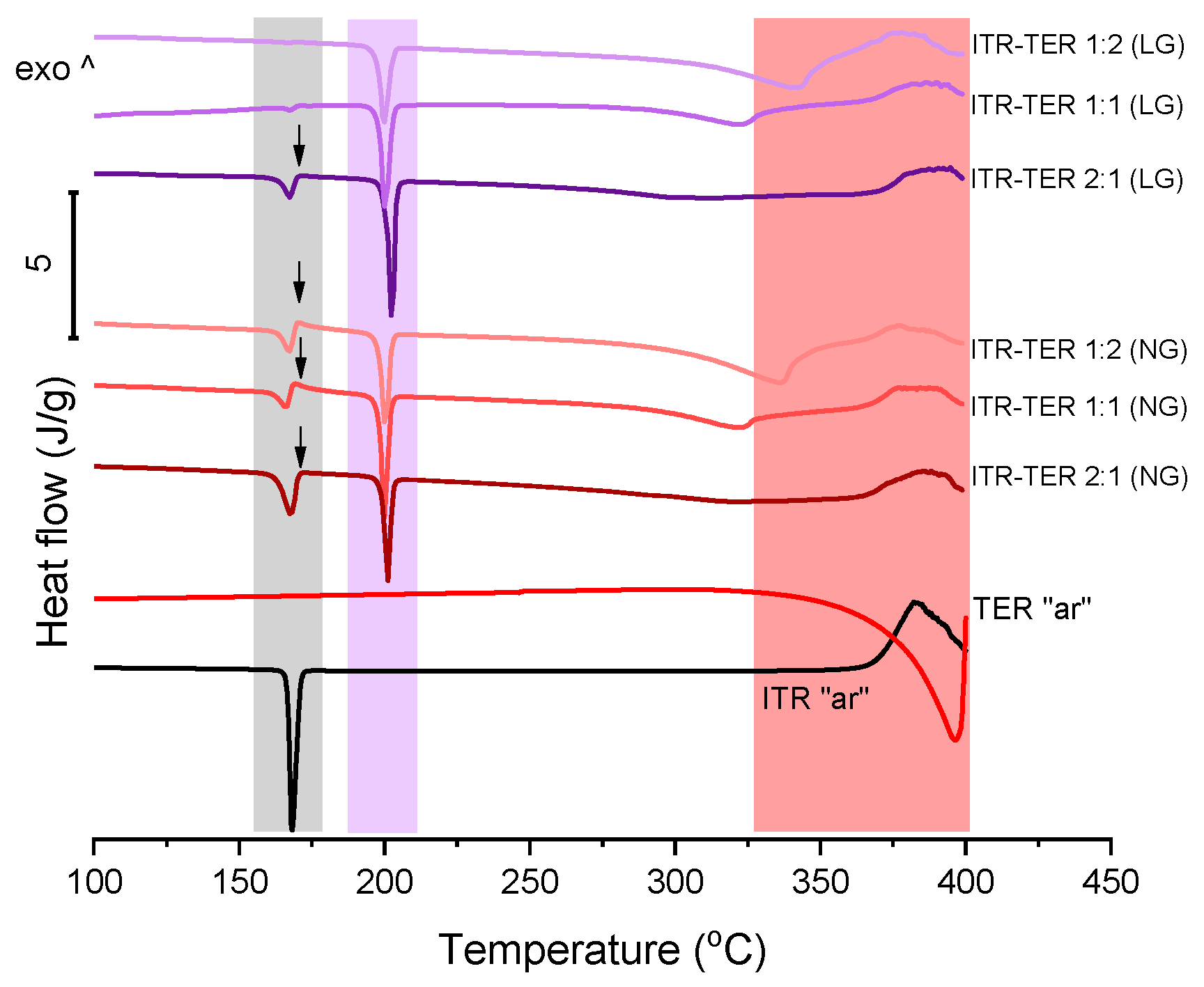
3.1. Characterization of ITR and Terephthalic Acid (TER) Mixtures Following Neat and Liquid-Assisted Grinding
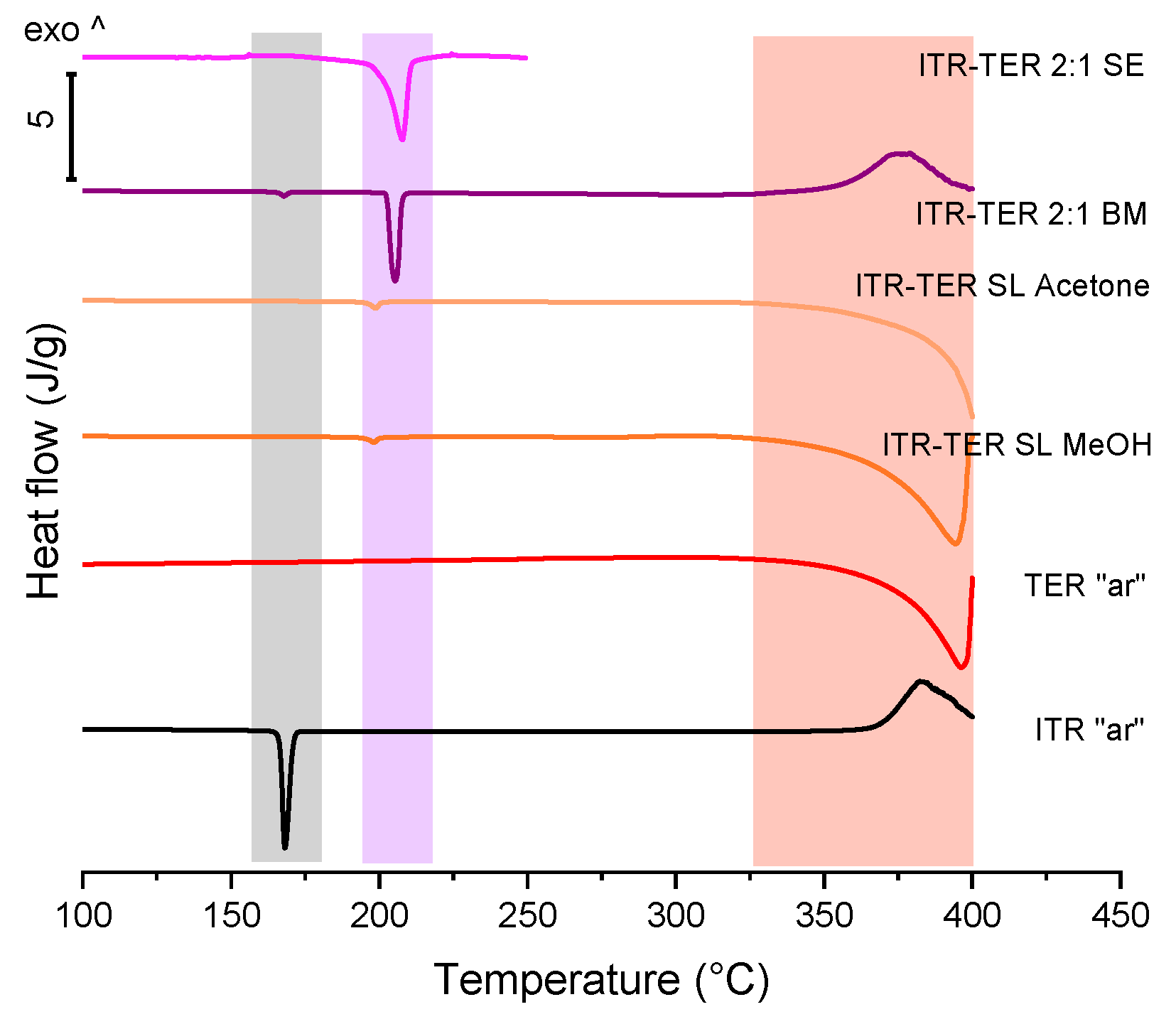
3.2. Properties of ITR and TER Samples Made by Slurring, Evaporation and Ball Milling Methods
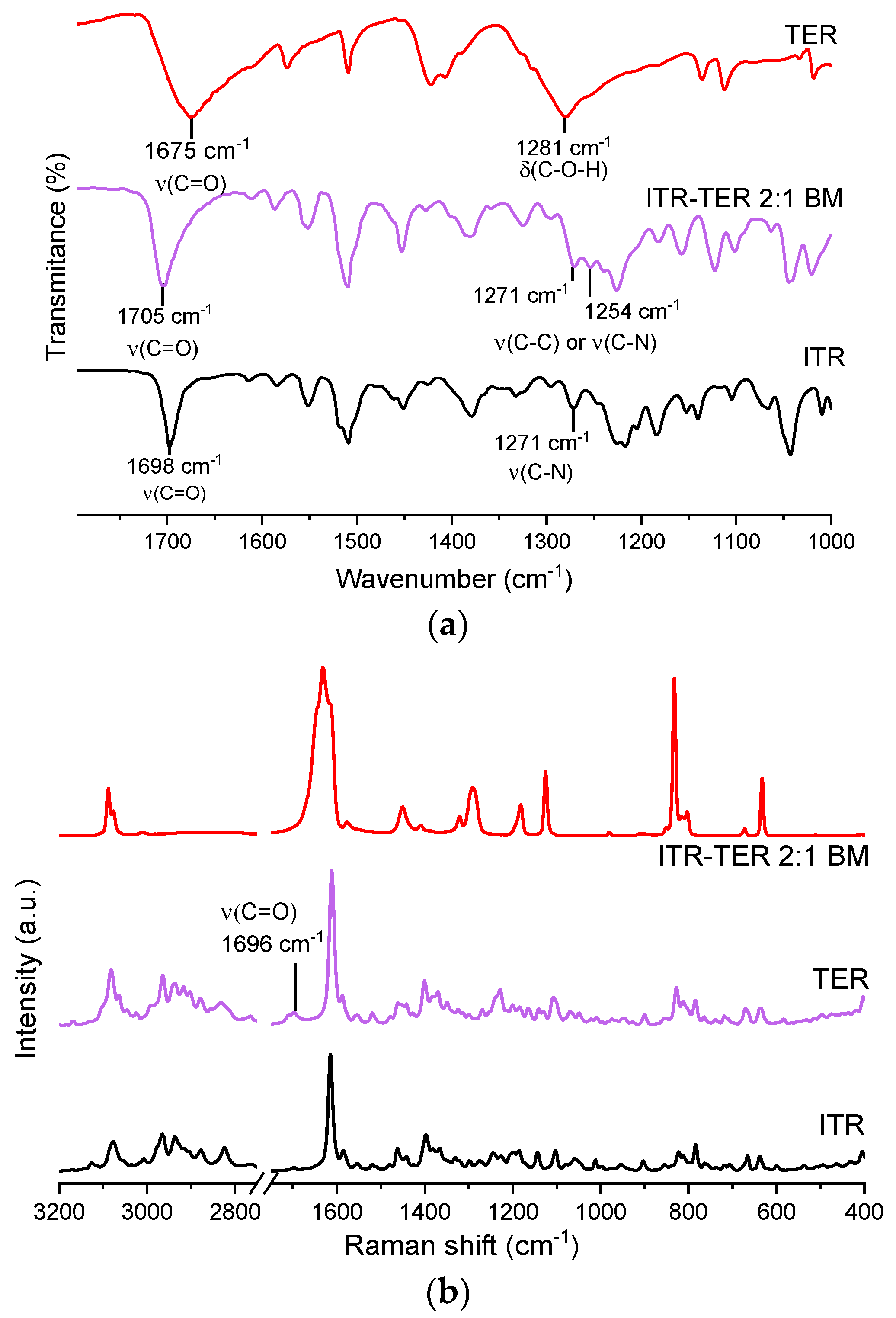
3.3. Infrared and Raman Spectroscopy
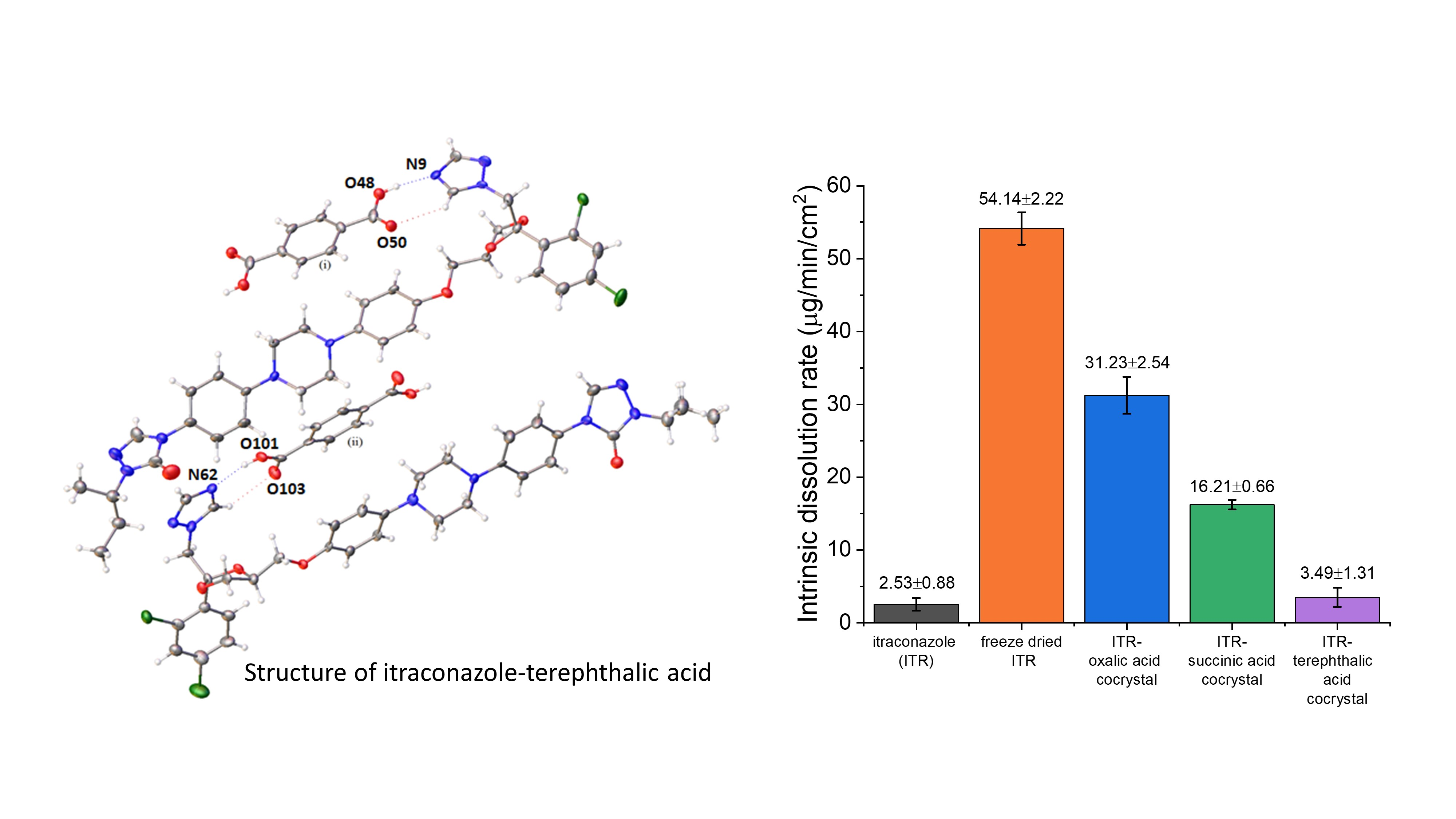
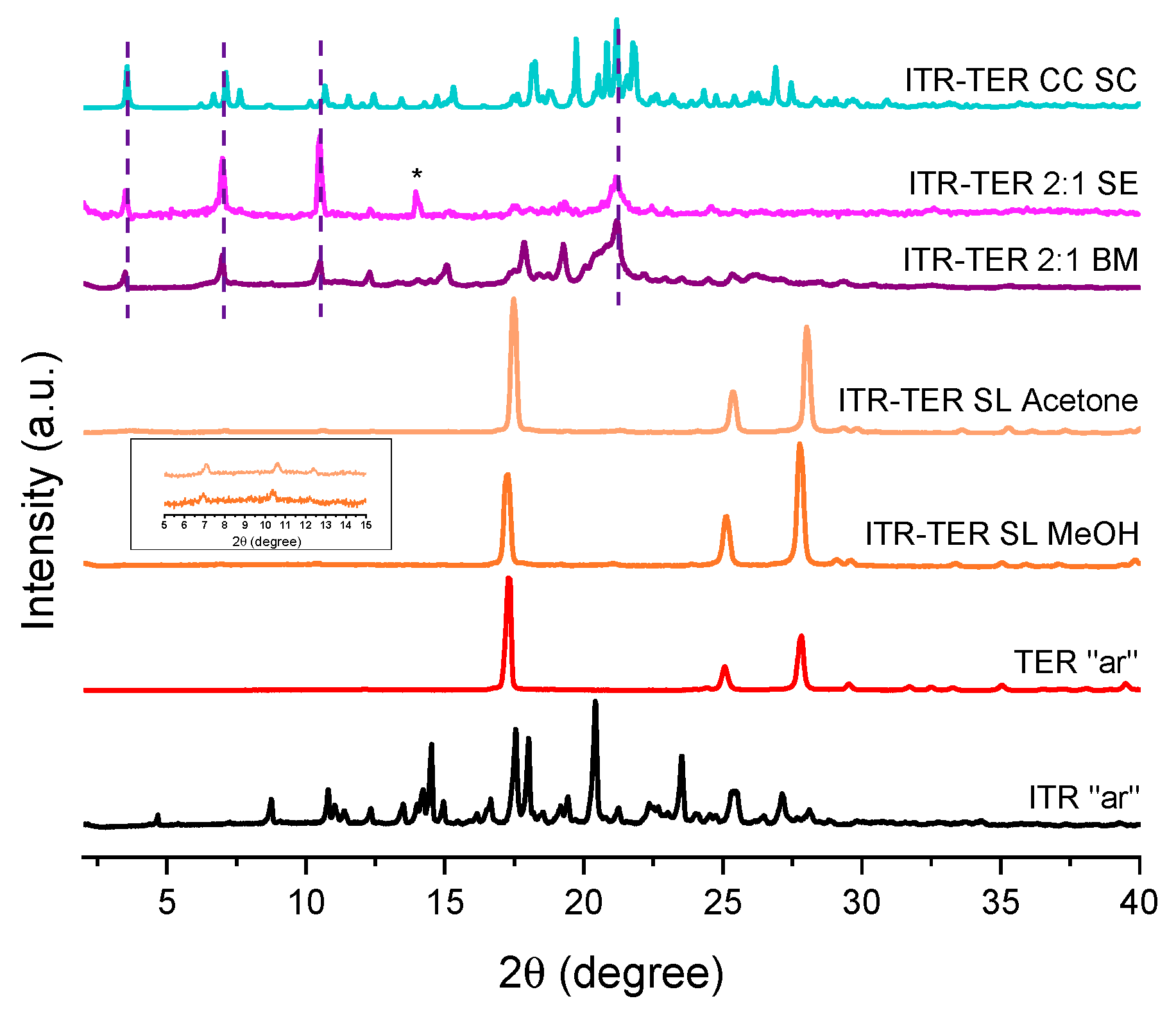
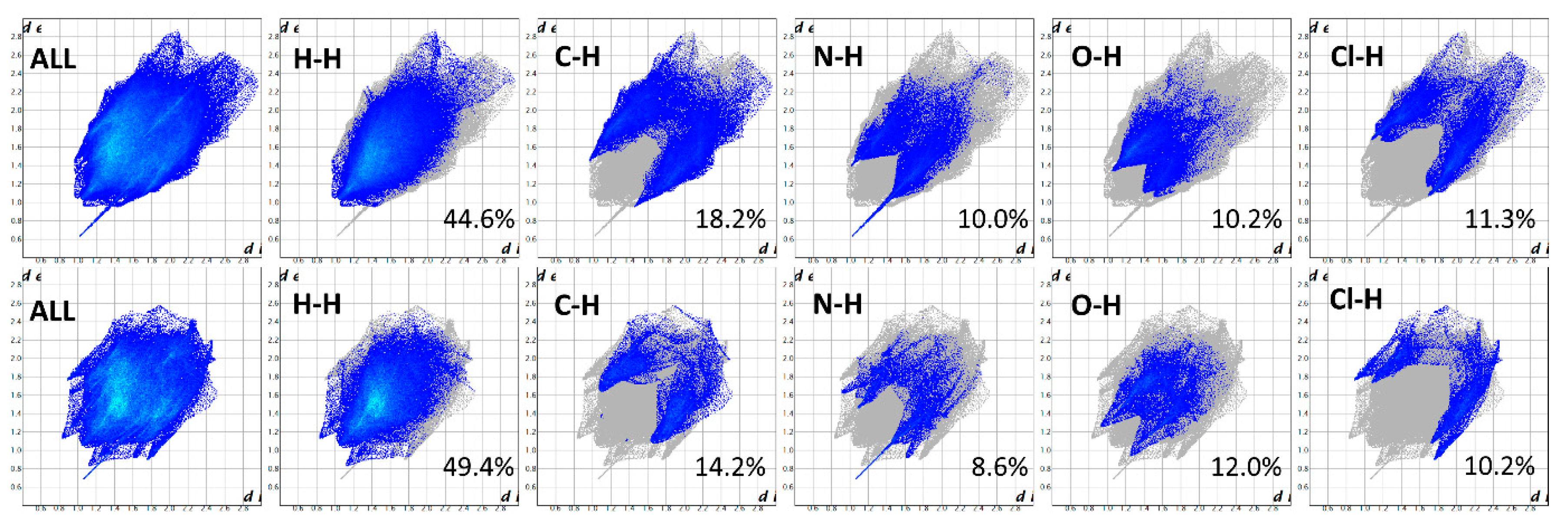
3.4. Crystallographic Analysis
3.5. Morphological Analysis
3.6. Pharmaceutical Evaluation of ITR Cocrystals
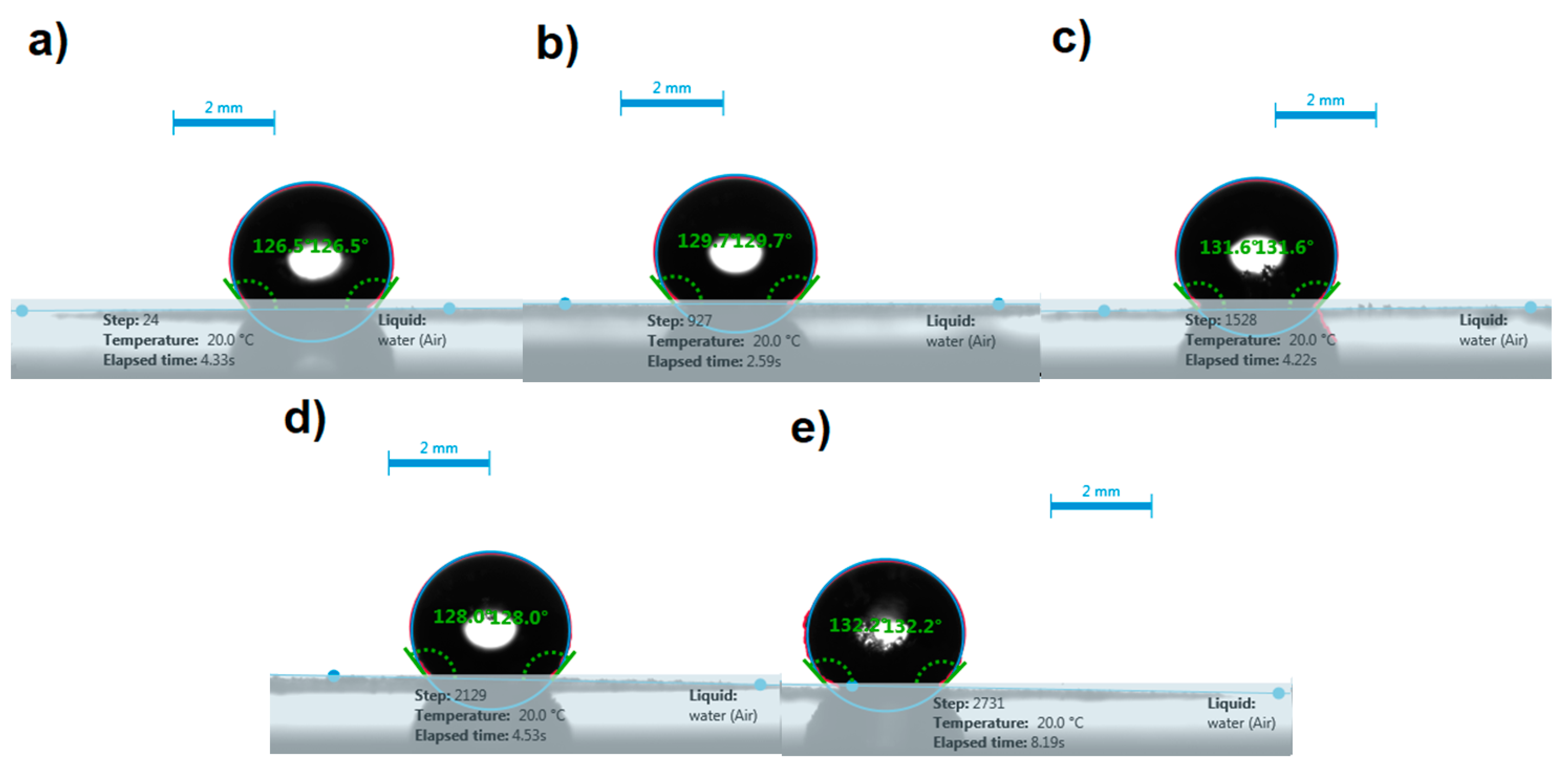
3.6.1. Contact Angle Analysis
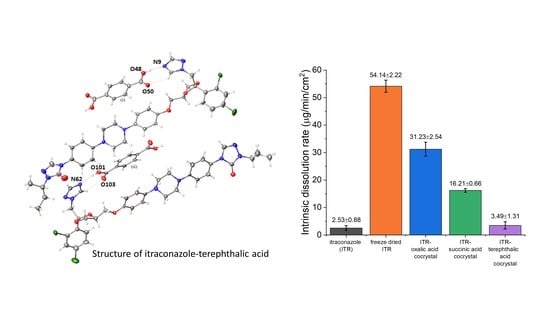
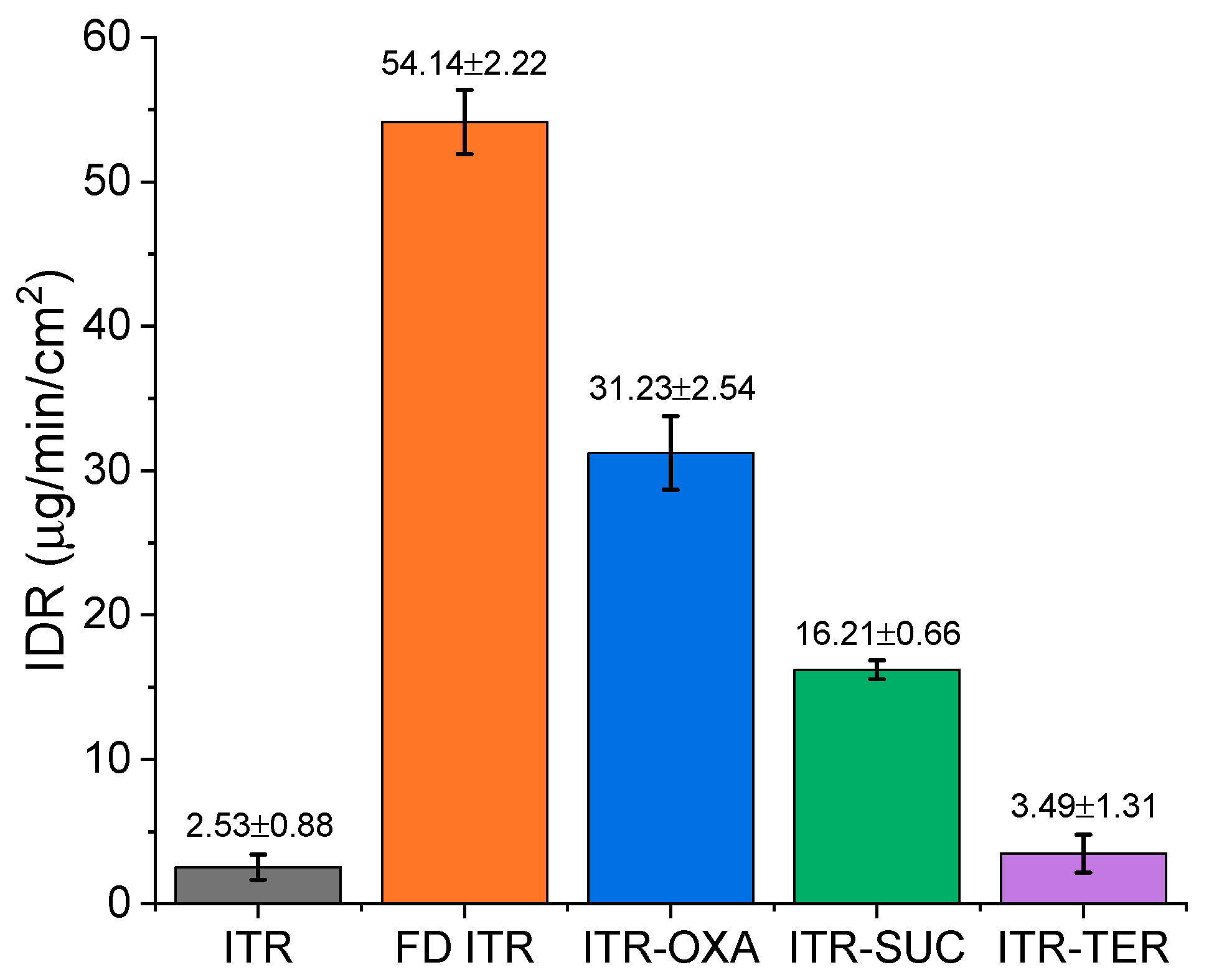
3.6.2. Intrinsic Dissolution Rate (IDR)
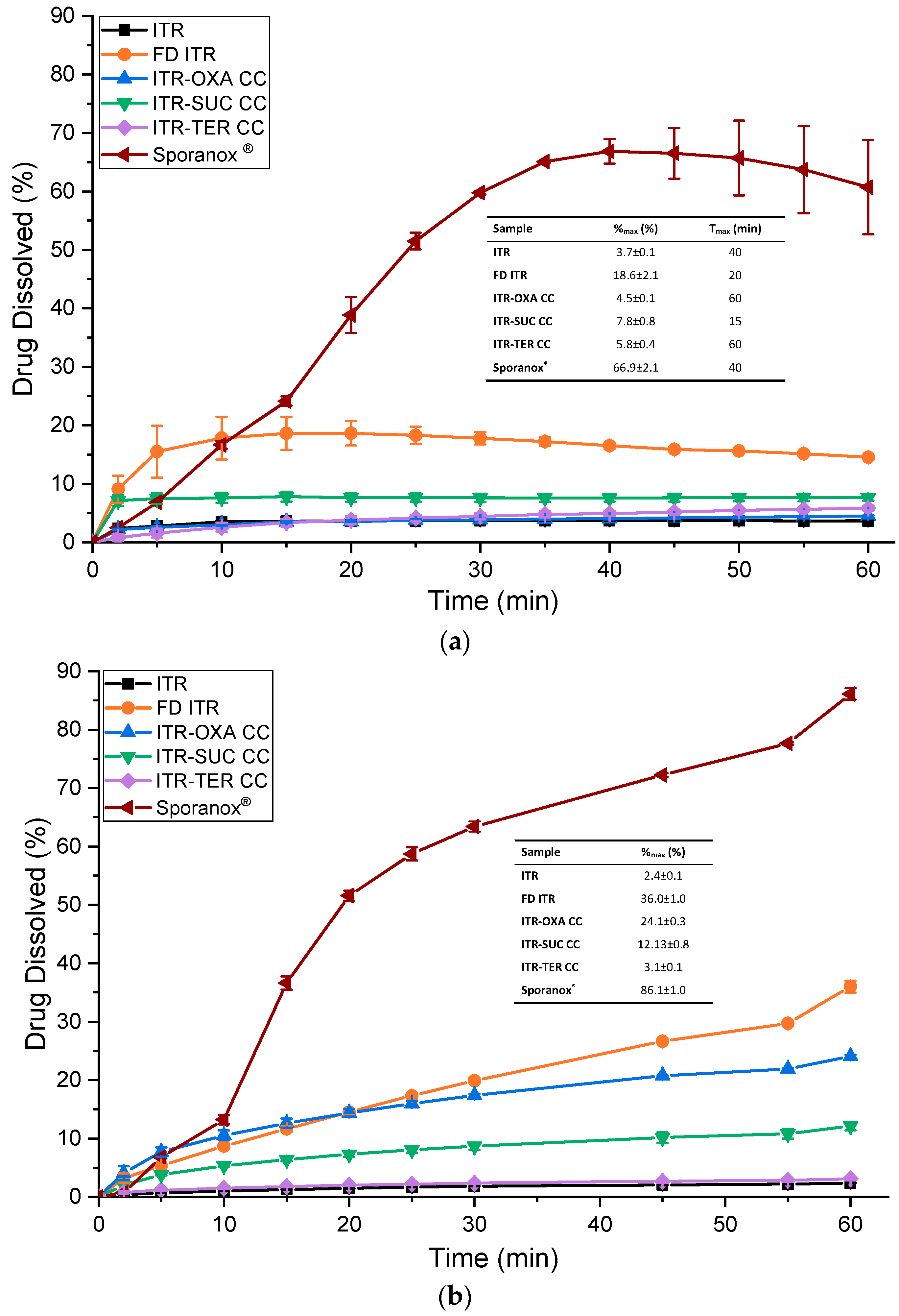
3.6.3. Powder/Formulation Dissolution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schultheiss, N.; Newman, A. Pharmaceutical cocrystals and their physicochemical properties. Cryst. Growth Des. 2009, 9, 2950–2967. [Google Scholar] [CrossRef] [Green Version]
- Thakuria, R.; Delori, A.; Jones, W.; Lipert, M.P.; Roy, L.; Rodríguez-Hornedo, N. Pharmaceutical cocrystals and poorly soluble drugs. Int. J. Pharm. 2013, 453, 101–125. [Google Scholar] [CrossRef]
- Berry, D.J.; Steed, J.W. Pharmaceutical cocrystals, salts and multicomponent systems; intermolecular interactions and property based design. Adv. Drug Deliv. Rev. 2017, 117, 3–24. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Cerreia Vioglio, P.; Chierotti, M.R.; Gobetto, R. Pharmaceutical aspects of salt and cocrystal forms of APIs and characterization challenges. Adv. Drug Deliv. Rev. 2017, 117, 86–110. [Google Scholar] [CrossRef]
