
Nanoparticle-Guided Brain Drug Delivery: Expanding the Therapeutic Approach to Neurodegenerative Diseases

 ,
, 
 , and
, and 
Abstract
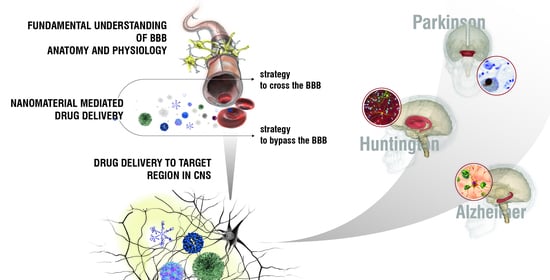
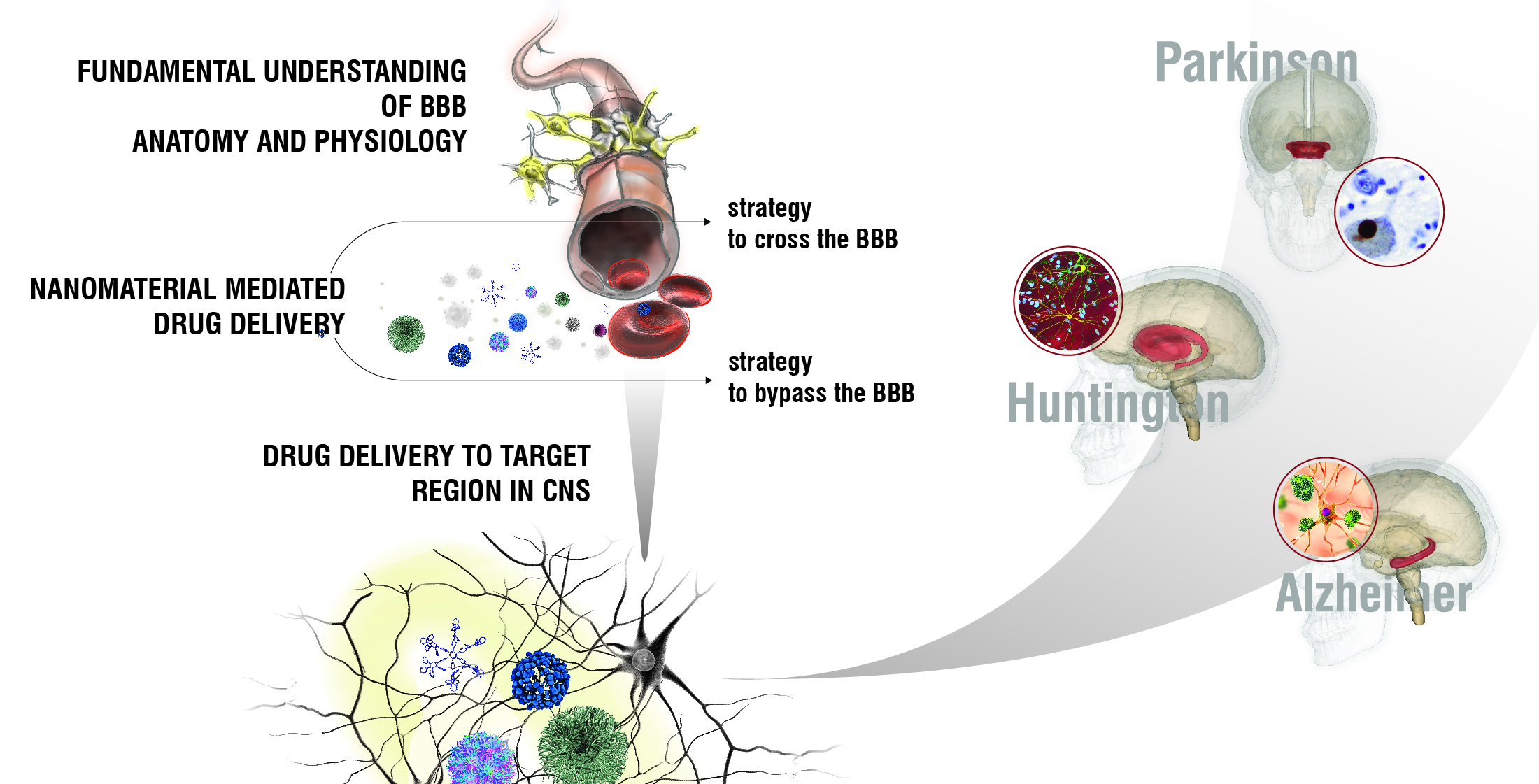
:
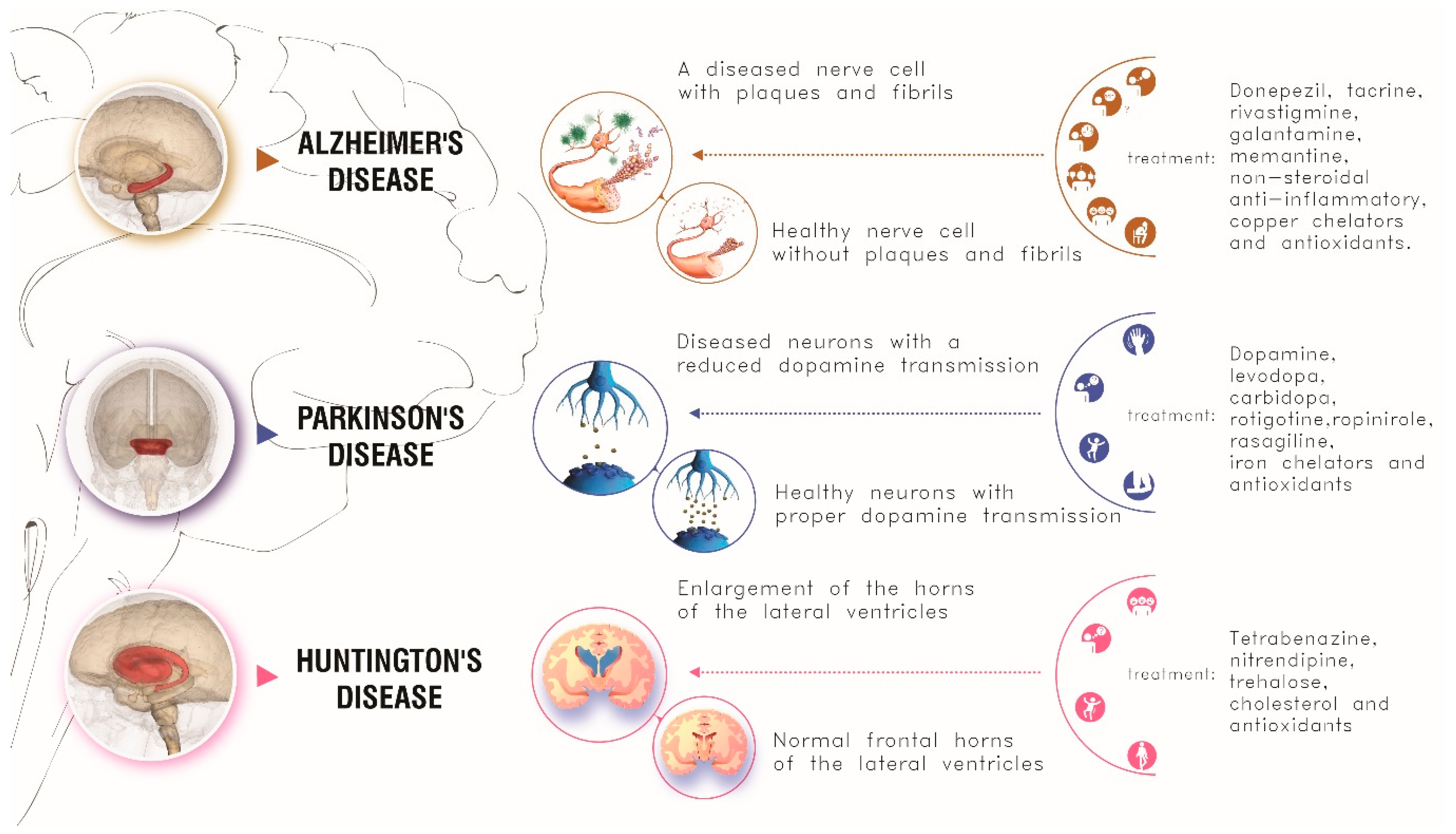
1. Introduction: Neurodegenerative Diseases
1.1. Alzheimer’s Disease
1.2. Parkinson’s Disease
1.3. Huntington’s Disease
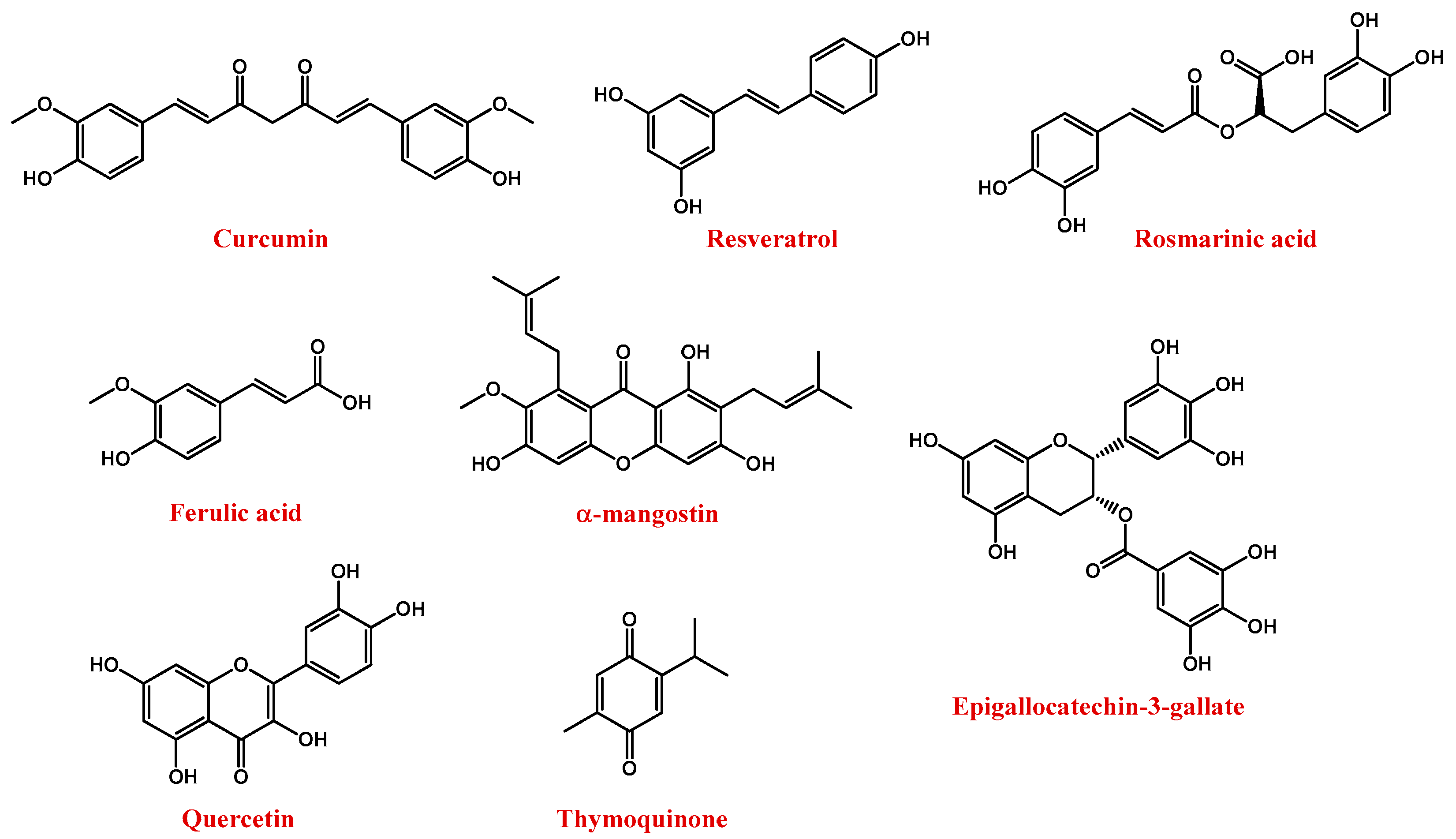
2. Oxidative Stress and Polyphenol Compounds in Neurodegenerative Diseases
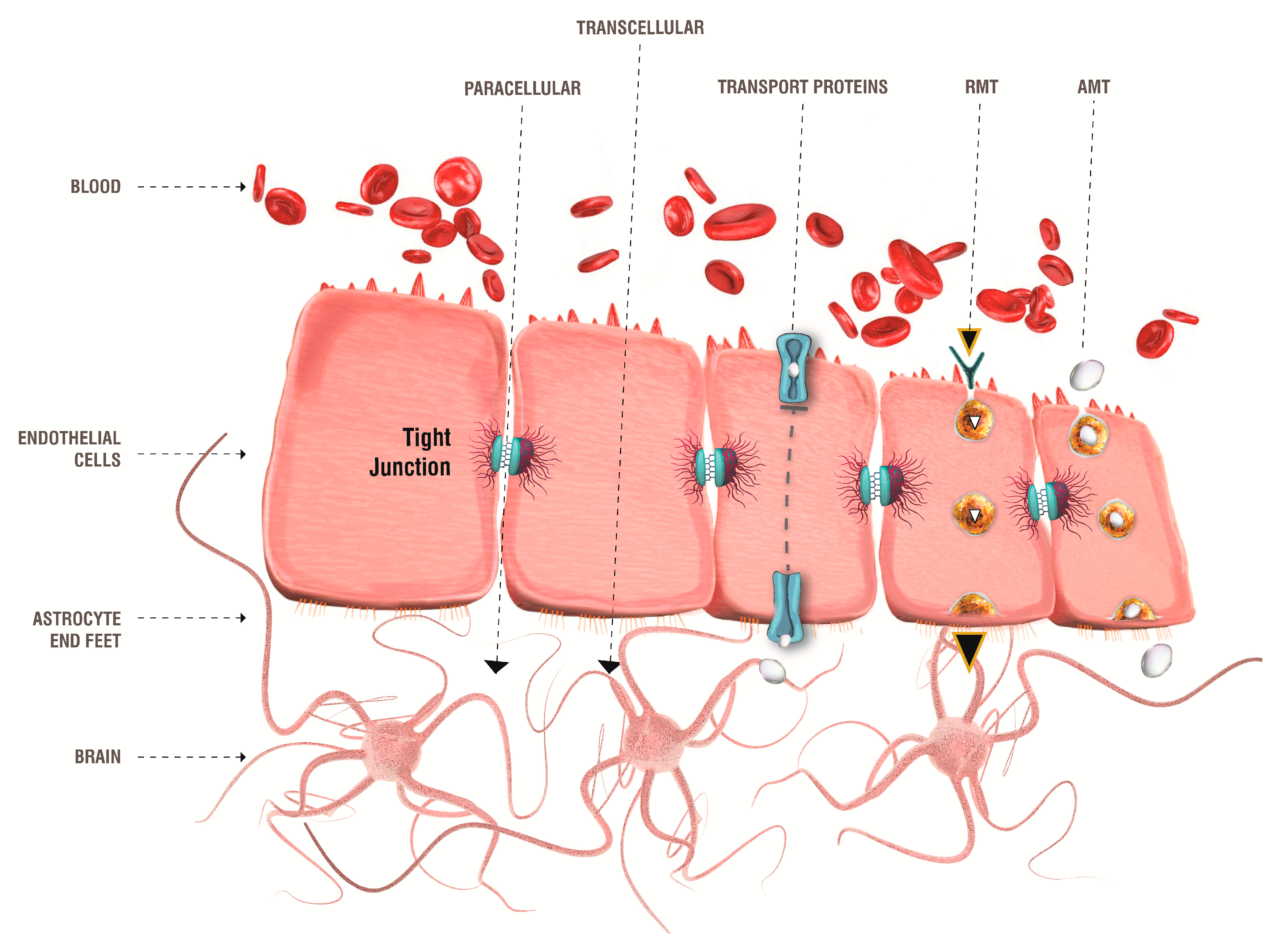
3. Targeting Brain: The Blood–Brain Barrier
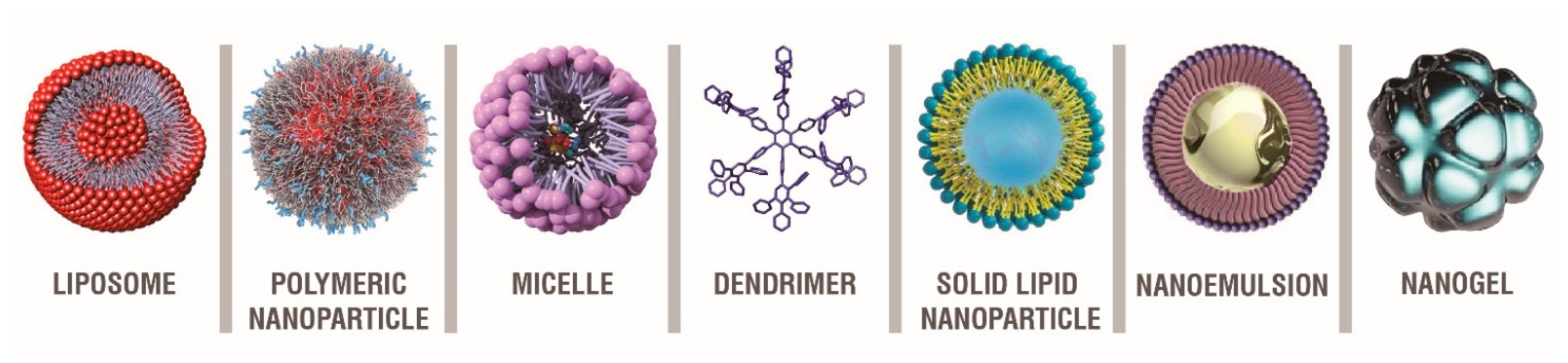
4. Nanotechnology as a Tool to Reach the CNS in Neurodegenerative Diseases
5. Nanosystems in the Treatment of Neurodegenerative Diseases
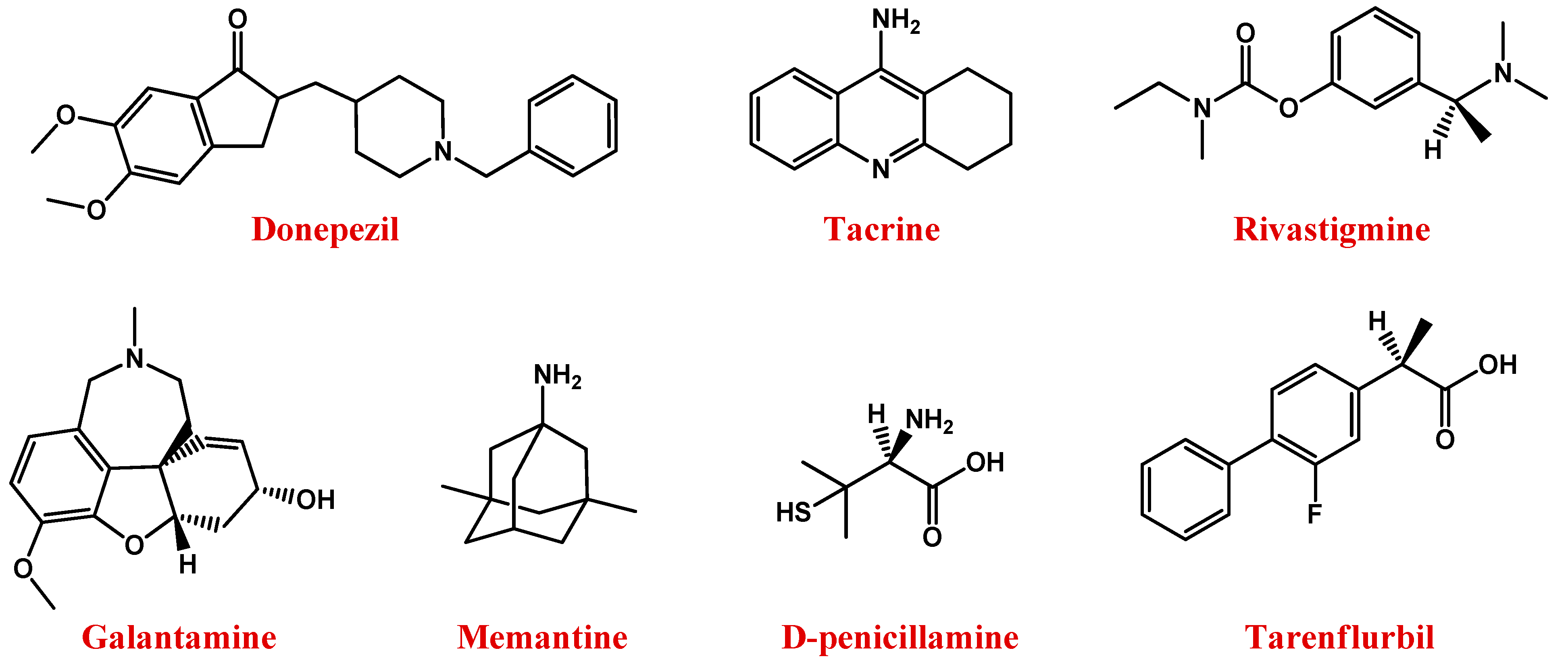
5.1. Alzheimer’s Disease
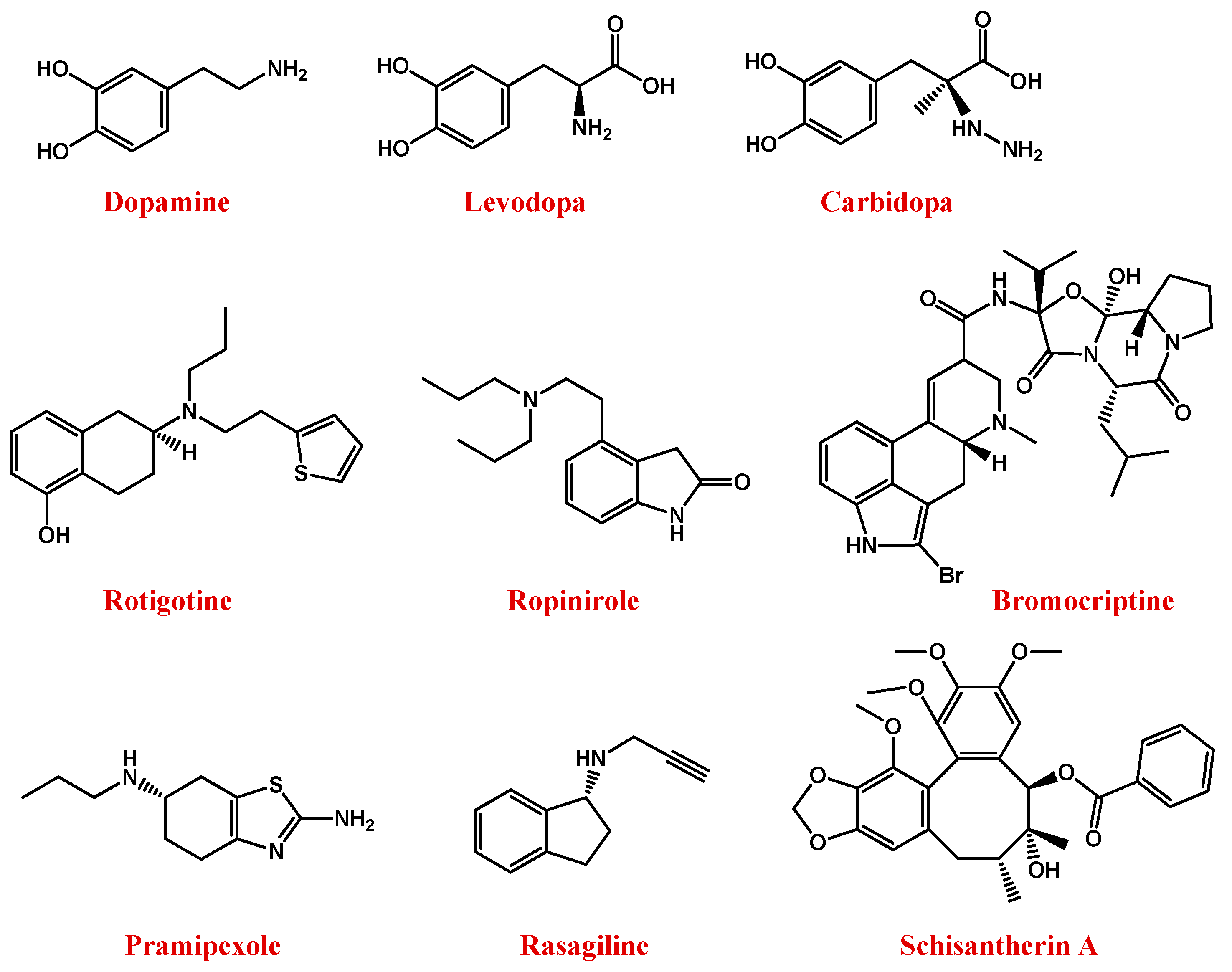
5.2. Parkinson’s Disease
5.3. Huntington’s Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Nanocarrier Platform | Composition | Bioactive Agent | Active Targeting Ligand | Ref. |
|---|---|---|---|---|---|
| Alzheimer | Polymeric NPs | PS80-coated PLGA NPs | Donepezil, cholinesterase inhibitor | [325] | |
| Polymeric NPs | PS80-coated PBCA NPs | Tacrine, cholinesterase inhibitor | [326] | ||
| Polymeric NPs | PS80-coated PBCA NPs | Rivastigmine, cholinesterase inhibitor | [327] | ||
| Polymeric NPs | PLGA and PBCA NPs | Rivastigmine, cholinesterase inhibitor | [328] | ||
| Micelles | PHEA-EDA-Sq17-PS80 amphiphilic copolymer | Rivastigmine, cholinesterase inhibitor | [329] | ||
| Polymeric NPs | PEG-PLGA NPs | Memantine, glutamate antagonist | [330] | ||
| Polymeric NPs | Chitosan | F(ab’) portion of the anti-amyloid antibody IgG4.1 | [331] | ||
| Liposomes | PEG-DMPC and PEG-EYPC | Amyloid beta binding llama single-domain antibody fragments (VHH-pa2H) | GSH | [333] | |
| Liposomes | Sm/Chol in 1:1 molar ratio | PA and CL | [334] | ||
| SLNs | Stearic acid (internal phase), phospholipon 90G (surfactant) and sodium taurocholate (co-surfactant) | PA and CL | [334] | ||
| Liposomes | Sm/Chol in 1:1 molar ratio | PA and mApoE peptide | [335,336] | ||
| Polymeric NPs | PEG-PLA NPs | TGN and QSH peptides | [337] | ||
| Polymeric NPs | PLGA NPs with pluronicF127 (0.1%) as stabilizer | iAβ5 peptide, Aβ aggregation inhibitor | Anti-TfR mAb OX26 and anti-Aβ mAb DE2B4 | [341] | |
| Polymeric NPs | PLGA NPs | Ac-LVFFARK-NH2, Aβ aggregation inhibitor | [343] | ||
| Dendrimers | KLVFF peptide, Aβ aggregation inhibitor | [344] | |||
| Gold NPs | LCA10 and VCD10 peptides, Aβ aggregation inhibitors | [346] | |||
| Polymeric NPs | PLGA NPs | Vitamin d-binding protein | [352] | ||
| Nanospheres | Oxidized mesoporous carbon nanospheres | Protoporphyrin IX, Aβ and tau aggregation inhibitor | RVG peptide | [354] | |
| Nanocrystals | CeNC/IONC/MSN-T807 | Methylene blue, tau aggregation inhibitor | T807 ligand | [357] | |
| Magnetic NPs | Dextran coated-Fe3O4 NPs | Osmotin protein, neuroprotective | [359] | ||
| Liposomes | MPB-PE or PDP-PE | d-penicillamine, copper chelator | [362] | ||
| Polymeric micelles | PEG-PLA | R-flurbiprofen (or tarenflurbil), anti-inflammatory | FBA, RNA aptamer | [364] | |
| Liposomes | DOTAP/DOPE/Chol/DSPE-PEG (4.5:4.5:2:4 molar ratio) | BDNF | Mannose and penetratin or rabies virus glycoprotein | [365] | |
| Polymeric NPs | PLGA NPs | Curcumin | [371,372] | ||
| Polymeric NPs | PLGA and PEG-PLGA NPs | Curcumin | [373] | ||
| Polymeric NPs | PS80-coated PBCA NPs | Curcumin | ApoE3 peptide | [374] | |
| Polymeric NPs | PLGA NPs | Curcumin | Tet-1 peptide | [375] | |
| Polymeric NPs | PLGA NPs | Curcumin | g7 glycopeptide | [376] | |
| Polymeric NPs | PEG-PLGA NPs | Curcumin | B6 peptide | [377] | |
| Polymeric NPs | PLGA NPs | Curcumin | NN2, RNA aptamer | [378] | |
| Liposomes | DPPC/Chol (2:1 molar ratio) with DPS–curcumin (20 mol %) | Curcumin | [385] | ||
| Liposomes | Sm/Chol in 1:1 molar ratio | Curcumin | PA or CL or GM1 ganglioside | [386] | |
| Liposomes | DPPC/DPPG/Chol/Y (8:2:10:1 or 2 molar ratio) | Curcumin | [386] | ||
| Liposomes | DSPC/Chol (2:1 molar ratio) with DPS-PEG2000-Cur (10 or 20 mol %) | Curcumin | Anti-TfR mAb OX26 | [387] | |
| Micelles | PS80 | Curcumin | [388] | ||
| Gold NPs | Silica-coated Au NPs | Curcumin | [389] | ||
| NLCs | PC/Chol oleate/glycerol trioleate (1:0.06:0.21 molar ratio) | Curcumin | Lactoferrin | [390] | |
| Polymeric NPs | PAAM-CL-PLGA NPs | Curcumin and rosmarinic acid | 83–14 mAb | [393] | |
| SLNs | Cetyl palmitate and PS80 as stabilizer | Resveratrol, grape skin and seed extracts | Anti-TfR mAb OX26 | [397] | |
| SLNs | Cetyl palmitate and PS80 as stabilizer | Resveratrol | ApoE peptide | [398] | |
| SLNs | Compritol 888 ATO (lipid matrix), Epikuron 200 (surfactant) and sodium taurocholate (co-surfactant) | Ferulic acid | [399] | ||
| Liposomes | DSPC/Chol/DSPE-PEG2000/DSPE-PEG2000-COOH (2:1:0.11:0.021 molar ratio) | α-mangostin | Transferrin | [400] | |
| Gold NPs | PEG-coated AuNPS | Anthocyanins | [401] | ||
| Polymeric NPs | PEG-PLGA NPs | Anthocyanins | [402] | ||
| Selenium NPs | EGCG | Tet-1 peptide | [403] | ||
| Polymeric NPs | PEG-PLGA NPs | EGCG and ascorbic acid | [404] | ||
| Liposomes | DPPC/Chol/DHDP (5:4:1 molar ratio) | Quercetin and rosmarinic acid | ApoE peptide and PA | [405] | |
| Parkinson | Liposomes | PC/Chol (7:3 molar ratio) with DSPE-PEG2000-COOH (2.5 mol %) | Dopamine | Transferrin | [409] |
| Polymeric NPs | Chitosan | Dopamine | [410] | ||
| Polymeric NPs | PLGA NPs | Dopamine | [411] | ||
| Polymeric NPs | PBCA NPs and poloxamer 188 as stabilizer | Dopamine | [412] | ||
| Nanogels | PVP/PAAc | Dopamine | [413] | ||
| Liposomes | HSPC/Chol/DSPE-PEG 20:10:2 molar ratio | l-DOPA, dopamine precursor | Chlorotoxin peptide | [415] | |
| Microspheres | PLGA NPs | Rotigotine, dopamine agonist | [416] | ||
| Polymeric NPs | PS80-coated chitosan | Ropinirole, dopamine agonist | [417] | ||
| SLNs | Dynasan-114 (solid lipid), soylecithin (primary surfactant) and poloxamer 188 (secondary surfactant) | Ropinirole, dopamine agonist | [418] | ||
| NLCs | Dynasan-114 (solid lipid), Caproyl 90 (liquid lipid) soylecithin (primary surfactant) and poloxamer 188 (secondary surfactant) | Ropinirole, dopamine agonist | [418] | ||
| MADs | Glyceryl monooleate and poloxamer 407 | Bromocriptine, dopamine agonist | [419] | ||
| NLCs | Tristearin/Miglyol 2:1 molar ratio with poloxamer | Bromocriptine, dopamine agonist | [419] | ||
| Polymeric NPs | PLGA and PEG-PLGA NPs | Urocortin | Lactoferrin | [420] | |
| Zwitterionic polimers | PMPC-coated acrylated BSA | Non-Fe hemin, iron chelator | TAT peptide | [421] | |
| Micelles | PTS | Coenzyme Q10, antioxidant | [422] | ||
| Nanocrystals | Pluronic F68 | schisantherin A, antioxidant | [423] | ||
| Polymeric NPs | mPEG–PLGA NPs | schisantherin A, antioxidant | [424] | ||
| Micelles | Curcumin | Lactoferrin | [425] | ||
| Cerasomes | PS80-modified cerasome-forming lipid N-[N-(3-triethoxysilyl)propylsuccinamoyl]-di-hexadecylamine | Curcumin | [426] | ||
| Polymeric NPs | Sodium alginate | Curcumin | [427] | ||
| Liposomes | Glyceryl monooleate NPs coated with Pluronic F-68 and vitamin E−TPGS | Curcumin and piperine | [430] | ||
| Spongosomes and cubosomes | Monoolein | Curcumin and fish oil | [431] | ||
| Polymeric NPs | PS80-coated PLA NPs | Resveratrol | [432] | ||
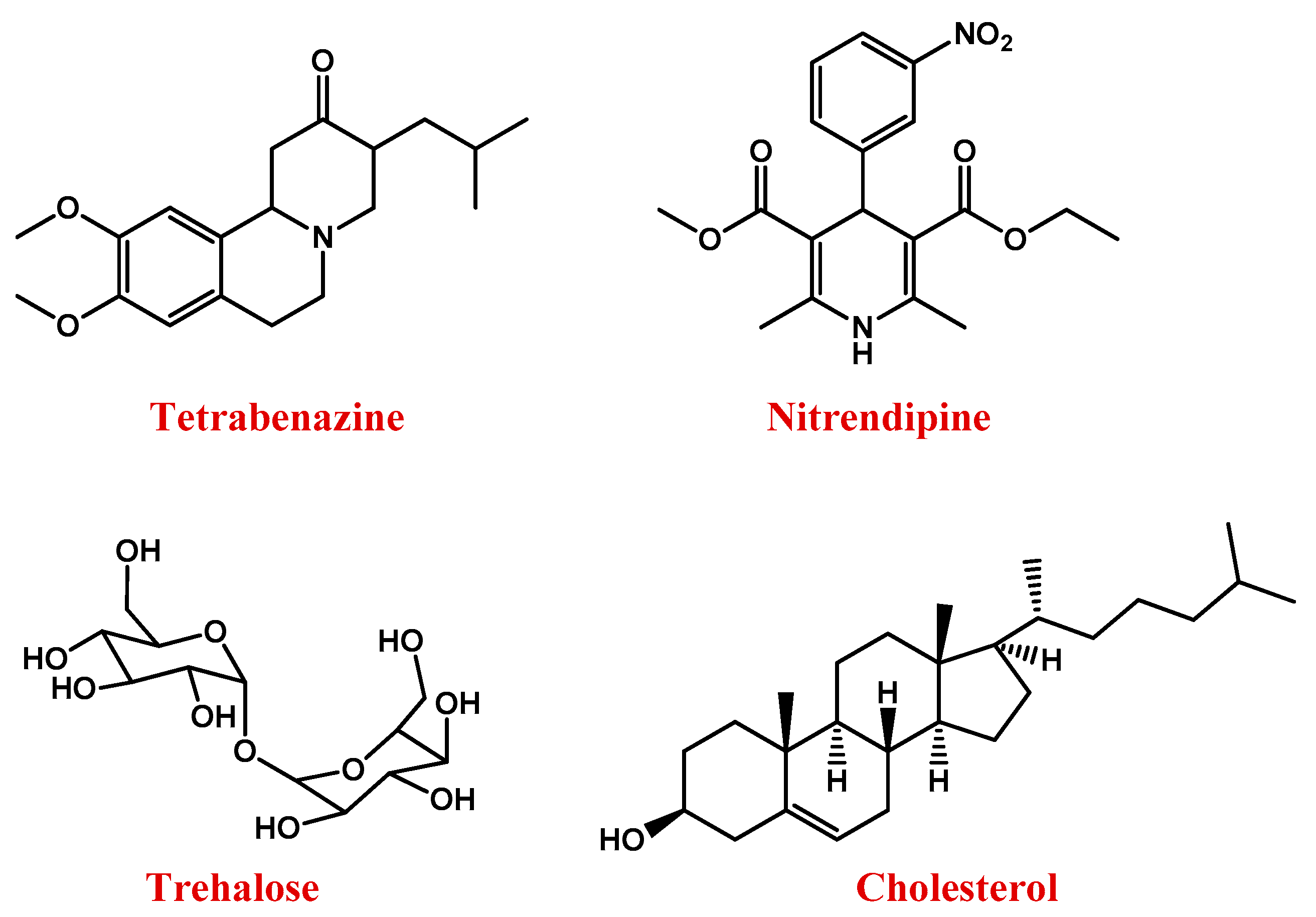
| Huntington | SLNs | Triglycerides (trimyristin, tripalmitin and tristearin), soy PC 95%, poloxamer 188 and charge modifiers (dicetyl phosphate and stearylamine) | Nitrendipine | [433] | |
| Magnetic NPs | Iron oxide core and switterionic polyacrylate shell | Trehalose | [434] | ||
| Polymeric NPs | PLGA NPs | Cholesterol | g7 glycopeptide | [435] | |
| SLNs | Stearic acid (solid core), lecithin (surfactant) and taurocholate (co-surfactant) | Curcumin | [436] | ||
| SLNs | Stearic acid (solid core), lecithin (surfactant) and taurocholate (co-surfactant) | Thymoquinone | [438,439] | ||
| Polymeric micelles | Polyaspartic acid | Quercetin | [440] | ||
| Polymeric NPs | PL NPs modified with trehalose/dopamine/arginine | Catechin | [441] | ||
| Polymeric NPs | PLGA NPs | PGQ9 peptide | [443] | ||
| Oligosaccharide-based NPs | β-cyclodextrin | siRNA | [445] |
6. Nanosystems for Nose-to-Brain Drug Delivery in the Treatment of Neurodegenerative Diseases
6.1. Alzheimer’s Disease
6.2. Parkinson’s Disease
| Disease | Nanocarrier Platform | Composition | Bioactive Agent | Active Targeting Ligand | Ref. |
|---|---|---|---|---|---|
| Alzheimer | Nanosuspensions | Chitosan | Donepezil, cholinesterase inhibitor | [473] | |
| Liposomes | DSPC/Chol/PEG (1:2:0.5 molar ratio) | Donepezil, cholinesterase inhibitor | [474] | ||
| Liposomes | Chol and EYPC, partly enriched with α-tocopherol and/or omega-3 fatty acids | Tacrine, cholinesterase inhibitor | [475] | ||
| Albumin NPs | β-CD, hydroxypropyl β-CD or sulphobutylether β-CD | Tacrine, cholinesterase inhibitor | [476] | ||
| Liposomes | EPC/Chol/DSPE (1:1:0.06 molar ratio) | Rivastigmine, cholinesterase inhibitor | CPP: GLPRRRRRRRRR | [477] | |
| Nanoemulsions | Capmul MCM (oil phase), PS80 (surfactant), transcutol P (co-surfactant) | Rivastigmine, cholinesterase inhibitor | [478] | ||
| Polymeric NPs | Chitosan | Rivastigmine, cholinesterase inhibitor | [479] | ||
| Liposomes | Soy PC/Chol (30:0.2 molar ratio) | Galantamine, cholinesterase inhibitor | [480] | ||
| Polymeric NPs | PLGA NPs | R-flurbiprofen (or tarenflurbil), anti-inflammatory | [481] | ||
| SLNs | Glyceryl monostearate/stearic acid/soya lecithin (8:2.5:3.5 molar ratio) and PS20 (surfactant) | R-flurbiprofen (or tarenflurbil), anti-inflammatory | [481] | ||
| Polymeric NPs | PEG-PLA NPs | VIP peptide | Wheat germ agglutinin | [483] | |
| Liposomes | EPC/DSPE-PEG2000/Chol (20:1:5 molar ratio) | H102 peptide | [485] | ||
| Polymeric NPs | PEG-PLGA NPs | bFGF | Solanum tuberosum lectin | [486] | |
| SLNs | Chitosan-coated and uncoated SLNs composed of Witepsol E 85 solid triglycerides | BACE1 siRNA | RVG-9R | [490] | |
| Nanoemulsions | Mixture of Capmul MCM and Captex 500 (oil phase), Cremophor EL and PS80 (surfactants); PEG400 and transcutol P(co-surfactants) | Curcumin | [491] | ||
| Polymeric NPs | Chitosan with Poloxamer 188 as stabilizer | Piperine | [492] | ||
| Parkinson | SLNs | Glycol chitosan with 0.01% of PS85 | Dopamine | [493] | |
| Polymeric NPs | PEG-PLGA NPs | Dopamine | Lactoferrin and borneol | [494] | |
| Polymeric NPs | PLGA NPs | l-DOPA, dopamine precursor | Wheat germ agglutinin | [496] | |
| Polymeric NPs | PEG-PLGA NPs | Rotigotine, dopamine agonist | Lactoferrin | [497,498] | |
| Polymeric NPs | Chitosan | Rotigotine, dopamine agonist | [499] | ||
| Microparticles | PLGA/DPPC/TMC | Ropinirole, dopamine agonist | [500] | ||
| Polymeric NPs | Chitosan-coated and uncoated PLGA NPs | Ropinirole, dopamine agonist | [501] | ||
| Polymeric NPs | Chitosan and sodium tripolyphosphate (6:1 molar ratio) | Pramipexole, dopamine agonist | [502] | ||
| Polymeric NPs | Chitosan-coated PLGA NPs | Rasagiline, MAO-B inhibitor | [503] | ||
| Polymeric NPs | PEG-PLGA NPs | Urocortine | Odorranalectin | [505] | |
| Polymeric NPs | Chitosan | Naringenin | [507] | ||
| Nanoemulsions | vitamin E/Sefsol (oil phase, 1:1 molar ratio), PS80 (surfactant) and transcutol P (co-surfactant) | Resveratrol | [508] | ||
| Polymeric NPs | PLGA NPs | GER-UDCA | [509] | ||
| SLNs | ATO 888/Span 85 (3:1 molar ratio) | GER-UDCA | [509] | ||
| Liposomes | DOPC/Chol/SA (50:30:5 molar ratio) | GDNF | [510] | ||
| NLCs | Precirol ATO/Mygliol (1:1 molar ratio) as lipids, PS80 and poloxamer 188 as surfactants | GDNF | TAT peptide | [511] | |
| NLCs | 2% gelatin solution and 20% poloxamer 188 | bFGF | [512] | ||
| Huntington | Nanoemulsions | Capmul MCM (oil phase), PS80 (surfactant) and transcutol P (co-surfactant) | Tetrabenazine | [513] | |
| SLNs | Glycerol monostearate (lipid), PS80 and soya lecithin (surfactants), HSPC as stabilizer | Rosmarinic acid | [514] | ||
| Liposomes | PC | Cholesterol | [515] | ||
| Polymeric NPs | Chitosan | siRNA | [516] |
6.3. Huntington’s Disease
6.4. Clinical Trials
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dugger, B.N.; Dickson, D.W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, J.R.; Sriramoju, B.; Kanwar, R.K. Neurological disorders and therapeutics targeted to surmount the blood-brain barrier. Int. J. Nanomed. 2012, 7, 3259–3278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poovaiah, N.; Davoudi, Z.; Peng, H.; Schlichtmann, B.; Mallapragada, S.; Narasimhan, B.; Wang, Q. Treatment of neurodegenerative disorders through the blood-brain barrier using nanocarriers. Nanoscale 2018, 10, 16962–16983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Prim. 2015, 1, 15056. [Google Scholar] [CrossRef]
- Soria Lopez, J.A.; González, H.M.; Léger, G.C. Alzheimer’s disease. Handb. Clin. Neurol. 2019, 167, 231–255. [Google Scholar] [CrossRef]
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s disease: Past, present, and future. J. Int. Neuropsychol. Soc. 2017, 23, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Bellenguez, C.; Grenier-Boley, B.; Lambert, J.C. Genetics of Alzheimer’s disease: Where we are, and where we are going. Curr. Opin. Neurobiol. 2020, 61, 40–48. [Google Scholar] [CrossRef]
- Allgaier, M.; Allgaier, C. An update on drug treatment options of Alzheimer’s disease. Front. Biosci. Landmark 2014, 19, 1345–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug treatments in Alzheimer’s disease. Clin. Med. J. R. Coll. Physicians Lond. 2016, 16, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7, F1000. [Google Scholar] [CrossRef] [Green Version]
- Husna Ibrahim, N.; Yahaya, M.F.; Mohamed, W.; Teoh, S.L.; Hui, C.K.; Kumar, J. Pharmacotherapy of Alzheimer’s disease: Seeking clarity in a time of uncertainty. Front. Pharmacol. 2020, 11, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ittner, L.M.; Götz, J. Amyloid-β and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Nisbet, R.M.; Götz, J. Amyloid-β and tau in Alzheimer’s disease: Novel pathomechanisms and non-pharmacological treatment strategies. J. Alzheimers Dis. 2018, 64, S517–S527. [Google Scholar] [CrossRef]
- Gallardo, G.; Holtzman, D.M. Amyloid-β and tau at the crossroads of Alzheimer’s disease. Adv. Exp. Med. Biol. 2019, 1184, 187–203. [Google Scholar] [CrossRef]
- Shafiei, S.S.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. Tau oligomers: Cytotoxicity, propagation, and mitochondrial damage. Front. Aging Neurosci. 2017, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.H.; Zhang, Y.; Hong, X.Y.; Zhang, J.F.; Wang, J.Z.; Liu, G.P. Role of microtubule-associated protein tau phosphorylation in Alzheimer’s disease. J. Huazhong Univ. Sci. Technol. Med. Sci. 2017, 37, 307–312. [Google Scholar] [CrossRef]
- Gao, Y.; Tan, L.; Yu, J.-T.; Tan, L. Tau in Alzheimer’s disease: Mechanisms and therapeutic strategies. Curr. Alzheimer Res. 2018, 15, 283–300. [Google Scholar] [CrossRef] [PubMed]
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W. The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 2019, 705, 183–194. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [Green Version]
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid toxicity in Alzheimer’s disease. Rev. Neurosci. 2018, 29, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Amyloid β oligomers (AβOs) in Alzheimer’s disease. J. Neural Transm. 2018, 125, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.P.; Clark, I.A.; Vissel, B. Questions concerning the role of amyloid-β in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol. 2018, 136, 663–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkoe, D.J. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid β-protein. J. Alzheimers Dis. 2001, 3, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Walter, J. Phosphorylation of amyloid beta (Aβ) peptides—A trigger for formation of toxic aggregates in Alzheimer’s disease. Aging 2011, 3, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Nisha, C.M.; Silakari, C.; Sharma, I.; Anusha, K.; Gupta, N.; Nair, P.; Tripathi, T.; Kumar, A. Current and novel therapeutic molecules and targets in Alzheimer’s disease. J. Formos. Med. Assoc. 2016, 115, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment combinations for Alzheimer’s disease: Current and future pharmacotherapy options. J. Alzheimers Dis. 2019, 67, 779–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haake, A.; Nguyen, K.; Friedman, L.; Chakkamparambil, B.; Grossberg, G.T. An update on the utility and safety of cholinesterase inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Drug Saf. 2020, 19, 147–157. [Google Scholar] [CrossRef]
- Christodoulou, C.; Melville, P.; Scherl, W.F.; MacAllister, W.S.; Elkins, L.E.; Krupp, L.B. Effects of donepezil on memory and cognition in multiple sclerosis. J. Neurol. Sci. 2006, 245, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Summers, W.K. Tacrine, and Alzheimer’s treatments. J. Alzheimers Dis. 2006, 9, 439–445. [Google Scholar] [CrossRef]
- Birks, J.S.; Chong, L.Y.; Grimley Evans, J. Rivastigmine for Alzheimer’s disease. Cochrane Database Syst. Rev. 2015, CD001191. [Google Scholar] [CrossRef]
- Prvulovic, D.; Hampel, H.; Pantel, J. Galantamine for Alzheimer’s disease. Expert Opin. Drug Metab. Toxicol. 2010, 6, 345–354. [Google Scholar] [CrossRef]
- Daulatzai, M.A. Pharmacotherpy and Alzheimer’s disease: The M-drugs (melatonin, minocycline, modafinil, and memantine) approach. Curr. Pharm. Des. 2016, 22, 2411–2430. [Google Scholar] [CrossRef]
- Alam, S.; Lingenfelter, K.S.; Bender, A.M.; Lindsley, C.W. Classics in chemical neuroscience: Memantine. ACS Chem. Neurosci. 2017, 8, 1823–1829. [Google Scholar] [CrossRef]
- Matsunaga, S.; Kishi, T.; Nomura, I.; Sakuma, K.; Okuya, M.; Ikuta, T.; Iwata, N. The efficacy and safety of memantine for the treatment of Alzheimer’s disease. Expert Opin. Drug Saf. 2018, 17, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Vicidomini, C.; Cioffi, F.; Broersen, K.; Roviello, V.; Riccardi, C.; Montesarchio, D.; Capasso, D.; Gaetano, S.D.; Roviello, G.N. Benzodifurans for biomedical applications: BZ4, a selective anti-proliferative and anti-amyloid lead compound. Future Med. Chem. 2019, 11, 285–302. [Google Scholar] [CrossRef] [PubMed]
- Oliver, D.M.A.; Reddy, P.H. Small molecules as therapeutic drugs for Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 96, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, K.L.C.; Alcântara, M.G.D.S.; de Aquino, T.M.; da Silva-Júnior, E.F. Tau protein aggregation in Alzheimer’s disease: Recent advances in the development of novel therapeutic agents. Curr. Pharm. Des. 2020, 26, 1682–1692. [Google Scholar] [CrossRef]
- Jouanne, M.; Rault, S.; Voisin-Chiret, A.S. Tau protein aggregation in Alzheimer’s disease: An attractive target for the development of novel therapeutic agents. Eur. J. Med. Chem. 2017, 139, 153–167. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Pinheiro, L.; Faustino, C. Therapeutic strategies targeting amyloid-β in Alzheimer’s disease. Curr. Alzheimer Res. 2019, 16, 418–452. [Google Scholar] [CrossRef]
- Soto, C.; Kindy, M.S.; Baumann, M.; Frangione, B. Inhibition of Alzheimer’s amyloidosis by peptides that prevent β-sheet conformation. Biochem. Biophys. Res. Commun. 1996, 226, 672–680. [Google Scholar] [CrossRef]
- Soto, C.; Sigurdsson, E.M.; Morelli, L.; Kumar, R.A.; Castaño, E.M.; Frangione, B. β-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: Implications for Alzheimer’s therapy. Nat. Med. 1998, 4, 822–826. [Google Scholar] [CrossRef]
- Wood, S.J.; Wetzel, R.; Martin, J.D.; Hurle, M.R. Prolines and amyloidogenicity in fragments of the Alzheimer’s peptide β/A4. Biochemistry 1995, 34, 724–730. [Google Scholar] [CrossRef]
- Tjernberg, L.O.; Näslundt, J.; Lindqvist, F.; Johansson, J.; Karlström, A.R.; Thyberg, J.; Tereniust, L.; Nordstedt, C. Arrest of β-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 1996, 271, 8545–8548. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.L.; Strzelec, A.; Kiessling, L.L.; Murphy, R.M. Structure-function relationships for inhibitors of β-Amyloid toxicity containing the recognition sequence KLVFF. Biochemistry 2001, 40, 7882–7889. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.I.; Nakamura, K.; Akikusa, S.; Okada, T.; Kodaka, M.; Okuno, H.; Watanabe, K.I.; Akikusa, S.; Konakahara, T. Inhibitors of fibril formation and cytotoxicity of β-amyloid peptide composed of KLVFF recognition element and flexible hydrophilic disrupting element. Biochem. Biophys. Res. Commun. 2002, 290, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Chacón, M.A.; Barría, M.I.; Soto, C.; Inestrosa, N.C. β-sheet breaker peptide prevents Aβ-induced spatial memory impairments with partial reduction of amyloid deposits. Mol. Psychiatry 2004, 9, 953–961. [Google Scholar] [CrossRef] [Green Version]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Lu, J.; Combs, G.F. Penicillamine: Pharmacokinetics and differential effects on zinc and copper status in chicks. J. Nutr. 1992, 122, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Cherny, R.A.; Barnham, K.J.; Lynch, T.; Volitakis, I.; Li, Q.X.; McLean, C.A.; Multhaup, G.; Beyreuther, K.; Tanzi, R.E.; Masters, C.L.; et al. Chelation and intercalation: Complementary properties in a compound for the treatment of Alzheimer’s disease. J. Struct. Biol. 2000, 130, 209–216. [Google Scholar] [CrossRef]
- Huang, X.; Atwood, C.S.; Hartshorn, M.A.; Multhaup, G.; Goldstein, L.E.; Scarpa, R.C.; Cuajungco, M.P.; Gray, D.N.; Lim, J.; Moir, R.D.; et al. The Aβ peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 1999, 38, 7609–7616. [Google Scholar] [CrossRef]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.S.; et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef] [Green Version]
- Gasparini, L.; Ongini, E.; Wenk, G. Non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer’s disease: Old and new mechanisms of action. J. Neurochem. 2004, 91, 521–536. [Google Scholar] [CrossRef]
- Etminan, M.; Gill, S.; Samii, A. Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer’s disease: Systematic review and meta-analysis of observational studies. Br. Med. J. 2003, 327, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.A.; Lee, H.Y.; Bressan, R.A.; Yun, D.J.; Kim, M.O. Novel osmotin attenuates glutamate-induced synaptic dysfunction and neurodegeneration via the JNK/PI3K/Akt pathway in postnatal rat brain. Cell Death Dis. 2014, 5, e1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naseer, M.I.; Ullah, I.; Narasimhan, M.L.; Lee, H.Y.; Bressan, R.A.; Yoon, G.H.; Yun, D.J.; Kim, M.O. Neuroprotective effect of osmotin against ethanol-induced apoptotic neurodegeneration in the developing rat brain. Cell Death Dis. 2014, 5, e1150. [Google Scholar] [CrossRef] [Green Version]
- Ali, T.; Yoon, G.H.; Shah, S.A.; Lee, H.Y.; Kim, M.O. Osmotin attenuates amyloid beta-induced memory impairment, tau phosphorylation and neurodegeneration in the mouse hippocampus. Sci. Rep. 2015, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rüb, U.; De Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Radhakrishnan, D.M.; Goyal, V. Parkinson’s disease: A review. Neurol. India 2018, 66, S26–S35. [Google Scholar] [CrossRef]
- Mehra, S.; Sahay, S.; Maji, S.K. α-synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta-Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef]
- Reich, S.G.; Savitt, J.M. Parkinson’s Disease. Med. Clin. N. Am. 2019, 103, 337–350. [Google Scholar] [CrossRef]
- Rizzi, G.; Tan, K.R. Dopamine and acetylcholine, a circuit point of view in Parkinson’s disease. Front. Neural Circuits 2017, 11, 110. [Google Scholar] [CrossRef] [Green Version]
- Hayes, M.W.; Fung, V.S.C.; Kimber, T.E.; O’Sullivan, J.D. Updates and advances in the treatment of Parkinson disease. Med. J. Aust. 2019, 211, 277–283. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.L.E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; Stenroos, E.S. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 12, 170–178. [Google Scholar] [CrossRef]
- McNaught, K.S.P.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 2003, 179, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Archibald, N.; Miller, N.; Rochester, L. Neurorehabilitation in Parkinson disease. Handb. Clin. Neurol. 2013, 110, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Segura-Aguilar, J.; Paris, I.; Muñoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef]
- Alexoudi, A.; Alexoudi, I.; Gatzonis, S. Parkinson’s disease pathogenesis, evolution and alternative pathways: A review. Rev. Neurol. 2018, 174, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Meder, D.; Herz, D.M.; Rowe, J.B.; Lehéricy, S.; Siebner, H.R. The role of dopamine in the brain—lessons learned from Parkinson’s disease. Neuroimage 2019, 190, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, K.R.; Schapira, A.H. Non-motor symptoms of Parkinson’s disease: Dopaminergic pathophysiology and treatment. Lancet Neurol. 2009, 8, 464–474. [Google Scholar] [CrossRef]
- Schrag, A.; Sauerbier, A.; Chaudhuri, K.R. New clinical trials for nonmotor manifestations of Parkinson’s disease. Mov. Disord. 2015, 30, 1490–1504. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Shephard, B.C.; Daniel, S.E. Is Parkinson’s disease a primary olfactory disorder? QJM Mon. J. Assoc. Physicians 1999, 92, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Pillay, V.; Choonara, Y.E. Advances in the treatment of Parkinson’s disease. Prog. Neurobiol. 2007, 81, 29–44. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and treatment of Parkinson disease: A review. JAMA J. Am. Med. Assoc. 2020, 323, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, A.C.; Moore, B.T.; Daniels, R.N. Classics in chemical neuroscience: Levodopa. ACS Chem. Neurosci. 2014, 5, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S.; Poewe, W. Levodopa: 50 years of a revolutionary drug for Parkinson disease. Mov. Disord. 2015, 30, 1–3. [Google Scholar] [CrossRef]
- Nagatsua, T.; Sawadab, M. l-dopa therapy for Parkinson’s disease: Past, present, and future. Park. Relat. Disord. 2009, 15, S3–S8. [Google Scholar] [CrossRef]
- Haddad, F.; Sawalha, M.; Khawaja, Y.; Najjar, A.; Karaman, R. Dopamine and levodopa prodrugs for the treatment of Parkinson’s disease. Molecules 2018, 23, 40. [Google Scholar] [CrossRef] [Green Version]
- Tambasco, N.; Romoli, M.; Calabresi, P. Levodopa in Parkinson’s disease: Current status and future developments. Curr. Neuropharmacol. 2017, 16, 1239–1252. [Google Scholar] [CrossRef]
- Lewitt, P.A. Levodopa therapy for Parkinson’s disease: Pharmacokinetics and pharmacodynamics. Mov. Disord. 2015, 30, 64–72. [Google Scholar] [CrossRef]
- McAfee, D.A.; Hadgraft, J.; Lane, M.E. Rotigotine: The first new chemical entity for transdermal drug delivery. Eur. J. Pharm. Biopharm. 2014, 88, 586–593. [Google Scholar] [CrossRef]
- Chen, F.; Jin, L.; Nie, Z. Safety and efficacy of rotigotine for treating Parkinson’s disease: A meta-analysis of randomised controlled trials. J. Pharm. Pharm. Sci. 2017, 20, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Contin, M.; Lopane, G.; Mohamed, S.; Calandra-Buonaura, G.; Capellari, S.; De Massis, P.; Nassetti, S.; Perrone, A.; Riva, R.; Sambati, L.; et al. Clinical pharmacokinetics of pramipexole, ropinirole and rotigotine in patients with Parkinson’s disease. Park. Relat. Disord. 2019, 61, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Pahwa, R.; Lyons, K.E.; Hauser, R.A. Ropinirole therapy for Parkinson’s disease. Expert Rev. Neurother. 2004, 4, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Jost, W.H.; Angersbach, D. Ropinirole, a non-ergoline dopamine agonist. CNS Drug Rev. 2005, 11, 253–272. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, A.N.; Goldstein, M. Bromocriptine in Parkinson disease. Pharmacol. Rev. 1985, 37, 217–227. [Google Scholar] [PubMed]
- Antonini, A.; Barone, P.; Ceravolo, R.; Fabbrini, G.; Tinazzi, M.; Abbruzzese, G. Role of pramipexole in the management of Parkinsons disease. CNS Drugs 2010, 24, 829–841. [Google Scholar] [CrossRef]
- Shen, T.; Ye, R.; Zhang, B. Efficacy and safety of pramipexole extended-release in Parkinson’s disease: A review based on meta-analysis of randomized controlled trials. Eur. J. Neurol. 2017, 24, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Swope, D.M.; Dashtipour, K. Comprehensive review of rasagiline, a second-generation monoamine oxidase inhibitor, for the treatment of Parkinson’s disease. Clin. Ther. 2007, 29, 1825–1849. [Google Scholar] [CrossRef]
- McCormack, P.L. Rasagiline: A review of its use in the treatment of idiopathic parkinson’s disease. CNS Drugs 2014, 28, 1083–1097. [Google Scholar] [CrossRef]
- Schettino, C.; Dato, C.; Capaldo, G.; Sampaolo, S.; Di Iorio, G.; Melone, M.A.B. Rasagiline for sleep disorders in patients with Parkinson’s disease: A prospective observational study. Neuropsychiatr. Dis. Treat. 2016, 12, 2497–2502. [Google Scholar] [CrossRef] [Green Version]
- Facci, L.; Stevens, D.A.; Pangallo, M.; Franceschini, D.; Skaper, S.D.; Strijbos, P.J.L.M. Corticotropin-releasing factor (CRF) and related peptides confer neuroprotection via type 1 CRF receptors. Neuropharmacology 2003, 45, 623–636. [Google Scholar] [CrossRef]
- Choi, J.S.; Pham, T.T.H.; Jang, Y.J.; Bao, C.B.; Lee, B.H.; Joo, K.M.; Cha, C.I.; Lee, K.H. Corticotropin-releasing factor (CRF) and urocortin promote the survival of cultured cerebellar GABAergic neurons through the Type 1 CRF receptor. J. Korean Med. Sci. 2006, 21, 518–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.S.; Gertler, T.S.; Surmeier, D.J. A molecular basis for the increased vulnerability of substantia nigra dopamine neurons in aging and Parkinson’s disease. Mov. Disord. 2010, 25, S63–S70. [Google Scholar] [CrossRef]
- Das, B.; Vedachalam, S.; Luo, D.; Antonio, T.; Reith, M.E.A.; Dutta, A.K. Development of a highly pPotent D2/D3 agonist and a partial agonist from structure-activity relationship study of N6-(2-(4-(1H-indol-5-yl)piperazin-1-yl)ethyl)-N6-propyl-4,5,6,7-tetrahydrobenzo[d]thiazole-2,6-diamine analogues: Implication in the treatmen. J. Med. Chem. 2015, 58, 9179–9195. [Google Scholar] [CrossRef]
- Xu, Q.; Kanthasamy, A.G.; Reddy, M.B. Neuroprotective effect of the natural iron chelator, phytic acid in a cell culture model of Parkinson’s disease. Toxicology 2008, 245, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Q.; Sa, F.; Chong, C.M.; Wang, Y.; Zhou, Z.Y.; Chang, R.C.C.; Chan, S.W.; Hoi, P.M.; Yuen Lee, S.M. Schisantherin A protects against 6-OHDA-induced dopaminergic neuron damage in zebrafish and cytotoxicity in SH-SY5Y cells through the ROS/NO and AKT/GSK3β pathways. J. Ethnopharmacol. 2015, 170, 8–15. [Google Scholar] [CrossRef]