- Campoy, S.; Adrio, J.L. Antifungals. Biochem. Pharmacol. 2017, 133, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Denning, D.W.; Hope, W.W. Therapy for fungal diseases: Opportunities and priorities. Trends Microbiol. 2010, 18, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, V.; Valsami, G.; Dokoumetzidis, A.; Macheras, P. Biopharmaceutics classification systems for new molecular entities (BCS-NMEs) and marketed drugs (BCS-MD): Theoretical basis and practical examples. Int. J. Pharm. 2008, 361, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Hardin, T.C.; Graybill, J.R.; Fetchick, R.; Woestenborghs, R.; Rinaldi, M.G.; Kuhn, J.G. Pharmacokinetics of itraconazole following oral administration to normal volunteers. Antimicrob. Agents Chemother. 1988, 32, 1310–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mugheirbi, N.A.; Paluch, K.J.; Tajber, L. Heat induced evaporative antisolvent nanoprecipitation (HIEAN) of itraconazole. Int. J. Pharm. 2014, 471, 400–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mugheirbi, N.A.; Tajber, L. Mesophase and size manipulation of itraconazole liquid crystalline nanoparticles produced via quasi nanoemulsion precipitation. Eur. J. Pharm. Biopharm. 2015, 96, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Kozyra, A.; Mugheirbi, N.A.; Paluch, K.J.; Garbacz, G.; Tajber, L. Phase Diagrams of Polymer-Dispersed Liquid Crystal Systems of Itraconazole/Component Immiscibility Induced by Molecular Anisotropy. Mol. Pharm. 2018, 15, 5192–5206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remenar, J.F.; Morissette, S.L.; Peterson, M.L.; Moulton, B.; MacPhee, J.M.; Guzmán, H.R.; Almarsson, Ö. Crystal engineering of novel cocrystals of a triazole drug with 1,4-dicarboxylic acids. J. Am. Chem. Soc. 2003, 125, 8456–8457. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Miroshnyk, I.; Pietilä, L.O.; Haarala, J.; Salmia, J.; Sinervo, K.; Mirza, S.; Van Veen, B.; Kolehmainen, E.; Nonappa; et al. Diversity in itraconazole cocrystals with aliphatic dicarboxylic acids of varying chain length. Cryst. Growth Des. 2013, 13, 4877–4884. [Google Scholar] [CrossRef]
- Weng, J.; Wong, S.N.; Xu, X.; Xuan, B.; Wang, C.; Chen, R.; Sun, C.C.; Lakerveld, R.; Kwok, P.C.L.; Chow, S.F. Cocrystal Engineering of Itraconazole with Suberic Acid via Rotary Evaporation and Spray Drying. Cryst. Growth Des. 2019, 19, 2736–2745. [Google Scholar] [CrossRef]
- Ma, Y.H.; Ge, S.W.; Wang, W.; Sun, B.W. Studies on the synthesis, structural characterization, Hirshfeld analysis and stability of apovincamine (API) and its co-crystal (terephthalic acid: Apovincamine = 1:2). J. Mol. Struct. 2015, 1097, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Mikhailovskaya, A.V.; Myz, S.A.; Bulina, N.V.; Gerasimov, K.B.; Kuznetsova, S.A.; Shakhtshneider, T.P. Screening and characterization of cocrystal formation between betulin and terephthalic acid. Mater. Today Proc. 2020, 25, 381–383. [Google Scholar] [CrossRef]
- Lemmerer, A.; Bernstein, J.; Kahlenberg, V. Hydrogen bonding patterns of the co-crystal containing the pharmaceutically active ingredient isoniazid and terephthalic acid. J. Chem. Crystallogr. 2011, 41, 991–997. [Google Scholar] [CrossRef]
- André, V.; Fernandes, A.; Santos, P.P.; Duarte, M.T. On the track of new multicomponent gabapentin crystal forms: Synthon competition and pH stability. Cryst. Growth Des. 2011, 11, 2325–2334. [Google Scholar] [CrossRef]
- Rodríguez-Spong, B.; Price, C.P.; Jayasankar, A.; Matzger, A.J.; Rodríguez-Hornedo, N. General principles of pharmaceutical solid polymorphism: A supramolecular perspective. Adv. Drug Deliv. Rev. 2004, 56, 241–274. [Google Scholar] [CrossRef] [PubMed]
- Paluch, K.J.; Tajber, L.; McCabe, T.; O’Brien, J.E.; Corrigan, O.I.; Healy, A.M. Preparation and solid state characterisation of chlorothiazide sodium intermolecular self-assembly suprastructure. Eur. J. Pharm. Sci. 2010, 41, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Machado Cruz, R.; Santos-Martinez, M.J.; Tajber, L. Impact of polyethylene glycol polymers on the physicochemical properties and mucoadhesivity of itraconazole nanoparticles. Eur. J. Pharm. Biopharm. 2019, 144, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; McKinnon, J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer 17. 2017. Available online: http://hirshfeldsurface.net (accessed on 1 July 2020).