- Sa, F.; Zhang, L.Q.; Chong, C.M.; Guo, B.J.; Li, S.; Zhang, Z.J.; Zheng, Y.; Hoi, P.M.; Lee, S.M.Y. Discovery of novel anti-parkinsonian effect of schisantherin A in In Vitro and In Vivo. Neurosci. Lett. 2015, 593, 7–12. [Google Scholar] [CrossRef]
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal huntingtin function: An alternative approach to Huntington’s disease. Nat. Rev. Neurosci. 2005, 6, 919–930. [Google Scholar] [CrossRef]
- Melone, M.A.B.; Jori, F.; Peluso, G. Huntingtons disease: New frontiers for molecular and cell therapy. Curr. Drug Targets 2005, 6, 43–56. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Huntington’s disease. Handb. Exp. Pharmacol. 2015, 220, 357–409. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. The Huntington’s paradox. Sci. Am. 2016, 315, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Caterino, M.; Squillaro, T.; Montesarchio, D.; Giordano, A.; Giancola, C.; Melone, M.A.B. Huntingtin protein: A new option for fixing the Huntington’s disease countdown clock. Neuropharmacology 2018, 135, 126–138. [Google Scholar] [CrossRef]
- Cattaneo, E.; Rigamonti, D.; Goffredo, D.; Zuccato, C.; Squitieri, F.; Sipione, S. Loss of normal huntingtin function: New developments in Huntington’s disease research. Trends Neurosci. 2001, 24, 182–188. [Google Scholar] [CrossRef]
- Cattaneo, E. Dysfunction of wild-type huntingtin in Huntington disease. News Physiol. Sci. 2003, 18, 34–37. [Google Scholar] [CrossRef] [Green Version]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef]
- Melone, M.A.B.; Calarco, A.; Petillo, O.; Margarucci, S.; Colucci-D’Amato, L.; Galderisi, U.; Koverech, G.; Peluso, G. Mutant huntingtin regulates EGF receptor fate in non-neuronal cells lacking wild-type protein. Biochim. Biophys. Acta-Mol. Basis Dis. 2013, 1832, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kay, C.; Collins, J.A.; Wright, G.E.B.; Baine, F.; Miedzybrodzka, Z.; Aminkeng, F.; Semaka, A.J.; McDonald, C.; Davidson, M.; Madore, S.J.; et al. The molecular epidemiology of Huntington disease is related to intermediate allele frequency and haplotype in the general population. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2018, 177, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, M.T.; Fiocchetti, M.; Totta, P.; Melone, M.A.B.; Cardinale, A.; Fusco, F.R.; Gustincich, S.; Persichetti, F.; Ascenzi, P.; Marino, M. Huntingtin polyQ mutation impairs the 17β-estradiol/neuroglobin pathway devoted to neuron survival. Mol. Neurobiol. 2017, 54, 6634–6646. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Prim. 2015, 1, 15005. [Google Scholar] [CrossRef]
- Zheng, J.; Winderickx, J.; Franssens, V.; Liu, B. A mitochondria-associated oxidative stress perspective on Huntington’s disease. Front. Mol. Neurosci. 2018, 11, 329. [Google Scholar] [CrossRef]
- Vonsattel, J.P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P. Neuropathological classification of huntington’s disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Anzilotti, S.; Giampà, C.; Laurenti, D.; Perrone, L.; Bernardi, G.; Melone, M.A.B.; Fusco, F.R. Immunohistochemical localization of receptor for advanced glycation end (RAGE) products in the R6/2 mouse model of Huntington’s disease. Brain Res. Bull. 2012, 87, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Leuti, A.; Laurenti, D.; Giampà, C.; Montagna, E.; Dato, C.; Anzilotti, S.; Melone, M.A.B.; Bernardi, G.; Fusco, F.R. Phosphodiesterase 10A (PDE10A) localization in the R6/2 mouse model of Huntington’s disease. Neurobiol. Dis. 2013, 52, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, A.; Fusco, F.R.; Paldino, E.; Giampà, C.; Marino, M.; Nuzzo, M.T.; D’Angelo, V.; Laurenti, D.; Straccia, G.; Fasano, D.; et al. Localization of neuroglobin in the brain of R6/2 mouse model of Huntington’s disease. Neurol. Sci. 2018, 39, 275–285. [Google Scholar] [CrossRef]
- Perrone, L.; Melone, M.A.B. New targets for therapy in polyglutamine (polyQ) expansion diseases. Curr. Drug Ther. 2008, 3, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Fusco, F.R.; Anzilotti, S.; Giampà, C.; Dato, C.; Laurenti, D.; Leuti, A.; Colucci D’Amato, L.; Perrone, L.; Bernardi, G.; Melone, M.A.B. Changes in the expression of extracellular regulated kinase (ERK 1/2) in the R6/2 mouse model of Huntington’s disease after phosphodiesterase IV inhibition. Neurobiol. Dis. 2012, 46, 225–233. [Google Scholar] [CrossRef]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 5, 1–8. [Google Scholar] [CrossRef]
- Venuto, C.S.; Mcgarry, A.; Ma, Q.; Kieburtz, K. Pharmacologic approaches to the treatment of Huntington’s disease. Mov. Disord. 2012, 27, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Caron, N.S.; Dorsey, E.R.; Hayden, M.R. Therapeutic approaches to huntington disease: From the bench to the clinic. Nat. Rev. Drug Discov. 2018, 17, 729–750. [Google Scholar] [CrossRef]
- Poon, L.H.; Kang, G.A.; Lee, A.J. Role of tetrabenazine for Huntington’s disease-associated chorea. Ann. Pharmacother. 2010, 44, 1080–1089. [Google Scholar] [CrossRef]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 2004, 10, 148–154. [Google Scholar] [CrossRef]
- Leoni, V.; Mariotti, C.; Tabrizi, S.J.; Valenza, M.; Wild, E.J.; Henley, S.M.D.; Hobbs, N.Z.; Mandelli, M.L.; Grisoli, M.; Björkhem, I.; et al. Plasma 24S-hydroxycholesterol and caudate MRI in pre-manifest and early Huntington’s disease. Brain 2008, 131, 2851–2859. [Google Scholar] [CrossRef]
- Leoni, V.; Long, J.D.; Mills, J.A.; Di Donato, S.; Paulsen, J.S. Plasma 24S-hydroxycholesterol correlation with markers of Huntington disease progression. Neurobiol. Dis. 2013, 55, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Valenza, M.; Marullo, M.; Di Paolo, E.; Cesana, E.; Zuccato, C.; Biella, G.; Cattaneo, E. Disruption of astrocyte-neuron cholesterol cross talk affects neuronal function in Huntington’s disease. Cell Death Differ. 2015, 22, 690–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenza, M.; Leoni, V.; Karasinska, J.M.; Petricca, L.; Fan, J.; Carroll, J.; Pouladi, M.A.; Fossale, E.; Nguyen, H.P.; Riess, O.; et al. Cholesterol defect is marked across multiple rodent models of Huntington’s disease and is manifest in astrocytes. J. Neurosci. 2010, 30, 10844–10850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenza, M.; Rigamonti, D.; Goffredo, D.; Zuccato, C.; Fenu, S.; Jamot, L.; Strand, A.; Tarditi, A.; Woodman, B.; Racchi, M.; et al. Dysfunction of the cholesterol biosynthetic pathway in Huntington’s disease. J. Neurosci. 2005, 25, 9932–9939. [Google Scholar] [CrossRef]
- Valenza, M.; Leoni, V.; Tarditi, A.; Mariotti, C.; Björkhem, I.; Di Donato, S.; Cattaneo, E. Progressive dysfunction of the cholesterol biosynthesis pathway in the R6/2 mouse model of Huntington’s disease. Neurobiol. Dis. 2007, 28, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Valenza, M.; Carroll, J.B.; Leoni, V.; Bertram, L.N.; Björkhem, I.; Singaraja, R.R.; Di Donato, S.; Lutjohann, D.; Hayden, M.R.; Cattaneo, E. Cholesterol biosynthesis pathway is disturbed in YAC128 mice and is modulated by huntingtin mutation. Hum. Mol. Genet. 2007, 16, 2187–2198. [Google Scholar] [CrossRef] [Green Version]
- Valenza, M.; Cattaneo, E. Cholesterol dysfunction in neurodegenerative diseases: Is Huntington’s disease in the list? Prog. Neurobiol. 2006, 80, 165–176. [Google Scholar] [CrossRef]
- Valenza, M.; Cattaneo, E. Emerging roles for cholesterol in Huntington’s disease. Trends Neurosci. 2011, 6, 919–930. [Google Scholar] [CrossRef]
- Valenza, M.; Cattaneo, E. Neuroprotection and brain cholesterol biosynthesis in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, E143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, D.K.; Scharfman, H.E. Brain-derived neurotrophic factor. Growth Factors 2004, 22, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog. Neurobiol. 2007, 81, 294–330. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Giampà, C.; Montagna, E.; Dato, C.; Melone, M.A.B.; Bernardi, G.; Fusco, F.R. Systemic delivery of recombinant brain derived neurotrophic factor (BDNF) in the R6/2 mouse model of Huntington’s disease. PLoS ONE 2013, 8, e64037. [Google Scholar] [CrossRef] [PubMed]
- Axelsen, T.M.; Woldbye, D.P.D. Gene therapy for Parkinson’s disease, an update. J. Parkinsons. Dis. 2018, 8, 195–215. [Google Scholar] [CrossRef] [Green Version]
- Allen, S.J.; Watson, J.J.; Shoemark, D.K.; Barua, N.U.; Patel, N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013, 138, 155–175. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative stress in neurodegenerative diseases: From molecular mechanisms to clinical applications. Oxidative Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidatives stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative stress in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Linseman, D.A. Therapeutic antioxidants for neurodegenerative disease. Recent Pat. CNS Drug Discov. 2012, 7, 183. [Google Scholar] [CrossRef] [PubMed]
- Vidoni, C.; Castiglioni, A.; Seca, C.; Secomandi, E.; Melone, M.A.B.; Isidoro, C. Dopamine exacerbates mutant Huntingtin toxicity via oxidative-mediated inhibition of autophagy in SH-SY5Y neuroblastoma cells: Beneficial effects of anti-oxidant therapeutics. Neurochem. Int. 2016, 101, 132–143. [Google Scholar] [CrossRef]
- Squillaro, T.; Cimini, A.; Peluso, G.; Giordano, A.; Melone, M.A.B. Nano-delivery systems for encapsulation of dietary polyphenols: An experimental approach for neurodegenerative diseases and brain tumors. Biochem. Pharmacol. 2018, 154, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Squillaro, T.; Schettino, C.; Sampaolo, S.; Galderisi, U.; Di Iorio, G.; Giordano, A.; Melone, M.A.B. Adult-onset brain tumors and neurodegeneration: Are polyphenols protective? J. Cell. Physiol. 2018, 233, 3955–3967. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.; Sampaolo, S.; Melone, M.A.B. Bioactive phenolic compounds in the modulation of central and peripheral nervous system cancers: Facts and misdeeds. Cancers 2020, 12, 454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, M.S.; Al Mamun, A.; Kabir, M.T.; Ahmad, J.; Jeandet, P.; Sarwar, M.S.; Ashraf, G.M.; Aleya, L. Neuroprotective role of polyphenols against oxidative stress-mediated neurodegeneration. Eur. J. Pharmacol. 2020, 886, 173412. [Google Scholar] [CrossRef]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.E.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (poly)phenolics in human health: Structures, bioavailability, and evidence of protective effects against chronic diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892. [Google Scholar] [CrossRef] [Green Version]
- Fraga, C.G.; Croft, K.D.; Kennedy, D.O.; Tomás-Barberán, F.A. The effects of polyphenols and other bioactives on human health. Food Funct. 2019, 10, 514–528. [Google Scholar] [CrossRef] [Green Version]
- Sandur, S.K.; Pandey, M.K.; Sung, B.; Ahn, K.S.; Murakami, A.; Sethi, G.; Limtrakul, P.; Badmaev, V.; Aggarwal, B.B. Curcumin, demethoxycurcumin, bisdemethoxycurcumin, tetrahydrocurcumin and turmerones differentially regulate anti-inflammatory and anti-proliferative responses through a ROS-independent mechanism. Carcinogenesis 2007, 28, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Perrone, D.; Ardito, F.; Giannatempo, G.; Dioguardi, M.; Troiano, G.; Lo Russo, L.; De Lillo, A.; Laino, L.; Lo Muzio, L. Biological and therapeutic activities, and anticancer properties of curcumin. Exp. Ther. Med. 2015, 10, 1615–1623. [Google Scholar] [CrossRef] [Green Version]
- Stanić, Z. Curcumin, a compound from natural sources, a true scientific challenge—A review. Plant Foods Hum. Nutr. 2017, 72, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.; Squillaro, T.; Napolitano, F.; Terracciano, C.; Sampaolo, S.; Melone, M.A.B. The autophagy signaling pathway: A potential multifunctional therapeutic target of curcumin in neurological and neuromuscular diseases. Nutrients 2019, 11, 1881. [Google Scholar] [CrossRef] [Green Version]
- Goozee, K.G.; Shah, T.M.; Sohrabi, H.R.; Rainey-Smith, S.R.; Brown, B.; Verdile, G.; Martins, R.N. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br. J. Nutr. 2016, 115, 449–465. [Google Scholar] [CrossRef]
- Tang, M.; Taghibiglou, C.; Liu, J. The mechanisms of action of curcumin in Alzheimer’s disease. J. Alzheimers Dis. 2017, 58, 1003–1016. [Google Scholar] [CrossRef]
- Hickey, M.A.; Zhu, C.; Medvedeva, V.; Lerner, R.P.; Patassini, S.; Franich, N.R.; Maiti, P.; Frautschy, S.A.; Zeitlin, S.; Levine, M.S.; et al. Improvement of neuropathology and transcriptional deficits in CAG 140 knock-in mice supports a beneficial effect of dietary curcumin in Huntington’s disease. Mol. Neurodegener. 2012, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Finicelli, M.; Squillaro, T.; Di Cristo, F.; Di Salle, A.; Melone, M.A.B.; Galderisi, U.; Peluso, G. Metabolic syndrome, mediterranean diet, and polyphenols: Evidence and perspectives. J. Cell. Physiol. 2019, 234, 5807–5826. [Google Scholar] [CrossRef]
- Yang, K.Y.; Lin, L.C.; Tseng, T.Y.; Wang, S.C.; Tsai, T.H. Oral bioavailability of curcumin in rat and the herbal analysis from curcuma longa by LC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 853, 183–189. [Google Scholar] [CrossRef] [PubMed]
- R Neves, A.; Lucio, M.; LC Lima, J.; Reis, S. Resveratrol in medicinal chemistry: A critical review of its pharmacokinetics, drug-delivery, and membrane interactions. Curr. Med. Chem. 2012, 19, 1663–1681. [Google Scholar] [CrossRef]
- Markus, M.A.; Morris, B.J. Resveratrol in prevention and treatment of common clinical conditions of aging. Clin. Interv. Aging 2008, 3, 331–339. [Google Scholar] [CrossRef]
- Wu, C.F.; Yang, J.Y.; Wang, F.; Wang, X.X. Resveratrol: Botanical origin, pharmacological activity and applications. Chin. J. Nat. Med. 2013, 11, 1–15. [Google Scholar] [CrossRef]
- Tellone, E.; Galtieri, A.; Russo, A.; Giardina, B.; Ficarra, S. Resveratrol: A focus on several neurodegenerative diseases. Oxidative Med. Cell. Longev. 2015, 2015, 392169. [Google Scholar] [CrossRef] [Green Version]
- Vidoni, C.; Secomandi, E.; Castiglioni, A.; Melone, M.A.B.; Isidoro, C. Resveratrol protects neuronal-like cells expressing mutant Huntingtin from dopamine toxicity by rescuing ATG4-mediated autophagosome formation. Neurochem. Int. 2018, 117, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Anekonda, T.S. Resveratrol-A boon for treating Alzheimer’s disease? Brain Res. Rev. 2006, 52, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Alkam, T.; Nitta, A.; Mizoguchi, H.; Itoh, A.; Nabeshima, T. A natural scavenger of peroxynitrites, rosmarinic acid, protects against impairment of memory induced by Aβ(25–35). Behav. Brain Res. 2007, 180, 139–145. [Google Scholar] [CrossRef]
- Srinivasan, M.; Sudheer, A.R.; Menon, V.P. Ferulic acid: Therapeutic potential through its antioxidant property. J. Clin. Biochem. Nutr. 2007, 40, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, C.; Santangelo, R. Ferulic acid: Pharmacological and toxicological aspects. Food Chem. Toxicol. 2014, 65, 185–195. [Google Scholar] [CrossRef]
- Chen, G.; Li, Y.; Wang, W.; Deng, L. Bioactivity and pharmacological properties of α-mangostin from the mangosteen fruit: A review. Expert Opin. Ther. Pat. 2018, 28, 415–427. [Google Scholar] [CrossRef]