- Frisić, T.; Childs, S.L.; Rizvi, S.A.A.; Jones, W. The role of solvent in mechanochemical and sonochemical cocrystal formation: A solubility-based approach for predicting cocrystallisation outcome. CrystEngComm 2009, 11, 418–426. [Google Scholar] [CrossRef]
- Braga, D.; Maini, L.; Grepioni, F. Mechanochemical preparation of co-crystals. Chem. Soc. Rev. 2013, 42, 7638–7648. [Google Scholar] [CrossRef]
- Zhang, S.; Lee, T.W.Y.; Chow, A.H.L. Crystallization of Itraconazole Polymorphs from Melt. Cryst. Growth Des. 2016, 16, 3791–3801. [Google Scholar] [CrossRef]
- Karagianni, A.; Malamatari, M.; Kachrimanis, K. Pharmaceutical cocrystals: New solid phase modification approaches for the formulation of APIs. Pharmaceutics 2018, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Elmas Kimyonok, A.B.; Ulutürk, M. Determination of the Thermal Decomposition Products of Terephthalic Acid by Using Curie-Point Pyrolyzer. J. Energ. Mater. 2016, 34, 113–122. [Google Scholar] [CrossRef]
- Yamashita, H.; Hirakura, Y.; Yuda, M.; Terada, K. Coformer screening using thermal analysis based on binary phase diagrams. Pharm. Res. 2014, 31, 1946–1957. [Google Scholar] [CrossRef]
- Williams, D.H.; Fleming, I. Spectroscopic Methods in Organic Chemistry, 5th ed.; McGraw-Hill Education: New York, NY, USA, 1995; ISBN 9780077091477. [Google Scholar]
- Karthikeyan, N.; Joseph Prince, J.; Ramalingam, S.; Periandy, S. Electronic [UV–Visible] and vibrational [FT-IR, FT-Raman] investigation and NMR–mass spectroscopic analysis of terephthalic acid using quantum Gaussian calculations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 139, 229–242. [Google Scholar] [CrossRef]
- Lahtinen, M.; Kolehmainen, E.; Haarala, J.; Shevchenko, A. Evidence of weak halogen bonding: New insights on itraconazole and its succinic acid cocrystal. Cryst. Growth Des. 2013, 13, 346–351. [Google Scholar] [CrossRef]
- Delori, A.; Frišić, T.; Jones, W. The role of mechanochemistry and supramolecular design in the development of pharmaceutical materials. CrystEngComm 2012, 14, 2350–2362. [Google Scholar] [CrossRef]
- Shevchenko, A.; Bimbo, L.M.; Miroshnyk, I.; Haarala, J.; Jelínková, K.; Syrjänen, K.; Van Veen, B.; Kiesvaara, J.; Santos, H.A.; Yliruusi, J. A new cocrystal and salts of itraconazole: Comparison of solid-state properties, stability and dissolution behavior. Int. J. Pharm. 2012, 436, 403–409. [Google Scholar] [CrossRef]
- Florence, A.T.; Attwood, D. Physicochemical Principles of Pharmacy, 4th ed.; Pharmaceutical Press: London, UK, 2006. [Google Scholar]
- Mugheirbi, N.A.; O’Connell, P.; Serrano, D.R.; Healy, A.M.; Taylor, L.S.; Tajber, L. A Comparative Study on the Performance of Inert and Functionalized Spheres Coated with Solid Dispersions Made of Two Structurally Related Antifungal Drugs. Mol. Pharm. 2017, 14, 3718–3728. [Google Scholar] [CrossRef] [PubMed]
- Park, C.-M.; Sheehan, R.J. Phthalic Acids and Other Benzenepolycarboxylic Acids. In Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2000. [Google Scholar]
- Stevens, D. A Itraconazole in Cyclodextrin Solution. Pharmacotherapy 1999, 19, 603–611. [Google Scholar] [CrossRef]