- Chen, L.G.; Yang, L.L.; Wang, C.C. Anti-inflammatory activity of mangostins from Garcinia mangostana. Food Chem. Toxicol. 2008, 46, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Pedraza-Chaverrí, J.; Reyes-Fermín, L.M.; Nolasco-Amaya, E.G.; Orozco-Ibarra, M.; Medina-Campos, O.N.; González-Cuahutencos, O.; Rivero-Cruz, I.; Mata, R. ROS scavenging capacity and neuroprotective effect of α-mangostin against 3-nitropropionic acid in cerebellar granule neurons. Exp. Toxicol. Pathol. 2009, 61, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xia, Z.; Xu, J.R.; Wang, Y.X.; Hou, L.N.; Qiu, Y.; Chen, H.Z. α-mangostin, a polyphenolic xanthone derivative from mangosteen, attenuates β-amyloid oligomers-induced neurotoxicity by inhibiting amyloid aggregation. Neuropharmacology 2012, 62, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Do, H.T.T.; Cho, J. Mangosteen pericarp and its bioactive xanthones: Potential therapeutic value in Alzheimer’s disease, Parkinson’s disease, and depression with pharmacokinetic and safety profiles. Int. J. Mol. Sci. 2020, 21, 6211. [Google Scholar] [CrossRef] [PubMed]
- Giusti, M.M.; Wrolstad, R.E. Acylated anthocyanins from edible sources and their applications in food systems. Biochem. Eng. J. 2003, 14, 217–225. [Google Scholar] [CrossRef]
- Zafra-Stone, S.; Yasmin, T.; Bagchi, M.; Chatterjee, A.; Vinson, J.A.; Bagchi, D. Berry anthocyanins as novel antioxidants in human health and disease prevention. Mol. Nutr. Food Res. 2007, 51, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Konishi, T. Anthocyanins and anthocyanin-rich extracts: Role in diabetes and eye function. Asia Pac. J. Clin. Nutr. 2007, 16, 200–208. [Google Scholar] [CrossRef]
- Weinreb, O.; Amit, T.; Mandel, S.; Youdim, M.B.H. Neuroprotective molecular mechanisms of (-)-epigallocatechin-3-gallate: A reflective outcome of its antioxidant, iron chelating and neuritogenic properties. Genes Nutr. 2009, 4, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Ehrnhoefer, D.E.; Duennwald, M.; Markovic, P.; Wacker, J.L.; Engemann, S.; Roark, M.; Legleiter, J.; Marsh, J.L.; Thompson, L.M.; Lindquist, S.; et al. Green tea (-)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Hum. Mol. Genet. 2006, 15, 2743–2751. [Google Scholar] [CrossRef]
- Rezai-Zadeh, K.; Arendash, G.W.; Hou, H.; Fernandez, F.; Jensen, M.; Runfeldt, M.; Shytle, R.D.; Tan, J. Green tea epigallocatechin-3-gallate (EGCG) reduces β-amyloid mediated cognitive impairment and modulates tau pathology in Alzheimer transgenic mice. Brain Res. 2008, 1214, 177–187. [Google Scholar] [CrossRef]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, G. Quercetin: A flavonol with multifaceted therapeutic applications? Fitoterapia 2015, 106, 256–271. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.G.; Garrick, J.M.; Roquè, P.J.; Pellacani, C. Mechanisms of neuroprotection by quercetin: Counteracting oxidative stress and more. Oxidative Med. Cell. Longev. 2016, 2016, 2986796. [Google Scholar] [CrossRef] [Green Version]
- Elumalai, P.; Lakshmi, S. Role of quercetin benefits in neurodegeneration. Adv. Neurobiol. 2016, 12, 229–245. [Google Scholar] [CrossRef]
- Amanzadeh, E.; Esmaeili, A.; Rahgozar, S.; Nourbakhshnia, M. Application of quercetin in neurological disorders: From nutrition to nanomedicine. Rev. Neurosci. 2019, 30, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Kanter, M. Nigella sativa and derived thymoquinone prevents hippocampal neurodegeneration after chronic toluene exposure in rats. Neurochem. Res. 2008, 33, 579–588. [Google Scholar] [CrossRef]
- Radad, K.; Moldzio, R.; Taha, M.; Rausch, W.D. Thymoquinone protects dopaminergic neurons against MPP+ and rotenone. Phyther. Res. 2009, 23, 696–700. [Google Scholar] [CrossRef]
- Abdelmeguid, N.E.; Fakhoury, R.; Kamal, S.M.; Al Wafai, R.J. Effects of Nigella sativa and thymoquinone on biochemical and subcellular changes in pancreatic β-cells of streptozotocin-induced diabetic rats. J. Diabetes 2010, 84, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Gilhotra, N.; Dhingra, D. Thymoquinone produced antianxiety-like effects in mice through modulation of GABA and NO levels. Pharmacol. Rep. 2011, 63, 660–669. [Google Scholar] [CrossRef]
- Islam, F.; Khan, A.; Vaibhav, K.; Javed, H.; Moshahid Khan, M.; Tabassum, R.; Ahmed, M.E.; Srivastava, P.; Khuwaja, G.; Islam, F.; et al. Attenuation of Aβ-induced neurotoxicity by thymoquinone via inhibition of mitochondrial dysfunction and oxidative stress. Mol. Cell. Biochem. 2012, 369, 55–65. [Google Scholar] [CrossRef]
- Ismail, N.; Ismail, M.; Azmi, N.H.; Abu Bakar, M.F.; Basri, H.; Abdullah, M.A. Modulation of hydrogen peroxide-induced oxidative stress in human neuronal cells by thymoquinone-rich fraction and thymoquinone via transcriptomic regulation of antioxidant and apoptotic signaling genes. Oxidative Med. Cell. Longev. 2016, 2016, 2528935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- De Boer, A.G.; Gaillard, P.J. Drug targeting to the brain. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 323–355. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-brain barrier drug targeting: The future of brain drug development. Mol. Interv. 2003, 3, 90–105. [Google Scholar] [CrossRef]
- Begley, D.J. Delivery of therapeutic agents to the central nervous system: The problems and the possibilities. Pharmacol. Ther. 2004, 104, 29–45. [Google Scholar] [CrossRef]
- Bors, L.A.; Erdö, F. Overcoming the blood-brain barrier. Challenges and tricks for CNS drug delivery. Sci. Pharm. 2019, 87, 6. [Google Scholar] [CrossRef] [Green Version]
- Begley, D.J.; Brightman, M.W. Structural and functional aspects of the blood-brain barrier. Prog. Drug Res. 2003, 37, 13–25. [Google Scholar] [CrossRef]
- Wolburg, H.; Lippoldt, A. Tight junctions of the blood-brain barrier: Development, composition and regulation. Vascul. Pharmacol. 2002, 38, 323–337. [Google Scholar] [CrossRef]
- Persidsky, Y.; Ramirez, S.H.; Haorah, J.; Kanmogne, G.D. Blood-brain barrier: Structural components and function under physiologic and pathologic conditions. J. Neuroimmune Pharmacol. 2006, 1, 223–236. [Google Scholar] [CrossRef]
- Harilal, S.; Jose, J.; Parambi, D.G.T.; Kumar, R.; Unnikrishnan, M.K.; Uddin, M.S.; Mathew, G.E.; Pratap, R.; Marathakam, A.; Mathew, B. Revisiting the blood-brain barrier: A hard nut to crack in the transportation of drug molecules. Brain Res. Bull. 2020, 160, 121–140. [Google Scholar] [CrossRef]
- Bauer, H.C.; Krizbai, I.A.; Bauer, H.; Traweger, A. “You shall not pass”—tight junctions of the blood brain barrier. Front. Neurosci. 2014, 8, 392. [Google Scholar] [CrossRef]
- Serlin, Y.; Shelef, I.; Knyazer, B.; Friedman, A. Anatomy and physiology of the blood-brain barrier. Semin. Cell Dev. Biol. 2015, 38, 2–6. [Google Scholar] [CrossRef] [Green Version]
- Sharif, Y.; Jumah, F.; Coplan, L.; Krosser, A.; Sharif, K.; Tubbs, R.S. Blood brain barrier: A review of its anatomy and physiology in health and disease. Clin. Anat. 2018, 31, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-brain barrier: From physiology to disease and back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Teleanu, D.M.; Negut, I.; Grumezescu, V.; Grumezescu, A.M.; Teleanu, R.I. Nanomaterials for drug delivery to the central nervous system. Nanomaterials 2019, 9, 371. [Google Scholar] [CrossRef] [Green Version]
- Nuriya, M.; Shinotsuka, T.; Yasui, M. Diffusion properties of molecules at the blood-brain interface: Potential contributions of astrocyte endfeet to diffusion barrier functions. Cereb. Cortex 2013, 23, 2118–2126. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, S.; Archunan, G.; Sivakumar, M.; Selvan, S.T.; Fred, A.L.; Kumar, S.; Gulyás, B.; Padmanabhan, P. Theranostic applications of nanoparticles in neurodegenerative disorders. Int. J. Nanomed. 2018, 13, 5561–5576. [Google Scholar] [CrossRef] [Green Version]
- Gabathuler, R. Approaches to transport therapeutic drugs across the blood-brain barrier to treat brain diseases. Neurobiol. Dis. 2010, 37, 48–57. [Google Scholar] [CrossRef]
- Matsumoto, J.; Stewart, T.; Banks, W.A.; Zhang, J. The transport mechanism of extracellular vesicles at the blood-brain barrier. Curr. Pharm. Des. 2018, 23, 6206–6214. [Google Scholar] [CrossRef]
- Juillerat-Jeanneret, L. The targeted delivery of cancer drugs across the blood-brain barrier: Chemical modifications of drugs or drug-nanoparticles? Drug Discov. Today 2008, 13, 1099–1106. [Google Scholar] [CrossRef]
- McEwen, B.S.; Reagan, L.P. Glucose transporter expression in the central nervous system: Relationship to synaptic function. Eur. J. Pharmacol. 2004, 490, 13–24. [Google Scholar] [CrossRef]
- Devraj, K.; Klinger, M.E.; Myers, R.L.; Mokashi, A.; Hawkins, R.A.; Simpson, I.A. GLUT-1 glucose transporters in the blood-brain barrier: Differential phosphorylation. J. Neurosci. Res. 2011, 89, 1913–1925. [Google Scholar] [CrossRef] [Green Version]
- del Amo, E.M.; Urtti, A.; Yliperttula, M. Pharmacokinetic role of L-type amino acid transporters LAT1 and LAT2. Eur. J. Pharm. Sci. 2008, 35, 161–174. [Google Scholar] [CrossRef]
- Gliddon, C.M.; Shao, Z.; LeMaistre, J.L.; Anderson, C.M. Cellular distribution of the neutral amino acid transporter subtype ASCT2 in mouse brain. J. Neurochem. 2009, 108, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Neuwelt, E.A.; Bauer, B.; Fahlke, C.; Fricker, G.; Iadecola, C.; Janigro, D.; Leybaert, L.; Molnár, Z.; O’Donnell, M.E.; Povlishock, J.T.; et al. Engaging neuroscience to advance translational research in brain barrier biology. Nat. Rev. Neurosci. 2011, 12, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharom, F.J. The P-glycoprotein multidrug transporter. Essays Biochem. 2011, 50, 161–178. [Google Scholar] [CrossRef] [Green Version]
- Taylor, E.M. The impact of efflux transporters in the brain on the development of drugs for CNS disorders. Clin. Pharmacokinet. 2002, 41, 81–92. [Google Scholar] [CrossRef]
- Potschka, H. Role of CNS efflux drug transporters in antiepileptic drug delivery: Overcoming CNS efflux drug transport. Adv. Drug Deliv. Rev. 2012, 64, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.T.; Zhao, Y.Z.; Wong, H.L.; Cai, J.; Peng, L.; Tian, X.Q. Current approaches to enhance CNS delivery of drugs across the brain barriers. Int. J. Nanomed. 2014, 9, 2241–2257. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, C.; Musumeci, D.; Russo Krauss, I.; Piccolo, M.; Irace, C.; Paduano, L.; Montesarchio, D. Exploring the conformational behaviour and aggregation properties of lipid-conjugated AS1411 aptamers. Int. J. Biol. Macromol. 2018, 118, 1384–1399. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, C.; Wang, L.; Chen, Y. A comprehensive review in improving delivery of small-molecule chemotherapeutic agents overcoming the blood-brain/brain tumor barriers for glioblastoma treatment. Drug Deliv. 2019, 26, 551–565. [Google Scholar] [CrossRef]
- Rueda, F.; Cruz, L.J. Targeting the brain with nanomedicine. Curr. Pharm. Des. 2017, 23, 1879–1896. [Google Scholar] [CrossRef]
- Patel, M.M.; Patel, B.M. Crossing the blood–brain barrier: Recent advances in drug delivery to the brain. CNS Drugs 2017, 31, 109–133. [Google Scholar] [CrossRef]
- Reynolds, J.L.; Mahato, R.I. Nanomedicines for the treatment of CNS diseases. J. Neuroimmune Pharmacol. 2017, 12, 1–5. [Google Scholar] [CrossRef]
- Zhou, Y.; Peng, Z.; Seven, E.S.; Leblanc, R.M. Crossing the blood-brain barrier with nanoparticles. J. Control. Release 2018, 270, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.T.; Lee, H.S.; Lee, J.J.; Park, E.K.; Lee, B.S.; Lee, J.Y.; Bae, J.S. Nanodelivery systems for overcoming limited transportation of therapeutic molecules through the blood-brain barrier. Future Med. Chem. 2018, 10, 2659–2674. [Google Scholar] [CrossRef]
- Saeedi, M.; Eslamifar, M.; Khezri, K.; Dizaj, S.M. Applications of nanotechnology in drug delivery to the central nervous system. Biomed. Pharmacother. 2019, 111, 666–675. [Google Scholar] [CrossRef]
- Sharma, G.; Sharma, A.R.; Lee, S.S.; Bhattacharya, M.; Nam, J.S.; Chakraborty, C. Advances in nanocarriers enabled brain targeted drug delivery across blood brain barrier. Int. J. Pharm. 2019, 559, 360–372. [Google Scholar] [CrossRef]
- Feng, L.; Wang, H.; Xue, X. Recent progress of nanomedicine in the treatment of central nervous system diseases. Adv. Ther. 2020, 3, 1900159. [Google Scholar] [CrossRef]
- Naqvi, S.; Panghal, A.; Flora, S.J.S. Nanotechnology: A promising approach for delivery of neuroprotective drugs. Front. Neurosci. 2020, 14, 494. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Lillard, J.W. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, S.A.A.; Saleh, A.M. Applications of nanoparticle systems in drug delivery technology. Saudi Pharm. J. 2018, 26, 64–70. [Google Scholar] [CrossRef]
- Choi, H.S.; Liu, W.; Misra, P.; Tanaka, E.; Zimmer, J.P.; Itty Ipe, B.; Bawendi, M.G.; Frangioni, J. V Renal clearance of nanoparticles. Nat. Biotechnol. 2007, 25, 1165–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betzer, O.; Shilo, M.; Opochinsky, R.; Barnoy, E.; Motiei, M.; Okun, E.; Yadid, G.; Popovtzer, R. The effect of nanoparticle size on the ability to cross the blood-brain barrier: An In Vivo study. Nanomedicine 2017, 12, 1533–1546. [Google Scholar] [CrossRef] [PubMed]
- Roduner, E. Size matters: Why nanomaterials are different. Chem. Soc. Rev. 2006, 35, 583–592. [Google Scholar] [CrossRef]
- Riehemann, K.; Schneider, S.W.; Luger, T.A.; Godin, B.; Ferrari, M.; Fuchs, H. Nanomedicine—Challenge and perspectives. Angew. Chem.-Int. Ed. Eng. 2009, 48, 872–897. [Google Scholar] [CrossRef] [Green Version]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; del Pilar Rodriguez-Torres, M.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Mout, R.; Moyano, D.F.; Rana, S.; Rotello, V.M. Surface functionalization of nanoparticles for nanomedicine. Chem. Soc. Rev. 2012, 41, 2539–2544. [Google Scholar] [CrossRef] [PubMed]
- Heinz, H.; Pramanik, C.; Heinz, O.; Ding, Y.; Mishra, R.K.; Marchon, D.; Flatt, R.J.; Estrela-Lopis, I.; Llop, J.; Moya, S.; et al. Nanoparticle decoration with surfactants: Molecular interactions, assembly, and applications. Surf. Sci. Rep. 2017, 72, 1–58. [Google Scholar] [CrossRef]
- Su, S.; Kang, P.M. Recent advances in nanocarrier-assisted therapeutics delivery systems. Pharmaceutics 2020, 12, 837. [Google Scholar] [CrossRef]
- Rabiei, M.; Kashanian, S.; Samavati, S.S.; Jamasb, S.; McInnes, S.J.P. Active targeting towards and inside the brain based on nanoparticles: A review. Curr. Pharm. Biotechnol. 2019, 21, 374–383. [Google Scholar] [CrossRef]
- Sharma, G.; Lakkadwala, S.; Modgil, A.; Singh, J. The role of cell-penetrating peptide and transferrin on enhanced delivery of drug to brain. Int. J. Mol. Sci. 2016, 17, 806. [Google Scholar] [CrossRef] [Green Version]
- Johnsen, K.B.; Burkhart, A.; Melander, F.; Kempen, P.J.; Vejlebo, J.B.; Siupka, P.; Nielsen, M.S.; Andresen, T.L.; Moos, T. Targeting transferrin receptors at the blood-brain barrier improves the uptake of immunoliposomes and subsequent cargo transport into the brain parenchyma. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Chen, J.; Gao, J. Nanocarriers as a powerful vehicle to overcome blood-brain barrier in treating neurodegenerative diseases: Focus on recent advances. Asian J. Pharm. Sci. 2019, 14, 480–496. [Google Scholar] [CrossRef]
- Johnsen, K.B.; Burkhart, A.; Thomsen, L.B.; Andresen, T.L.; Moos, T. Targeting the transferrin receptor for brain drug delivery. Prog. Neurobiol. 2019, 181, 101665. [Google Scholar] [CrossRef]
- Suzuki, Y.A.; Lopez, V.; Lönnerdal, B. Mammalian lactoferrin receptors: Structure and function. Cell. Mol. Life Sci. 2005, 62, 2560–2575. [Google Scholar] [CrossRef]
- Huang, R.Q.; Ke, W.L.; Qu, Y.H.; Zhu, J.H.; Pei, Y.Y.; Jiang, C. Characterization of lactoferrin receptor in brain endothelial capillary cells and mouse brain. J. Biomed. Sci. 2007, 14, 121–128. [Google Scholar] [CrossRef]
- Sabra, S.; Agwa, M.M. Lactoferrin, a unique molecule with diverse therapeutical and nanotechnological applications. Int. J. Biol. Macromol. 2020, 164, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, M.; Birch, D.; Nielsen, H.M. Applications and challenges for use of cell-penetrating peptides as delivery vectors for peptide and protein cargos. Int. J. Mol. Sci. 2016, 17, 185. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Wang, J.; Xu, D. Cell-penetrating peptides as noninvasive transmembrane vectors for the development of novel multifunctional drug-delivery systems. J. Control. Release 2016, 229, 130–139. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-penetrating peptides in diagnosis and treatment of human diseases: From preclinical research to clinical application. Front. Pharmacol. 2020, 11, 697. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. Selection In Vitro of single-stranded DNA molecules that fold into specific ligand-binding structures. Nature 1992, 355, 850–852. [Google Scholar] [CrossRef] [PubMed]
- Platella, C.; Riccardi, C.; Montesarchio, D.; Roviello, G.N.; Musumeci, D. G-quadruplex-based aptamers against protein targets in therapy and diagnostics. Biochim. Biophys. Acta-Gen. Subj. 2017, 1861, 1429–1447. [Google Scholar] [CrossRef]
- Musumeci, D.; Riccardi, C.; Montesarchio, D. G-quadruplex forming oligonucleotides as anti-HIV agents. Molecules 2015, 20, 17511–17532. [Google Scholar] [CrossRef] [Green Version]
- Nimjee, S.M.; White, R.R.; Becker, R.C.; Sullenger, B.A. Aptamers as therapeutics. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 61–79. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [Green Version]
- Ismail, S.I.; Alshaer, W. Therapeutic aptamers in discovery, preclinical and clinical stages. Adv. Drug Deliv. Rev. 2018, 134, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Morita, Y.; Leslie, M.; Kameyama, H.; Volk, D.E.; Tanaka, T. Aptamer therapeutics in cancer: Current and future. Cancers 2018, 10, 80. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.H.; Elsherbiny, M.E.; Emara, M. Updates on aptamer research. Int. J. Mol. Sci. 2019, 20, 2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lai, B.S.; Juhas, M. Recent advances in aptamer discovery and applications. Molecules 2019, 24, 941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riccardi, C.; Capasso, D.; Coppola, A.; Platella, C.; Montesarchio, D.; Di Gaetano, S.; Roviello, G.N.; Musumeci, D. Synthesis, antiproliferative activity and DNA binding studies of nucleoamino acid-containing Pt(II) complexes. Pharmaceuticals 2020, 13, 284. [Google Scholar] [CrossRef]
- Riccardi, C.; Napolitano, E.; Platella, C.; Musumeci, D.; Montesarchio, D. G-quadruplex-based aptamers targeting human thrombin: Discovery, chemical modifications and antithrombotic effects. Pharmacol. Ther. 2021, 217, 107649. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, C.; Napolitano, E.; Platella, C.; Musumeci, D.; Melone, M.A.B.; Montesarchio, D. Anti-VEGF DNA-based aptamers in cancer therapeutics and diagnostics. Med. Res. Rev. 2021, 41, 464–506. [Google Scholar] [CrossRef]
- Simone, R.; Fratta, P.; Neidle, S.; Parkinson, G.N.; Isaacs, A.M. G-quadruplexes: Emerging roles in neurodegenerative diseases and the non-coding transcriptome. FEBS Lett. 2015, 589, 1653–1668. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Yu, S.; Zheng, Y.; Zheng, Y.; Yang, H.; Zhang, J. Aptamer and its applications in neurodegenerative diseases. Cell. Mol. Life Sci. 2017, 74, 683–695. [Google Scholar] [CrossRef]
- Bouvier-Müller, A.; Ducongé, F. Nucleic acid aptamers for neurodegenerative diseases. Biochimie 2018, 145, 73–83. [Google Scholar] [CrossRef]
- Asamitsu, S.; Takeuchi, M.; Ikenoshita, S.; Imai, Y.; Kashiwagi, H.; Shioda, N. Perspectives for applying G-quadruplex structures in neurobiology and neuropharmacology. Int. J. Mol. Sci. 2019, 20, 2884. [Google Scholar] [CrossRef] [Green Version]
- Bhadra, D.; Bhadra, S.; Jain, P.; Jain, N.K. Pegnology: A review of PEG-ylated systems. Pharmazie 2002, 57, 5–29. [Google Scholar] [PubMed]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Xie, C.; Wang, H.; Hu, Y. Specific role of polysorbate 80 coating on the targeting of nanoparticles to the brain. Biomaterials 2004, 25, 3065–3071. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, J. Nanoparticulate systems for brain delivery of drugs. Adv. Drug Deliv. Rev. 2001, 47, 65–81. [Google Scholar] [CrossRef]
- Meng, H.; Leong, W.; Leong, K.W.; Chen, C.; Zhao, Y. Walking the line: The fate of nanomaterials at biological barriers. Biomaterials 2018, 174, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, C.; Praça, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-mediated brain drug delivery: Overcoming blood-brain barrier to treat neurodegenerative diseases. J. Control. Release 2016, 235, 34–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furtado, D.; Björnmalm, M.; Ayton, S.; Bush, A.I.; Kempe, K.; Caruso, F. Overcoming the blood–brain barrier: The role of nanomaterials in treating neurological diseases. Adv. Mater. 2018, 30, 1801362. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.R.; Yang, X.; Fu, M.; Zhai, G. Recent progress of drug nanoformulations targeting to brain. J. Control. Release 2018, 291, 37–64. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Cutler, E.G.; Cho, H. Therapeutic nanoplatforms and delivery strategies for neurological disorders. Nano Converg. 2018, 5, 1–15. [Google Scholar] [CrossRef]
- Masoudi Asil, S.; Ahlawat, J.; Guillama Barroso, G.; Narayan, M. Nanomaterial based drug delivery systems for the treatment of neurodegenerative diseases. Biomater. Sci. 2020, 8, 4088–4107. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.K.C.; Leong, L.I.; Liu, Y.; Luo, M.; Chan, H.Y.E.; Choi, C.H.J. Preclinical nanomedicines for polyglutamine-based neurodegenerative diseases. Mol. Pharm. 2020, 18, 610–626. [Google Scholar] [CrossRef]
- Patel, T.; Zhou, J.; Piepmeier, J.M.; Saltzman, W.M. Polymeric nanoparticles for drug delivery to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 701–705. [Google Scholar] [CrossRef] [Green Version]
- Kreuter, J. Drug delivery to the central nervous system by polymeric nanoparticles: What do we know? Adv. Drug Deliv. Rev. 2014, 71, 2–14. [Google Scholar] [CrossRef]
- Tonda-Turo, C.; Origlia, N.; Mattu, C.; Accorroni, A.; Chiono, V. Current limitations in the treatment of Parkinson’s and Alzheimer’s diseases: State-of-the-art and future perspective of polymeric carriers. Curr. Med. Chem. 2018, 25, 5755–5771. [Google Scholar] [CrossRef]
- Shakeri, S.; Ashrafizadeh, M.; Zarrabi, A.; Roghanian, R.; Afshar, E.G.; Pardakhty, A.; Mohammadinejad, R.; Kumar, A.; Thakur, V.K. Multifunctional polymeric nanoplatforms for brain diseases diagnosis, therapy and theranostics. Biomedicines 2020, 8, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Kim, C.S.; Saylor, D.M.; Koo, D. Polymer degradation and drug delivery in PLGA-based drug–polymer applications: A review of experiments and theories. J. Biomed. Mater. Res.-Part B Appl. Biomater. 2017, 105, 1692–1716. [Google Scholar] [CrossRef] [PubMed]
- Songjiang, Z.; Lixiang, W. Amyloid-beta associated with chitosan nano-carrier has favorable immunogenicity and permeates the BBB. AAPS PharmSciTech 2009, 10, 900–905. [Google Scholar] [CrossRef] [Green Version]
- Mao, S.; Sun, W.; Kissel, T. Chitosan-based formulations for delivery of DNA and siRNA. Adv. Drug Deliv. Rev. 2010, 61, 12–27. [Google Scholar] [CrossRef]
- Yu, S.; Xu, X.; Feng, J.; Liu, M.; Hu, K. Chitosan and chitosan coating nanoparticles for the treatment of brain disease. Int. J. Pharm. 2019, 560, 282–293. [Google Scholar] [CrossRef]
- Geszke-Moritz, M.; Moritz, M. Solid lipid nanoparticles as attractive drug vehicles: Composition, properties and therapeutic strategies. Mater. Sci. Eng. C 2016, 68, 982–994. [Google Scholar] [CrossRef]
- Cacciatore, I.; Ciulla, M.; Fornasari, E.; Marinelli, L.; Di Stefano, A. Solid lipid nanoparticles as a drug delivery system for the treatment of neurodegenerative diseases. Expert Opin. Drug Deliv. 2016, 13, 1121–1131. [Google Scholar] [CrossRef]
- Vieira, D.B.; Gamarra, L.F. Getting into the brain: Liposome-based strategies for effective drug delivery across the blood–brain barrier. Int. J. Nanomed. 2016, 11, 5381–5414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, M.I.; Lopes, C.M.; Amaral, M.H.; Costa, P.C. Current insights on lipid nanocarrier-assisted drug delivery in the treatment of neurodegenerative diseases. Eur. J. Pharm. Biopharm. 2020, 149, 192–217. [Google Scholar] [CrossRef] [PubMed]
- Pottoo, F.H.; Sharma, S.; Javed, M.N.; Barkat, M.A.; Harshita; Alam, M.S.; Naim, M.J.; Alam, O.; Ansari, M.A.; Barreto, G.E.; et al. Lipid-based nanoformulations in the treatment of neurological disorders. Drug Metab. Rev. 2020, 52, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Kaur, I.P.; Bhandari, R.; Bhandari, S.; Kakkar, V. Potential of solid lipid nanoparticles in brain targeting. J. Control. Release 2008, 127, 97–109. [Google Scholar] [CrossRef]
- Naseri, N.; Valizadeh, H.; Zakeri-Milani, P. Solid lipid nanoparticles and nanostructured lipid carriers: Structure preparation and application. Adv. Pharm. Bull. 2015, 5, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and challenges of liposome assisted drug delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [Green Version]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Alavi, M.; Karimi, N.; Safaei, M. Application of various types of liposomes in drug delivery systems. Adv. Pharm. Bull. 2017, 7, 3–9. [Google Scholar] [CrossRef]
- Yue, X.; Dai, Z. Recent advances in liposomal nanohybrid cerasomes as promising drug nanocarriers. Adv. Colloid Interface Sci. 2014, 207, 32–42. [Google Scholar] [CrossRef]
- Angelova, A.; Garamus, V.M.; Angelov, B.; Tian, Z.; Li, Y.; Zou, A. Advances in structural design of lipid-based nanoparticle carriers for delivery of macromolecular drugs, phytochemicals and anti-tumor agents. Adv. Colloid Interface Sci. 2017, 249, 331–345. [Google Scholar] [CrossRef]
- Mertins, O.; Mathews, P.D.; Angelova, A. Advances in the design of ph-sensitive cubosome liquid crystalline nanocarriers for drug delivery applications. Nanomaterials 2020, 10, 963. [Google Scholar] [CrossRef]
- Karami, Z.; Saghatchi Zanjani, M.R.; Hamidi, M. Nanoemulsions in CNS drug delivery: Recent developments, impacts and challenges. Drug Discov. Today 2019, 24, 1104–1115. [Google Scholar] [CrossRef]
- Nirale, P.; Paul, A.; Yadav, K.S. Nanoemulsions for targeting the neurodegenerative diseases: Alzheimer’s, Parkinson’s and Prion’s. Life Sci. 2020, 245, 117394. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Li, X.; Wang, Q.; Liao, L.; Zhang, C. Nanocomposite hydrogels and their applications in drug delivery and tissue engineering. J. Biomed. Nanotechnol. 2015, 11, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Neamtu, I.; Rusu, A.G.; Diaconu, A.; Nita, L.E.; Chiriac, A.P. Basic concepts and recent advances in nanogels as carriers for medical applications. Drug Deliv. 2017, 24, 539–557. [Google Scholar] [CrossRef] [Green Version]
- Singh, Y.; Meher, J.G.; Raval, K.; Khan, F.A.; Chaurasia, M.; Jain, N.K.; Chourasia, M.K. Nanoemulsion: Concepts, development and applications in drug delivery. J. Control. Release 2017, 252, 28–49. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, C.; Pang, Z. Dendrimer-based drug delivery systems for brain targeting. Biomolecules 2019, 9, 790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, Z.; Shah, A.; Siddiq, M.; Kraatz, H.B. Polymeric micelles as drug delivery vehicles. RSC Adv. 2014, 4, 17028–17038. [Google Scholar] [CrossRef]
- Agrawal, M.; Saraf, S.; Saraf, S.; Antimisiaris, S.G.; Hamano, N.; Li, S.D.; Chougule, M.; Shoyele, S.A.; Gupta, U.; Ajazuddin, A.; et al. Recent advancements in the field of nanotechnology for the delivery of anti-Alzheimer drug in the brain region. Expert Opin. Drug Deliv. 2018, 15, 589–617. [Google Scholar] [CrossRef]
- Karthivashan, G.; Ganesan, P.; Park, S.Y.; Kim, J.S.; Choi, D.K. Therapeutic strategies and nano-drug delivery applications in management of ageing alzheimer’s disease. Drug Deliv. 2018, 25, 307–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, J.; Fatima, M.T.; Islam, Z.; Khan, R.H.; Uversky, V.N.; Salahuddin, P. Nanoparticle formulations in the diagnosis and therapy of Alzheimer’s disease. Int. J. Biol. Macromol. 2019, 130, 515–526. [Google Scholar] [CrossRef]
- Arya, M.A.; Manoj Kumar, M.K.; Sabitha, M.; Krishnakumar Menon, K.; Nair, S.C. Nanotechnology approaches for enhanced CNS delivery in treating Alzheimer’s disease. J. Drug Deliv. Sci. Technol. 2019, 51, 297–309. [Google Scholar] [CrossRef]
- Bhavna, B.; Shadab, M.; Ali, M.; Baboota, S.; Sahni, J.K.; Bhatnagar, A.; Ali, J. Preparation, characterization, In Vivo biodistribution and pharmacokinetic studies of donepezil-loaded PLGA nanoparticles for brain targeting. Drug Dev. Ind. Pharm. 2014, 40, 278–287. [Google Scholar] [CrossRef]
- Wilson, B.; Samanta, M.K.; Santhi, K.; Kumar, K.P.S.; Paramakrishnan, N.; Suresh, B. Targeted delivery of tacrine into the brain with polysorbate 80-coated poly(n-butylcyanoacrylate) nanoparticles. Eur. J. Pharm. Biopharm. 2008, 70, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.; Samanta, M.K.; Santhi, K.; Kumar, K.P.S.; Paramakrishnan, N.; Suresh, B. Poly(n-butylcyanoacrylate) nanoparticles coated with polysorbate 80 for the targeted delivery of rivastigmine into the brain to treat Alzheimer’s disease. Brain Res. 2008, 1200, 159–168. [Google Scholar] [CrossRef]
- Joshi, S.A.; Chavhan, S.S.; Sawant, K.K. Rivastigmine-loaded PLGA and PBCA nanoparticles: Preparation, optimization, characterization, In Vitro and pharmacodynamic studies. Eur. J. Pharm. Biopharm. 2010, 76, 189–199. [Google Scholar] [CrossRef]
- Scialabba, C.; Rocco, F.; Licciardi, M.; Pitarresi, G.; Ceruti, M.; Giammona, G. Amphiphilic polyaspartamide copolymer-based micelles for rivastigmine delivery to neuronal cells. Drug Deliv. 2012, 19, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-López, E.; Ettcheto, M.; Egea, M.A.; Espina, M.; Cano, A.; Calpena, A.C.; Camins, A.; Carmona, N.; Silva, A.M.; Souto, E.B.; et al. Memantine loaded PLGA PEGylated nanoparticles for Alzheimer’s disease: In Vitro and In Vivo characterization. J. Nanobiotechnol. 2018, 16, 1–16. [Google Scholar] [CrossRef]
- Agyare, E.K.; Curran, G.L.; Ramakrishnan, M.; Yu, C.C.; Poduslo, J.F.; Kandimalla, K.K. Development of a smart nano-vehicle to target cerebrovascular amyloid deposits and brain parenchymal plaques observed in Alzheimer’s disease and cerebral amyloid angiopathy. Pharm. Res. 2008, 25, 2674–2684. [Google Scholar] [CrossRef]
- Kannan, R.; Chakrabarti, R.; Tang, D.; Kim, K.J.; Kaplowitz, N. GSH transport in human cerebrovascular endothelial cells and human astrocytes: Evidence for luminal localization of Na+-dependent GSH transport in HCEC. Brain Res. 2000, 852, 374–382. [Google Scholar] [CrossRef]