- Tyner, T.; Francis, J. Oxalic Acid Dihydrate. In ACS Reagent Chemicals; American Chemical Society: Washington, DC, USA, 2018. [Google Scholar]
- Litsanov, B.; Brocker, M.; Oldiges, M.; Bott, M. Succinic Acid. In Bioprocessing of Renewable Resources to Commodity Bioproducts; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2014; pp. 435–472. ISBN 9781118845394. [Google Scholar]
- Babu, N.J.; Nangia, A. Solubility Advantage of Amorphous Drugs and Pharmaceutical Cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- Grossjohann, C.; Eccles, K.S.; Maguire, A.R.; Lawrence, S.E.; Tajber, L.; Corrigan, O.I.; Healy, A.M. Characterisation, solubility and intrinsic dissolution behaviour of benzamide: Dibenzyl sulfoxide cocrystal. Int. J. Pharm. 2012, 422, 24–32. [Google Scholar] [CrossRef]
- Van Eerdenbrugh, B.; Van den Mooter, G.; Augustijns, P. Top-down production of drug nanocrystals: Nanosuspension stabilization, miniaturization and transformation into solid products. Int. J. Pharm. 2008, 364, 64–75. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Excipient (%, w/w) | Formulation | ||||
|---|---|---|---|---|---|
| ITR | FD ITR | ITR–OXA | ITR–SUC | ITR–TER | |
| API | 21.74 | 21.74 | |||
| Cocrystal | 23.22 | 23.13 | 23.70 | ||
| Sucrose | 41.74 | 41.74 | 40.95 | 41.0 | 40.69 |
| HPMC (*) | 32.61 | 32.61 | 31.99 | 32.03 | 31.79 |
| PEG (**) | 3.91 | 3.91 | 3.84 | 3.84 | 3.82 |
| Total (%) | 100 | 100 | 100 | 100 | 100 |
| Powder weight (mg) (***) | 460 | 460 | 501.1 | 498.32 | 514.2 |
| Sample | Initial (°) | Final * (°) | p-Value |