- Rotman, M.; Welling, M.M.; Bunschoten, A.; De Backer, M.E.; Rip, J.; Nabuurs, R.J.A.; Gaillard, P.J.; Van Buchem, M.A.; Van Der Maarel, S.M.; Van Der Weerd, L. Enhanced glutathione PEGylated liposomal brain delivery of an anti-amyloid single domain antibody fragment in a mouse model for Alzheimer’s disease. J. Control. Release 2015, 203, 40–50. [Google Scholar] [CrossRef]
- Gobbi, M.; Re, F.; Canovi, M.; Beeg, M.; Gregori, M.; Sesana, S.; Sonnino, S.; Brogioli, D.; Musicanti, C.; Gasco, P.; et al. Lipid-based nanoparticles with high binding affinity for amyloid-β1–42 peptide. Biomaterials 2010, 31, 6519–6529. [Google Scholar] [CrossRef]
- Balducci, C.; Mancini, S.; Minniti, S.; La Vitola, P.; Zotti, M.; Sancini, G.; Mauri, M.; Cagnotto, A.; Colombo, L.; Fiordaliso, F.; et al. Multifunctional liposomes reduce brain β-amyloid burden and ameliorate memory impairment in Alzheimer’s disease mouse models. J. Neurosci. 2014, 34, 14022–14031. [Google Scholar] [CrossRef] [PubMed]
- Bana, L.; Minniti, S.; Salvati, E.; Sesana, S.; Zambelli, V.; Cagnotto, A.; Orlando, A.; Cazzaniga, E.; Zwart, R.; Scheper, W.; et al. Liposomes bi-functionalized with phosphatidic acid and an ApoE-derived peptide affect Aβ aggregation features and cross the blood-brain-barrier: Implications for therapy of Alzheimer disease. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wan, X.; Zheng, X.; Shao, X.; Liu, Q.; Zhang, Q.; Qian, Y. Dual-functional nanoparticles targeting amyloid plaques in the brains of Alzheimer’s disease mice. Biomaterials 2014, 35, 456–465. [Google Scholar] [CrossRef]
- Li, J.; Feng, L.; Fan, L.; Zha, Y.; Guo, L.; Zhang, Q.; Chen, J.; Pang, Z.; Wang, Y.; Jiang, X.; et al. Targeting the brain with PEG-PLGA nanoparticles modified with phage-displayed peptides. Biomaterials 2011, 32, 4943–4950. [Google Scholar] [CrossRef] [Green Version]
- Wiesehan, K.; Buder, K.; Linke, R.P.; Patt, S.; Stoldt, M.; Unger, E.; Schmitt, B.; Bucci, E.; Willbold, D. Selection of D-amino-acid peptides that bind to Alzheimer’s disease amyloid peptide Aβ1–42 by mirror image phage display. ChemBioChem 2003, 4, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Bartnik, D.; Funke, S.A.; Andrei-Selmer, L.C.; Bacher, M.; Dodel, R.; Willbold, D. Differently selected d-enantiomeric peptides act on different aβ species. Rejuvenation Res. 2010, 13, 202–205. [Google Scholar] [CrossRef]
- Loureiro, J.A.; Gomes, B.; Fricker, G.; Coelho, M.A.N.; Rocha, S.; Pereira, M.C. Cellular uptake of PLGA nanoparticles targeted with anti-amyloid and anti-transferrin receptor antibodies for Alzheimer’s disease treatment. Colloids Surf. B Biointerfaces 2016, 145, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Moos, T.; Morgan, E.H. Restricted transport of anti-transferrin receptor antibody (OX26) through the blood-brain barrier in the rat. J. Neurochem. 2001, 79, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, N.; Dong, X.Y.; Zheng, J.; Liu, F.F.; Sun, Y. Design of LVFFARK and LVFFARK-Functionalized Nanoparticles for Inhibiting Amyloid β-Protein Fibrillation and Cytotoxicity. ACS Appl. Mater. Interfaces 2015, 7, 5650–5662. [Google Scholar] [CrossRef] [PubMed]
- Chafekar, S.M.; Malda, H.; Merkx, M.; Meijer, E.W.; Viertl, D.; Lashuel, H.A.; Baas, F.; Scheper, W. Branched KLVFF tetramers strongly potentiate inhibition of β-amyloid aggregation. ChemBioChem 2007, 8, 1857–1864. [Google Scholar] [CrossRef] [Green Version]
- Riccardi, C.; Russo Krauss, I.; Musumeci, D.; Morvan, F.; Meyer, A.; Vasseur, J.J.; Paduano, L.; Montesarchio, D. Fluorescent thrombin binding aptamer-tagged nanoparticles for an efficient and reversible control of thrombin activity. ACS Appl. Mater. Interfaces 2017, 9, 35574–35587. [Google Scholar] [CrossRef]
- Xiong, N.; Zhao, Y.; Dong, X.; Zheng, J.; Sun, Y. Design of a molecular hybrid of dual peptide inhibitors coupled on AuNPs for enhanced inhibition of amyloid β-protein aggregation and cytotoxicity. Small 2017, 13, 1601666. [Google Scholar] [CrossRef]
- Fradinger, E.A.; Monien, B.H.; Urbanc, B.; Lomakin, A.; Tan, M.; Li, H.; Spring, S.M.; Condron, M.M.; Cruz, L.; Xie, C.W.; et al. C-terminal peptides coassemble into Aβ42 oligomers and protect neurons against Aβ42-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 14175–14180. [Google Scholar] [CrossRef] [Green Version]
- Moretti, R.; Morelli, M.E.; Caruso, P. Vitamin D in neurological diseases: A rationale for a pathogenic impact. Int. J. Mol. Sci. 2018, 19, 2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riccardi, C.; Perrone, L.; Napolitano, F.; Sampaolo, S.; Melone, M.A.B. Understanding the biological activities of vitamin d in type 1 neurofibromatosis: New insights into disease pathogenesis and therapeutic design. Cancers 2020, 12, 2965. [Google Scholar] [CrossRef]
- Moon, M.; Song, H.; Hong, H.J.; Nam, D.W.; Cha, M.Y.; Oh, M.S.; Yu, J.; Ryu, H.; Mook-Jung, I. Vitamin D-binding protein interacts with Aβ and suppresses Aβ-mediated pathology. Cell Death Differ. 2013, 20, 630–638. [Google Scholar] [CrossRef] [Green Version]
- Bishnoi, R.J.; Palmer, R.F.; Royall, D.R. Vitamin D binding protein as a serum biomarker of Alzheimer’s disease. J. Alzheimers Dis. 2014, 43, 37–45. [Google Scholar] [CrossRef]
- Jeon, S.G.; Cha, M.Y.; Kim, J.I.; Hwang, T.W.; Kim, K.A.; Kim, T.H.; Song, K.C.; Kim, J.J.; Moon, M. Vitamin D-binding protein-loaded PLGA nanoparticles suppress Alzheimer’s disease-related pathology in 5XFAD mice. Nanomed. Nanotechnol. Biol. Med. 2019, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.I.; Lee, S.; Suh, Y.S.; Lee, J.S.; Kim, A.K.; Kwon, O.Y.; Yu, K.; Park, C.B. Photoexcited porphyrins as a strong suppressor of β-amyloid aggregation and synaptic toxicity. Angew. Chem.-Int. Ed. Eng. 2015, 54, 11472–11476. [Google Scholar] [CrossRef]
- Xu, M.; Zhou, H.; Liu, Y.; Sun, J.; Xie, W.; Zhao, P.; Liu, J. Ultrasound-excited protoporphyrin IX-modified multifunctional nanoparticles as a strong inhibitor of tau phosphorylation and β-Amyloid aggregation. ACS Appl. Mater. Interfaces 2018, 10, 32965–32980. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Wu, J.W.; Myeku, N.; Figueroa, Y.H.; Herman, M.; Marinec, P.S.; Gestwicki, J.E.; Dickey, C.A.; Yu, W.H.; Duff, K.E. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy In Vitro and In Vivo. Autophagy 2012, 8, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Hochgräfe, K.; Sydow, A.; Matenia, D.; Cadinu, D.; Könen, S.; Petrova, O.; Pickhardt, M.; Goll, P.; Morellini, F.; Mandelkow, E.; et al. Preventive methylene blue treatment preserves cognition in mice expressing full-length pro-aggregant human tau. Acta Neuropathol. Commun. 2015, 3, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Du, Y.; Zhang, K.; Liang, Z.; Li, J.; Yu, H.; Ren, R.; Feng, J.; Jin, Z.; Li, F.; et al. Tau-targeted multifunctional nanocomposite for combinational therapy of Alzheimer’s disease. ACS Nano 2018, 12, 1321–1338. [Google Scholar] [CrossRef]
- Xia, C.F.; Arteaga, J.; Chen, G.; Gangadharmath, U.; Gomez, L.F.; Kasi, D.; Lam, C.; Liang, Q.; Liu, C.; Mocharla, V.P.; et al. [18F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimers Dement. 2013, 9, 666–676. [Google Scholar] [CrossRef]
- Amin, F.U.; Hoshiar, A.K.; Do, T.D.; Noh, Y.; Shah, S.A.; Khan, M.S.; Yoon, J.; Kim, M.O. Osmotin-loaded magnetic nanoparticles with electromagnetic guidance for the treatment of Alzheimer’s disease. Nanoscale 2017, 9, 10619–10632. [Google Scholar] [CrossRef] [Green Version]
- Vangijzegem, T.; Stanicki, D.; Laurent, S. Magnetic iron oxide nanoparticles for drug delivery: Applications and characteristics. Expert Opin. Drug Deliv. 2019, 16, 69–78. [Google Scholar] [CrossRef]
- Berry, C.C.; Curtis, A.S.G. Functionalisation of magnetic nanoparticles for applications in biomedicine. J. Phys. D. Appl. Phys. 2003, 36, R198–R206. [Google Scholar] [CrossRef]
- Cui, Z.; Lockman, P.R.; Atwood, C.S.; Hsu, C.H.; Gupte, A.; Allen, D.D.; Mumper, R.J. Novel D-penicillamine carrying nanoparticles for metal chelation therapy in Alzheimer’s and other CNS diseases. Eur. J. Pharm. Biopharm. 2005, 59, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.B.; Dellamaggiore, K.R.; Ouellette, C.P.; Sedano, C.D.; Lizadjohry, M.; Chernis, G.A.; Gonzales, M.; Baltasar, F.E.; Fan, A.L.; Myerowitz, R.; et al. Aptamer-based endocytosis of a lysosomal enzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 15908–15913. [Google Scholar] [CrossRef] [Green Version]
- Mu, C.; Dave, N.; Hu, J.; Desai, P.; Pauletti, G.; Bai, S.; Hao, J. Solubilization of flurbiprofen into aptamer-modified PEG-PLA micelles for targeted delivery to brain-derived endothelial cells In Vitro. J. Microencapsul. 2013, 30, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Sharma, D.; Singh, J. GLUT-1: An effective target to deliver brain-derived neurotrophic factor gene across the blood brain barrier. ACS Chem. Neurosci. 2020, 11, 1620–1633. [Google Scholar] [CrossRef]
- Naksuriya, O.; Okonogi, S.; Schiffelers, R.M.; Hennink, W.E. Curcumin nanoformulations: A review of pharmaceutical properties and preclinical studies and clinical data related to cancer treatment. Biomaterials 2014, 35, 3365–3383. [Google Scholar] [CrossRef]
- Sun, M.; Su, X.; Ding, B.; He, X.; Liu, X.; Yu, A.; Lou, H.; Zhai, G. Advances in nanotechnology-based delivery systems for curcumin. Nanomedicine 2012, 7, 1085–1100. [Google Scholar] [CrossRef] [PubMed]
- Yallapu, M.M.; Nagesh, P.K.B.; Jaggi, M.; Chauhan, S.C. Therapeutic applications of curcumin nanoformulations. AAPS J. 2015, 17, 1341–1356. [Google Scholar] [CrossRef] [Green Version]
- Yavarpour-Bali, H.; Pirzadeh, M.; Ghasemi-Kasman, M. Curcumin-loaded nanoparticles: A novel therapeutic strategy in treatment of central nervous system disorders. Int. J. Nanomed. 2019, 14, 4449–4460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Rauf Khan, A.; Fu, M.; Zhai, Y.; Ji, J.; Bobrovskaya, L.; Zhai, G. Advances in curcumin-loaded nanopreparations: Improving bioavailability and overcoming inherent drawbacks. J. Drug Target. 2019, 27, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, J.; Ankola, D.D.; Beniwal, V.; Singh, D.; Kumar, M.N.V.R. Nanoparticle encapsulation improves oral bioavailability of curcumin by at least 9-fold when compared to curcumin administered with piperine as absorption enhancer. Eur. J. Pharm. Sci. 2009, 37, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.K.; Agarwal, S.; Seth, B.; Yadav, A.; Nair, S.; Bhatnagar, P.; Karmakar, M.; Kumari, M.; Chauhan, L.K.S.; Patel, D.K.; et al. Curcumin-loaded nanoparticles potently induce adult neurogenesis and reverse cognitive deficits in Alzheimer’s disease model via canonical Wnt/β-catenin pathway. ACS Nano 2014, 8, 76–103. [Google Scholar] [CrossRef] [PubMed]
- Khalil, N.M.; do Nascimento, T.C.F.; Casa, D.M.; Dalmolin, L.F.; de Mattos, A.C.; Hoss, I.; Romano, M.A.; Mainardes, R.M. Pharmacokinetics of curcumin-loaded PLGA and PLGA-PEG blend nanoparticles after oral administration in rats. Colloids Surf. B Biointerfaces 2013, 101, 353–360. [Google Scholar] [CrossRef]
- Mulik, R.S.; Mönkkönen, J.; Juvonen, R.O.; Mahadik, K.R.; Paradkar, A.R. ApoE3 mediated poly(butyl) cyanoacrylate nanoparticles containing curcumin: Study of enhanced activity of curcumin against beta amyloid induced cytotoxicity using In Vitro cell culture model. Mol. Pharm. 2010, 7, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Mathew, A.; Fukuda, T.; Nagaoka, Y.; Hasumura, T.; Morimoto, H.; Yoshida, Y.; Maekawa, T.; Venugopal, K.; Kumar, D.S. Curcumin loaded-PLGA nanoparticles conjugated with Tet-1 peptide for potential use in Alzheimer’s disease. PLoS ONE 2012, 7, e32616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbara, R.; Belletti, D.; Pederzoli, F.; Masoni, M.; Keller, J.; Ballestrazzi, A.; Vandelli, M.A.; Tosi, G.; Grabrucker, A.M. Novel curcumin loaded nanoparticles engineered for blood-brain barrier crossing and able to disrupt Abeta aggregates. Int. J. Pharm. 2017, 526, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Zheng, Y.; Liu, X.; Fang, W.; Chena, X.; Liao, W.; Jing, X.; Lei, M.; Tao, E.; Ma, Q.; et al. Curcumin-loaded plga-peg nanoparticles conjugated with b6 peptide for potential use in alzheimer’s disease. Drug Deliv. 2018, 25, 1091–1102. [Google Scholar] [CrossRef] [Green Version]
- Mathew, A.; Aravind, A.; Brahatheeswaran, D.; Fukuda, T.; Nagaoka, Y.; Hasumura, T.; Iwai, S.; Morimoto, H.; Yoshida, Y.; Maekawa, T.; et al. Amyloid-binding aptamer conjugated curcumin-PLGA nanoparticle for potential use in Alzheimer’s disease. Bionanoscience 2012, 2, 83–93. [Google Scholar] [CrossRef]
- Martel, C.L.; Mackic, J.B.; Matsubara, E.; Governale, S.; Miguel, C.; Miao, W.; McComb, J.G.; Frangione, B.; Ghiso, J.; Zlokovic, B.V. Isoform-specific effects of apolipoproteins E2, E3, and E4 on cerebral capillary sequestration and blood-brain barrier transport of circulating Alzheimer’s amyloid β. J. Neurochem. 1997, 69, 1995–2004. [Google Scholar] [CrossRef]
- Park, I.K.; Lasiene, J.; Chou, S.H.; Horner, P.J.; Pun, S.H. Neuron-specific delivery of nucleic acids mediated by Tet1-modified poly(ethylenimine). J. Gene Med. 2007, 9, 691–702. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.K.; Teng, Q.; Garrity-Moses, M.; Federici, T.; Tanase, D.; Imperiale, M.J.; Boulis, N.M. A novel peptide defined through phage display for therapeutic protein and vector neuronal targeting. Neurobiol. Dis. 2005, 19, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Tosi, G.; Vergoni, A.V.; Ruozi, B.; Bondioli, L.; Badiali, L.; Rivasi, F.; Costantino, L.; Forni, F.; Vandelli, M.A. Sialic acid and glycopeptides conjugated PLGA nanoparticles for central nervous system targeting: In Vivo pharmacological evidence and biodistribution. J. Control. Release 2010, 145, 49–57. [Google Scholar] [CrossRef]
- Tosi, G.; Fano, R.A.; Bondioli, L.; Badiali, L.; Benassi, R.; Rivasi, F.; Ruozi, B.; Forni, F.; Vandelli, M.A. Investigation on mechanisms of glycopeptide nanoparticles for drug delivery across the blood-brain barrier. Nanomedicine 2011, 6, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Tada, K.; Mihara, H. RNA aptamers selected against amyloid β-peptide (Aβ) inhibit the aggregation of Aβ. Mol. Biosyst. 2009, 5, 986–991. [Google Scholar] [CrossRef]
- Lazar, A.N.; Mourtas, S.; Youssef, I.; Parizot, C.; Dauphin, A.; Delatour, B.; Antimisiaris, S.G.; Duyckaerts, C. Curcumin-conjugated nanoliposomes with high affinity for Aβ deposits: Possible applications to Alzheimer disease. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 712–721. [Google Scholar] [CrossRef]
- Taylor, M.; Moore, S.; Mourtas, S.; Niarakis, A.; Re, F.; Zona, C.; La Ferla, B.; Nicotra, F.; Masserini, M.; Antimisiaris, S.G.; et al. Effect of curcumin-associated and lipid ligand-functionalized nanoliposomes on aggregation of the Alzheimer’s Aβ peptide. Nanomedicine 2011, 7, 541–550. [Google Scholar] [CrossRef]
- Mourtas, S.; Lazar, A.N.; Markoutsa, E.; Duyckaerts, C.; Antimisiaris, S.G. Multifunctional nanoliposomes with curcumin-lipid derivative and brain targeting functionality with potential applications for Alzheimer disease. Eur. J. Med. Chem. 2014, 80, 175–183. [Google Scholar] [CrossRef]
- Hagl, S.; Kocher, A.; Schiborr, C.; Kolesova, N.; Frank, J.; Eckert, G.P. Curcumin micelles improve mitochondrial function in neuronal PC12 cells and brains of NMRI mice—Impact on bioavailability. Neurochem. Int. 2015, 89, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Palmal, S.; Maity, A.R.; Singh, B.K.; Basu, S.; Jana, N.R.; Jana, N.R. Inhibition of amyloid fibril growth and dissolution of amyloid fibrils by curcumin-gold nanoparticles. Chem.-A Eur. J. 2014, 20, 6184–6191. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Asghar, S.; Gao, S.; Su, Z.; Song, J.; Huo, M.; Meng, W.; Ping, Q.; Xiao, Y. A novel LDL-mimic nanocarrier for the targeted delivery of curcumin into the brain to treat Alzheimer’s disease. Colloids Surf. B Biointerfaces 2015, 134, 88–97. [Google Scholar] [CrossRef]
- Rensen, P.C.N.; De Vrueh, R.L.A.; Kuiper, J.; Bijsterbosch, M.K.; Biessen, E.A.L.; Van Berkel, T.J.C. Recombinant lipoproteins: Lipoprotein-like lipid particles for drug targeting. Adv. Drug Deliv. Rev. 2001, 47, 251–276. [Google Scholar] [CrossRef]
- Sarkar, G.; Curran, G.L.; Mahlum, E.; Decklever, T.; Wengenack, T.M.; Blahnik, A.; Hoesley, B.; Lowe, V.J.; Poduslo, J.F.; Jenkins, R.B. A carrier for non-covalent delivery of functional beta-galactosidase and antibodies against amyloid plaques and IgM to the brain. PLoS ONE 2011, 6, e28881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, Y.C.; Tsai, H.C. Rosmarinic acid- and curcumin-loaded polyacrylamide-cardiolipin-poly(lactide-co-glycolide) nanoparticles with conjugated 83-14 monoclonal antibody to protect β-amyloid-insulted neurons. Mater. Sci. Eng. C 2018, 91, 445–457. [Google Scholar] [CrossRef]
- Bourignon, D.B.; Bezian, J.H.; Bordenave, L.; Bareille, R.; Latapie, M.J.; Baquey, C.; Ducassou, D. Biocompatibility of polyacrylamide microcapsules implanted in peritoneal cavity or spleen of the rat. Effect on various inflammatory reactions In Vitro. Biomater. Artif. Cells Artif. Organs 1990, 18, 25–42. [Google Scholar] [CrossRef]
- Boado, R.J.; Zhang, Y.; Zhang, Y.; Pardridge, W.M. Humanization of anti-human insulin receptor antibody for drug targeting across the human blood-brain barrier. Biotechnol. Bioeng. 2007, 96, 381–391. [Google Scholar] [CrossRef]
- Li, F.; Gong, Q.; Dong, H.; Shi, J. Resveratrol, a neuroprotective supplement for Alzheimer’s disease. Curr. Pharm. Des. 2012, 18, 27–33. [Google Scholar] [CrossRef]
- Loureiro, J.A.; Andrade, S.; Duarte, A.; Neves, A.R.; Queiroz, J.F.; Nunes, C.; Sevin, E.; Fenart, L.; Gosselet, F.; Coelho, M.A.N.; et al. Resveratrol and grape extract-loaded solid lipid nanoparticles for the treatment of Alzheimer’s disease. Molecules 2017, 22, 277. [Google Scholar] [CrossRef] [PubMed]
- Neves, A.R.; Queiroz, J.F.; Reis, S. Brain-targeted delivery of resveratrol using solid lipid nanoparticles functionalized with apolipoprotein E. J. Nanobiotechnol. 2016, 14, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Picone, P.; Bondi, M.L.; Montana, G.; Bruno, A.; Pitarresi, G.; Giammona, G.; Di Carlo, M. Ferulic acid inhibits oxidative stress and cell death induced by Ab oligomers: Improved delivery by solid lipid nanoparticles. Free Radic. Res. 2009, 43, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.L.; Huang, M.; Wang, X.R.; Fu, J.; Han, M.; Shen, Y.Q.; Xia, Z.; Gao, J.Q. Transferrin-modified liposome promotes α-mangostin to penetrate the blood-brain barrier. Nanomedicine 2016, 12, 421–430. [Google Scholar] [CrossRef]
- Kim, M.J.; Rehman, S.U.; Amin, F.U.; Kim, M.O. Enhanced neuroprotection of anthocyanin-loaded PEG-gold nanoparticles against Aβ1-42-induced neuroinflammation and neurodegeneration via the NF-KB /JNK/GSK3β signaling pathway. Nanomedicine 2017, 13, 2533–2544. [Google Scholar] [CrossRef] [PubMed]
- Amin, F.U.; Shah, S.A.; Badshah, H.; Khan, M.; Kim, M.O. Anthocyanins encapsulated by PLGA@PEG nanoparticles potentially improved its free radical scavenging capabilities via p38/JNK pathway against Aβ1-42-induced oxidative stress. J. Nanobiotechnol. 2017, 15, 12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhou, X.; Yu, Q.; Yang, L.; Sun, D.; Zhou, Y.; Liu, J. Epigallocatechin-3-gallate (EGCG)-stabilized selenium nanoparticles coated with Tet-1 peptide to reduce amyloid-β aggregation and cytotoxicity. ACS Appl. Mater. Interfaces 2014, 6, 8475–8487. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Ettcheto, M.; Chang, J.H.; Barroso, E.; Espina, M.; Kühne, B.A.; Barenys, M.; Auladell, C.; Folch, J.; Souto, E.B.; et al. Dual-drug loaded nanoparticles of Epigallocatechin-3-gallate (EGCG)/ascorbic acid enhance therapeutic efficacy of EGCG in a APPswe/PS1dE9 Alzheimer’s disease mice model. J. Control. Release 2019, 301, 62–75. [Google Scholar] [CrossRef]
- Kuo, Y.C.; Chen, I.Y.; Rajesh, R. Use of functionalized liposomes loaded with antioxidants to permeate the blood–brain barrier and inhibit β-amyloid-induced neurodegeneration in the brain. J. Taiwan Inst. Chem. Eng. 2018, 87, 1–14. [Google Scholar] [CrossRef]
- Ganesan, P.; Ko, H.M.; Kim, I.S.; Choi, D.K. Recent trends in the development of nanophytobioactive compounds and delivery systems for their possible role in reducing oxidative stress in Parkinson’s disease models. Int. J. Nanomed. 2015, 10, 6757–6772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Ortega, P.V.; Saludas, L.; Hanafy, A.S.; Garbayo, E.; Blanco-Prieto, M.J. Micro- and nanotechnology approaches to improve Parkinson’s disease therapy. J. Control. Release 2019, 295, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Karthivashan, G.; Ganesan, P.; Park, S.Y.; Lee, H.W.; Choi, D.K. Lipid-based nanodelivery approaches for dopamine-replacement therapies in Parkinson’s disease: From preclinical to translational studies. Biomaterials 2020, 232, 119704. [Google Scholar] [CrossRef]
- Lopalco, A.; Cutrignelli, A.; Denora, N.; Lopedota, A.; Franco, M.; Laquintana, V. Transferrin functionalized liposomes loading dopamine HCl: Development and permeability studies across an In Vitro model of human blood-brain barrier. Nanomaterials 2018, 8, 178. [Google Scholar] [CrossRef] [Green Version]
- Trapani, A.; De Giglio, E.; Cafagna, D.; Denora, N.; Agrimi, G.; Cassano, T.; Gaetani, S.; Cuomo, V.; Trapani, G. Characterization and evaluation of chitosan nanoparticles for dopamine brain delivery. Int. J. Pharm. 2011, 419, 296–307. [Google Scholar] [CrossRef]
- Pahuja, R.; Seth, K.; Shukla, A.; Shukla, R.K.; Bhatnagar, P.; Chauhan, L.K.S.; Saxena, P.N.; Arun, J.; Chaudhari, B.P.; Patel, D.K.; et al. Trans-blood brain barrier delivery of dopamine-loaded nanoparticles reverses functional deficits in parkinsonian rats. ACS Nano 2015, 9, 4850–4871. [Google Scholar] [CrossRef]
- Jahansooz, F.; Hosseinzade, B.E.; Zarmi, A.H.; Hadi, F.; Massood Hojjati, S.M.; Shahpasand, K. Dopamine-loaded poly (butyl cyanoacrylate) nanoparticles reverse behavioral deficits in Parkinson’s animal models. Ther. Deliv. 2020, 11, 387–399. [Google Scholar] [CrossRef]
- Rashed, E.R.; Abd El-Rehim, H.A.; El-Ghazaly, M.A. Potential efficacy of dopamine loaded-PVP/PAA nanogel in experimental models of Parkinsonism: Possible disease modifying activity. J. Biomed. Mater. Res.-Part A 2015, 103, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- DeBin, J.A.; Strichartz, G.R. Chloride channel inhibition by the venom of the scorpion Leiurus quinquestriatus. Toxicon 1991, 29, 1403–1408. [Google Scholar] [CrossRef]
- Xiang, Y.; Wu, Q.; Liang, L.; Wang, X.; Wang, J.; Zhang, X.; Pu, X.; Zhang, Q. Chlorotoxin-modified stealth liposomes encapsulating levodopa for the targeting delivery against the Parkinson’s disease in the MPTP-induced mice model. J. Drug Target. 2012, 20, 67–75. [Google Scholar] [CrossRef]
- Yu, X.; Yao, J.Y.; He, J.; Tian, J.W. Protection of MPTP-induced neuroinflammation and neurodegeneration by rotigotine-loaded microspheres. Life Sci. 2015, 124, 136–143. [Google Scholar] [CrossRef]
- Ray, S.; Sinha, P.; Laha, B.; Maiti, S.; Bhattacharyya, U.K.; Nayak, A.K. Polysorbate 80 coated crosslinked chitosan nanoparticles of ropinirole hydrochloride for brain targeting. J. Drug Deliv. Sci. Technol. 2018, 48, 21–29. [Google Scholar] [CrossRef]
- Dudhipala, N.; Gorre, T. Neuroprotective effect of ropinirole lipid nanoparticles enriched hydrogel for parkinson’s disease: In Vitro, ex vivo, pharmacokinetic and pharmacodynamic evaluation. Pharmaceutics 2020, 12, 448. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Mariani, P.; Ravani, L.; Contado, C.; Volta, M.; Bido, S.; Drechsler, M.; Mazzoni, S.; Menegatti, E.; Morari, M.; et al. Nanoparticulate lipid dispersions for bromocriptine delivery: Characterization and In Vivo study. Eur. J. Pharm. Biopharm. 2012, 80, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Shi, Y.; Jiang, W.; Han, J.; Huang, S.; Jiang, X. Lactoferrin conjugated PEG-PLGA nanoparticles for brain delivery: Preparation, characterization and efficacy in Parkinsons disease. Int. J. Pharm. 2011, 415, 273–283. [Google Scholar] [CrossRef]
- Wang, N.; Jin, X.; Guo, D.; Tong, G.; Zhu, X. Iron chelation nanoparticles with delayed saturation as an effective therapy for Parkinson disease. Biomacromolecules 2017, 18, 461–474. [Google Scholar] [CrossRef]
- Sikorska, M.; Lanthier, P.; Miller, H.; Beyers, M.; Sodja, C.; Zurakowski, B.; Gangaraju, S.; Pandey, S.; Sandhu, J.K. Nanomicellar formulation of coenzyme Q10 (Ubisol-Q10) effectively blocks ongoing neurodegeneration in the mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model: Potential use as an adjuvant treatment in Parkinson’s disease. Neurobiol. Aging 2014, 35, 2329–2346. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Li, C.; Li, Y.; Yi, X.; Lee, S.M.Y.; Zheng, Y. Oral delivery of a nanocrystal formulation of schisantherin a with improved bioavailability and brain delivery for the treatment of Parkinson’s disease. Mol. Pharm. 2016, 13, 3864–3875. [Google Scholar] [CrossRef]
- Chen, T.; Li, C.; Li, Y.; Yi, X.; Wang, R.; Lee, S.M.Y.; Zheng, Y. Small-sized mPEG-PLGA nanoparticles of schisantherin A with sustained release for enhanced brain uptake and anti-Parkinsonian activity. ACS Appl. Mater. Interfaces 2017, 9, 9516–9527. [Google Scholar] [CrossRef] [PubMed]
- Bollimpelli, V.S.; Kumar, P.; Kumari, S.; Kondapi, A.K. Neuroprotective effect of curcumin-loaded lactoferrin nano particles against rotenone induced neurotoxicity. Neurochem. Int. 2016, 95, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yan, F.; Liang, X.; Wu, M.; Shen, Y.; Chen, M.; Xu, Y.; Zou, G.; Jiang, P.; Tang, C.; et al. Localized delivery of curcumin into brain with polysorbate 80-modified cerasomes by ultrasound-targeted microbubble destruction for improved Parkinson’s disease therapy. Theranostics 2018, 8, 2264–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddique, Y.H.; Khan, W.; Singh, B.R.; Naqvi, A.H. Synthesis of alginate-curcumin nanocomposite and its protective role in transgenic Drosophila model of Parkinson’s disease. ISRN Pharmacol. 2013, 794582. [Google Scholar] [CrossRef] [Green Version]
- Al-Baghdadi, O.B.; Prater, N.I.; Van Der Schyf, C.J.; Geldenhuys, W.J. Inhibition of monoamine oxidase by derivatives of piperine, an alkaloid from the pepper plant Piper nigrum, for possible use in Parkinson’s disease. Bioorg. Med. Chem. Lett. 2012, 22, 7183–7188. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, P.; Vaibhav, K.; Tabassum, R.; Khan, A.; Ishrat, T.; Khan, M.M.; Ahmad, A.; Islam, F.; Safhi, M.M.; Islam, F. Anti-apoptotic and anti-inflammatory effect of piperine on 6-OHDA induced Parkinson’s rat model. J. Nutr. Biochem. 2013, 24, 680–687. [Google Scholar] [CrossRef]
- Kundu, P.; Das, M.; Tripathy, K.; Sahoo, S.K. Delivery of dual drug loaded lipid based nanoparticles across the blood–brain barrier impart enhanced neuroprotection in a rotenone induced mouse model of Parkinson’s disease. ACS Chem. Neurosci. 2016, 7, 1658–1670. [Google Scholar] [CrossRef]
- Rakotoarisoa, M.; Angelov, B.; Garamus, V.M.; Angelova, A. Curcumin-and fish oil-loaded spongosome and cubosome nanoparticles with neuroprotective potential against H2O2-induced oxidative stress in differentiated human SH-SY5Y cells. ACS Omega 2019, 4, 3061–3073. [Google Scholar] [CrossRef] [Green Version]
- Da Rocha Lindner, G.; Bonfanti Santos, D.; Colle, D.; Gasnhar Moreira, E.L.; Daniel Prediger, R.; Farina, M.; Khalil, N.M.; Mara Mainardes, R. Improved neuroprotective effects of resveratrol-loaded polysorbate 80-coated poly(lactide) nanoparticles in MPTP-induced Parkinsonism. Nanomedicine 2015, 10, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Manjunath, K.; Venkateswarlu, V. Pharmacokinetics, tissue distribution and bioavailability of nitrendipine solid lipid nanoparticles after intravenous and intraduodenal administration. J. Drug Target. 2006, 14, 632–645. [Google Scholar] [CrossRef]
- Debnath, K.; Pradhan, N.; Singh, B.K.; Jana, N.R.; Jana, N.R. Poly(trehalose) nanoparticles prevent amyloid aggregation and suppress polyglutamine aggregation in a Huntington’s disease. ACS Appl. Mater. Interfaces 2017, 9, 24126–24139. [Google Scholar] [CrossRef]
- Valenza, M.; Chen, J.Y.; Di Paolo, E.; Ruozi, B.; Belletti, D.; Ferrari Bardile, C.; Leoni, V.; Caccia, C.; Brilli, E.; Di Donato, S.; et al. Cholesterol-loaded nanoparticles ameliorate synaptic and cognitive function in H untington’s disease mice. EMBO Mol. Med. 2015, 7, 1547–1564. [Google Scholar] [CrossRef]
- Sandhir, R.; Yadav, A.; Mehrotra, A.; Sunkaria, A.; Singh, A.; Sharma, S. Curcumin nanoparticles attenuate neurochemical and neurobehavioral deficits in experimental model of Huntington’s disease. Neuromol. Med. 2014, 16, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Borlongan, C.V.; Koutouzis, T.K.; Sanberg, P.R. 3-Nitropropionic acid animal model and Huntington’s disease. Neurosci. Biobehav. Rev. 1997, 71, 2642–2644. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, S.; Thangarajan, S. A novel therapeutic application of solid lipid nanoparticles encapsulated thymoquinone (TQ-SLNs) on 3-nitroproponic acid induced Huntington’s disease-like symptoms in wistar rats. Chem. Biol. Interact. 2016, 256, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Thangarajan, S. Thymoquinone loaded solid lipid nanoparticles counteracts 3-nitropropionic acid induced motor impairments and neuroinflammation in rat model of Huntington’s disease. Metab. Brain Dis. 2018, 33, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Debnath, K.; Jana, N.R.; Jana, N.R. Quercetin encapsulated polymer nanoparticle for inhibiting intracellular polyglutamine aggregation. ACS Appl. Bio Mater. 2019, 2, 5298–5305. [Google Scholar] [CrossRef]
- Mandal, S.; Debnath, K.; Jana, N.R.; Jana, N.R. Trehalose-conjugated, catechin-loaded polylactide nanoparticles for improved neuroprotection against intracellular polyglutamine aggregates. Biomacromolecules 2020, 21, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Jayaraman, M.; Roland, B.P.; Landrum, E.; Fullam, T.; Kodali, R.; Thakur, A.K.; Arduini, I.; Wetzel, R. Inhibiting the nucleation of amyloid structure in a huntingtin fragment by targeting α-helix-rich oligomeric intermediates. J. Mol. Biol. 2012, 415, 900–917. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.S.; Thakur, A.K. Biodegradable delivery system containing a peptide inhibitor of polyglutamine aggregation: A step toward therapeutic development in Huntington’s disease. J. Pept. Sci. 2014, 20, 630–639. [Google Scholar] [CrossRef]