|---|---|---|---|
| ITR | 126.8 ± 0.4 | 126.5 ±0.3 | 0.27 |
| FD ITR | 128.9 ± 0.8 | 127.0 ± 1.7 | 0.16 |
| ITR–OXA | 128.7 ± 2.9 | 126.0 ± 3.5 | 0.35 |
| ITR–SUC | 126.5 ± 1.5 | 124.8 ± 2.0 | 0.30 |
| ITR–TER | 130.5 ± 1.7 | 129.7 ± 1.9 | 0.61 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado Cruz, R.; Boleslavská, T.; Beránek, J.; Tieger, E.; Twamley, B.; Santos-Martinez, M.J.; Dammer, O.; Tajber, L. Identification and Pharmaceutical Characterization of a New Itraconazole Terephthalic Acid Cocrystal. Pharmaceutics 2020, 12, 741. https://doi.org/10.3390/pharmaceutics12080741
Machado Cruz R, Boleslavská T, Beránek J, Tieger E, Twamley B, Santos-Martinez MJ, Dammer O, Tajber L. Identification and Pharmaceutical Characterization of a New Itraconazole Terephthalic Acid Cocrystal. Pharmaceutics. 2020; 12(8):741. https://doi.org/10.3390/pharmaceutics12080741
Chicago/Turabian StyleMachado Cruz, Ricardo, Tereza Boleslavská, Josef Beránek, Eszter Tieger, Brendan Twamley, Maria Jose Santos-Martinez, Ondřej Dammer, and Lidia Tajber. 2020. "Identification and Pharmaceutical Characterization of a New Itraconazole Terephthalic Acid Cocrystal" Pharmaceutics 12, no. 8: 741. https://doi.org/10.3390/pharmaceutics12080741
APA StyleMachado Cruz, R., Boleslavská, T., Beránek, J., Tieger, E., Twamley, B., Santos-Martinez, M. J., Dammer, O., & Tajber, L. (2020). Identification and Pharmaceutical Characterization of a New Itraconazole Terephthalic Acid Cocrystal. Pharmaceutics, 12(8), 741. https://doi.org/10.3390/pharmaceutics12080741