- Wang, Y.L.; Liu, W.; Wada, E.; Murata, M.; Wada, K.; Kanazawa, I. Clinico-pathological rescue of a model mouse of Huntington’s disease by siRNA. Neurosci. Res. 2005, 53, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Godinho, B.M.D.C.; Ogier, J.R.; Darcy, R.; O’Driscoll, C.M.; Cryan, J.F. Self-assembling modified β-cyclodextrin nanoparticles as neuronal siRNA delivery vectors: Focus on huntington’s disease. Mol. Pharm. 2013, 10, 640–649. [Google Scholar] [CrossRef]
- Chaturvedi, K.; Ganguly, K.; Kulkarni, A.R.; Kulkarni, V.H.; Nadagouda, M.N.; Rudzinski, W.E.; Aminabhavi, T.M. Cyclodextrin-based siRNA delivery nanocarriers: A state-of-the-art review. Expert Opin. Drug Deliv. 2011, 8, 1455–1468. [Google Scholar] [CrossRef]
- Irace, C.; Misso, G.; Capuozzo, A.; Piccolo, M.; Riccardi, C.; Luchini, A.; Caraglia, M.; Paduano, L.; Montesarchio, D.; Santamaria, R. Antiproliferative effects of ruthenium-based nucleolipidic nanoaggregates in human models of breast cancer In Vitro: Insights into their mode of action. Sci. Rep. 2017, 7, 45236–45249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riccardi, C.; Musumeci, D.; Irace, C.; Paduano, L.; Montesarchio, D. Ru(III) complexes for anticancer therapy: The importance of being nucleolipidic. Eur. J. Org. Chem. 2017, 2017, 1100–1119. [Google Scholar] [CrossRef]
- Riccardi, C.; Musumeci, D.; Capuozzo, A.; Irace, C.; King, S.; Russo Krauss, I.; Paduano, L.; Montesarchio, D. “Dressing up” an old drug: An aminoacyl lipid for the functionalization of Ru(III)-based anticancer agents. ACS Biomater. Sci. Eng. 2018, 4, 163–174. [Google Scholar] [CrossRef]
- Riccardi, C.; Fàbrega, C.; Grijalvo, S.; Vitiello, G.; D’Errico, G.; Eritja, R.; Montesarchio, D. AS1411-decorated niosomes as effective nanocarriers for Ru(III)-based drugs in anticancer strategies. J. Mater. Chem. B 2018, 6, 5368–5384. [Google Scholar] [CrossRef]
- Riccardi, C.; Musumeci, D.; Trifuoggi, M.; Irace, C.; Paduano, L.; Montesarchio, D. Anticancer ruthenium (III) complexes and Ru(III) containing nanoformulations: An update on the mechanism of action and biological activity. Pharmaceuticals 2019, 12, 146. [Google Scholar] [CrossRef] [Green Version]
- Piccolo, M.; Misso, G.; Ferraro, M.G.; Riccardi, C.; Capuozzo, A.; Zarone, M.R.; Maione, F.; Trifuoggi, M.; Stiuso, P.; D’Errico, G.; et al. Exploring cellular uptake, accumulation and mechanism of action of a cationic Ru-based nanosystem in human preclinical models of breast cancer. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.R.; Liu, M.; Khan, M.W.; Zhai, G. Progress in brain targeting drug delivery system by nasal route. J. Control. Release 2017, 268, 364–389. [Google Scholar] [CrossRef] [PubMed]
- Erdő, F.; Bors, L.A.; Farkas, D.; Bajza, Á.; Gizurarson, S. Evaluation of intranasal delivery route of drug administration for brain targeting. Brain Res. Bull. 2018, 143, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.I.; Beg, S.; Samad, A.; Baboota, S.; Kohli, K.; Ali, J.; Ahuja, A.; Akbar, M. Strategy for effective brain drug delivery. Eur. J. Pharm. Sci. 2010, 40, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Choonara, Y.E.; Kumar, P.; Modi, G.; Pillay, V. Improving drug delivery technology for treating neurodegenerative diseases. Expert Opin. Drug Deliv. 2016, 13, 1029–1043. [Google Scholar] [CrossRef] [PubMed]
- Pardeshi, C.V.; Belgamwar, V.S. Direct nose to brain drug delivery via integrated nerve pathways bypassing the blood-brain barrier: An excellent platform for brain targeting. Expert Opin. Drug Deliv. 2013, 10, 957–972. [Google Scholar] [CrossRef]
- Bourganis, V.; Kammona, O.; Alexopoulos, A.; Kiparissides, C. Recent advances in carrier mediated nose-to-brain delivery of pharmaceutics. Eur. J. Pharm. Biopharm. 2018, 128, 337–362. [Google Scholar] [CrossRef]
- Patel, A.A.; Patel, R.J.; Patel, S.R. Nanomedicine for intranasal delivery to improve brain uptake. Curr. Drug Deliv. 2017, 15, 461–469. [Google Scholar] [CrossRef]
- Agrawal, M.; Saraf, S.; Saraf, S.; Antimisiaris, S.G.; Chougule, M.B.; Shoyele, S.A.; Alexander, A. Nose-to-brain drug delivery: An update on clinical challenges and progress towards approval of anti-Alzheimer drugs. J. Control. Release 2018, 281, 139–177. [Google Scholar] [CrossRef]
- Md, S.; Bhattmisra, S.K.; Zeeshan, F.; Shahzad, N.; Mujtaba, M.A.; Srikanth Meka, V.; Radhakrishnan, A.; Kesharwani, P.; Baboota, S.; Ali, J. Nano-carrier enabled drug delivery systems for nose to brain targeting for the treatment of neurodegenerative disorders. J. Drug Deliv. Sci. Technol. 2018, 43, 295–310. [Google Scholar] [CrossRef]
- Dhas, N.L.; Kudarha, R.R.; Mehta, T.A. Intranasal delivery of nanotherapeutics/nanobiotherapeutics for the treatment of Alzheimer’s disease: A proficient approach. Crit. Rev. Ther. Drug Carr. Syst. 2019, 36, 373–447. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.; Nabi, B.; Zafar, A.; Baboota, S.; Ali, J. Intranasal delivery of mucoadhesive nanocarriers: A viable option for Parkinson’s disease treatment? Expert Opin. Drug Deliv. 2019, 16, 1355–1366. [Google Scholar] [CrossRef]
- Singh, R.; Brumlik, C.; Vaidya, M.; Choudhury, A. A patent review on nanotechnology-based nose-to-brain drug delivery. Recent Pat. Nanotechnol. 2020, 14, 174–192. [Google Scholar] [CrossRef] [PubMed]
- Bonferoni, M.C.; Rossi, S.; Sandri, G.; Ferrari, F.; Gavini, E.; Rassu, G.; Giunchedi, P. Nanoemulsions for “nose-to-brain” drug delivery. Pharmaceutics 2019, 11, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, B.; Gorain, B.; Mohananaidu, K.; Sengupta, P.; Mandal, U.K.; Choudhury, H. Targeted drug delivery to the brain via intranasal nanoemulsion: Available proof of concept and existing challenges. Int. J. Pharm. 2019, 565, 258–268. [Google Scholar] [CrossRef]
- Battaglia, L.; Panciani, P.P.; Muntoni, E.; Capucchio, M.T.; Biasibetti, E.; De Bonis, P.; Mioletti, S.; Fontanella, M.; Swaminathan, S. Lipid nanoparticles for intranasal administration: Application to nose-to-brain delivery. Expert Opin. Drug Deliv. 2018, 15, 369–378. [Google Scholar] [CrossRef]
- Hong, S.S.; Oh, K.T.; Choi, H.G.; Lim, S.J. Liposomal formulations for nose-to-brain delivery: Recent advances and future perspectives. Pharmaceutics 2019, 11, 540. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira Junior, E.R.; Santos, L.C.R.; Salomão, M.A.; Nascimento, T.L.; de Almeida Ribeiro Oliveira, G.; Lião, L.M.; Lima, E.M. Nose-to-brain drug delivery mediated by polymeric nanoparticles: Influence of PEG surface coating. Drug Deliv. Transl. Res. 2020. [Google Scholar] [CrossRef]
- Cunha, S.; Almeida, H.; Amaral, M.H.; Lobo, J.M.S.; Silva, A.C. Intranasal lipid nanoparticles for the treatment of neurodegenerative diseases. Curr. Pharm. Des. 2018, 23, 6553–6562. [Google Scholar] [CrossRef]
- Bies, C.; Lehr, C.M.; Woodley, J.F. Lectin-mediated drug targeting: History and applications. Adv. Drug Deliv. Rev. 2004, 56, 425–435. [Google Scholar] [CrossRef]
- Sonvico, F.; Clementino, A.; Buttini, F.; Colombo, G.; Pescina, S.; Guterres, S.S.; Pohlmann, A.R.; Nicoli, S. Surface-modified nanocarriers for nose-to-brain delivery: From bioadhesion to targeting. Pharmaceutics 2018, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Md, S.; Ali, M.; Ali, R.; Bhatnagar, A.; Baboota, S.; Ali, J. Donepezil nanosuspension intended for nose to brain targeting: In Vitro and In Vivo safety evaluation. Int. J. Biol. Macromol. 2014, 67, 418–425. [Google Scholar] [CrossRef]
- Al Asmari, A.K.; Ullah, Z.; Tariq, M.; Fatani, A. Preparation, characterization, and In Vivo evaluation of intranasally administered liposomal formulation of donepezil. Drug Des. Devel. Ther. 2016, 10, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Corace, G.; Angeloni, C.; Malaguti, M.; Hrelia, S.; Stein, P.C.; Brandl, M.; Gotti, R.; Luppi, B. Multifunctional liposomes for nasal delivery of the anti-Alzheimer drug tacrine hydrochloride. J. Liposome Res. 2014, 24, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Luppi, B.; Bigucci, F.; Corace, G.; Delucca, A.; Cerchiara, T.; Sorrenti, M.; Catenacci, L.; Di Pietra, A.M.; Zecchi, V. Albumin nanoparticles carrying cyclodextrins for nasal delivery of the anti-Alzheimer drug tacrine. Eur. J. Pharm. Sci. 2011, 44, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Z.; Zhang, Y.Q.; Wang, Z.Z.; Wu, K.; Lou, J.N.; Qi, X.R. Enhanced brain distribution and pharmacodynamics of rivastigmine by liposomes following intranasal administration. Int. J. Pharm. 2013, 452, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.F.; Khan, S.; Gaba, B.; Alam, T.; Baboota, S.; Ali, J.; Ali, A. Optimization of rivastigmine nanoemulsion for enhanced brain delivery: In-vivo and toxicity evaluation. J. Mol. Liq. 2018, 255, 384–396. [Google Scholar] [CrossRef]
- Fazil, M.; Md, S.; Haque, S.; Kumar, M.; Baboota, S.; Sahni, J.K.; Ali, J. Development and evaluation of rivastigmine loaded chitosan nanoparticles for brain targeting. Eur. J. Pharm. Sci. 2012, 47, 6–15. [Google Scholar] [CrossRef]
- Li, W.; Zhou, Y.; Zhao, N.; Hao, B.; Wang, X.; Kong, P. Pharmacokinetic behavior and efficiency of acetylcholinesterase inhibition in rat brain after intranasal administration of galanthamine hydrobromide loaded flexible liposomes. Environ. Toxicol. Pharmacol. 2012, 34, 272–279. [Google Scholar] [CrossRef]
- Muntimadugu, E.; Dhommati, R.; Jain, A.; Challa, V.G.S.; Shaheen, M.; Khan, W. Intranasal delivery of nanoparticle encapsulated tarenflurbil: A potential brain targeting strategy for Alzheimer’s disease. Eur. J. Pharm. Sci. 2016, 92, 224–234. [Google Scholar] [CrossRef]
- Muller, J.M.; Lelievre, V.; Becq-Giraudon, L.; Meunier, A.C. VIP as a cell-growth and differentiation neuromodulator role in neurodevelopment. Mol. Neurobiol. 1995, 10, 115–134. [Google Scholar] [CrossRef]
- Gao, X.; Wu, B.; Zhang, Q.; Chen, J.; Zhu, J.; Zhang, W.; Rong, Z.; Chen, H.; Jiang, X. Brain delivery of vasoactive intestinal peptide enhanced with the nanoparticles conjugated with wheat germ agglutinin following intranasal administration. J. Control. Release 2007, 121, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Takami, S.; Getchell, M.L.; Getchell, T.V. Lectin histochemical localization of galactose, N-acetylgalactosamine, and N-acetylglucosamine in glycoconjugates of the rat vomeronasal organ, with comparison to the olfactory and septal mucosae. Cell Tissue Res. 1994, 277, 211–230. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Shao, X.; Zhang, C.; Tan, Y.; Liu, Q.; Wan, X.; Zhang, Q.; Xu, S.; Jiang, X. Intranasal H102 peptide-loaded liposomes for brain delivery to treat Alzheimer’s disease. Pharm. Res. 2015, 32, 3837–3849. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, J.; Feng, C.; Shao, X.; Liu, Q.; Zhang, Q.; Pang, Z.; Jiang, X. Intranasal nanoparticles of basic fibroblast growth factor for brain delivery to treat Alzheimer’s disease. Int. J. Pharm. 2014, 461, 192–202. [Google Scholar] [CrossRef]
- Abe, K.; Saito, H. Effects of basic fibroblast growth factor on central nervous system functions. Pharmacol. Res. 2001, 43, 307–312. [Google Scholar] [CrossRef]
- Noshita, T.; Murayama, N.; Oka, T.; Ogino, R.; Nakamura, S.; Inoue, T. Effect of bFGF on neuronal damage induced by sequential treatment of amyloid β and excitatory amino acid In Vitro and In Vivo. Eur. J. Pharmacol. 2012, 695, 76–82. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, C.; Liu, Q.; Shao, X.; Feng, C.; Shen, Y.; Zhang, Q.; Jiang, X. Solanum tuberosum lectin-conjugated PLGA nanoparticles for nose-to-brain delivery: In Vivo and In Vitro evaluations. J. Drug Target. 2012, 20, 174–184. [Google Scholar] [CrossRef]
- Rassu, G.; Soddu, E.; Posadino, A.M.; Pintus, G.; Sarmento, B.; Giunchedi, P.; Gavini, E. Nose-to-brain delivery of BACE1 siRNA loaded in solid lipid nanoparticles for Alzheimer’s therapy. Colloids Surf. B Biointerfaces 2017, 152, 296–301. [Google Scholar] [CrossRef]
- Sood, S.; Jain, K.; Gowthamarajan, K. Optimization of curcumin nanoemulsion for intranasal delivery using design of experiment and its toxicity assessment. Colloids Surf. B Biointerfaces 2014, 113, 330–337. [Google Scholar] [CrossRef]
- Elnaggar, Y.S.R.; Etman, S.M.; Abdelmonsif, D.A.; Abdallah, O.Y. Intranasal piperine-loaded chitosan nanoparticles as Brain-targeted therapy in Alzheimer’s disease: Optimization, biological efficacy, and potential toxicity. J. Pharm. Sci. 2015, 104, 3544–3556. [Google Scholar] [CrossRef]
- Cometa, S.; Bonifacio, M.A.; Trapani, G.; Di Gioia, S.; Dazzi, L.; De Giglio, E.; Trapani, A. In Vitro investigations on dopamine loaded solid lipid nanoparticles. J. Pharm. Biomed. Anal. 2020, 185, 113257. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wang, A.; Yan, X.; Chu, L.; Yang, X.; Song, Y.; Sun, K.; Yu, X.; Liu, R.; Wu, Z.; et al. Brain-targeted intranasal delivery of dopamine with borneol and lactoferrin co-modified nanoparticles for treating Parkinson’s disease. Drug Deliv. 2019, 26, 700–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, H.P.; Gao, X.C.; Zhang, L.Q.; Wei, S.Q.; Bi, S.; Yang, Z.C.; Cui, H. In Vitro evaluation of enhancing effect of borneol on transcorneal permeation of compounds with different hydrophilicities and molecular sizes. Eur. J. Pharmacol. 2013, 705, 20–25. [Google Scholar] [CrossRef]
- Arisoy, S.; Sayiner, O.; Comoglu, T.; Onal, D.; Atalay, O.; Pehlivanoglu, B. In Vitro and In Vivo evaluation of levodopa-loaded nanoparticles for nose to brain delivery. Pharm. Dev. Technol. 2020, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.C.; Wang, A.P.; Chu, Y.C.; Liu, S.; Mu, H.J.; Liu, W.H.; Wu, Z.M.; Sun, K.X.; Li, Y.X. Intranasal delivery of rotigotine to the brain with lactoferrin-modified PEG-PLGA nanoparticles for Parkinson’s disease treatment. Int. J. Nanomed. 2016, 11, 6547–6559. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Xu, L.; Bi, C.; Duan, D.; Chu, L.; Yu, X.; Wu, Z.; Wang, A.; Sun, K. Lactoferrin-modified rotigotine nanoparticles for enhanced nose-to-brain delivery: LESA-MS/MS-based drug biodistribution, pharmacodynamics, and neuroprotective effects. Int. J. Nanomed. 2018, 13, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Bhattamisra, S.K.; Shak, A.T.; Xi, L.W.; Safian, N.H.; Choudhury, H.; Lim, W.M.; Shahzad, N.; Alhakamy, N.A.; Anwer, M.K.; Radhakrishnan, A.K.; et al. Nose to brain delivery of rotigotine loaded chitosan nanoparticles in human SH-SY5Y neuroblastoma cells and animal model of Parkinson’s disease. Int. J. Pharm. 2020, 579, 119148. [Google Scholar] [CrossRef] [PubMed]
- Karavasili, C.; Bouropoulos, N.; Sygellou, L.; Amanatiadou, E.P.; Vizirianakis, I.S.; Fatouros, D.G. PLGA/DPPC/trimethylchitosan spray-dried microparticles for the nasal delivery of ropinirole hydrochloride: In Vitro, ex vivo and cytocompatibility assessment. Mater. Sci. Eng. C 2016, 59, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Chatzitaki, A.-T.; Jesus, S.; Karavasili, C.; Andreadis, D.; Fatouros, D.G.; Borges, O. Chitosan-coated PLGA nanoparticles for the nasal delivery of ropinirole hydrochloride: In Vitro and ex vivo evaluation of efficacy and safety. Int. J. Pharm. 2020, 59, 119776. [Google Scholar] [CrossRef]
- Raj, R.; Wairkar, S.; Sridhar, V.; Gaud, R. Pramipexole dihydrochloride loaded chitosan nanoparticles for nose to brain delivery: Development, characterization and In Vivo anti-Parkinson activity. Int. J. Biol. Macromol. 2018, 109, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N. Rasagiline-encapsulated chitosan-coated PLGA nanoparticles targeted to the brain in the treatment of parkinson’s disease. J. Liq. Chromatogr. Relat. Technol. 2017, 40, 677–690. [Google Scholar] [CrossRef]
- Lundh, B.; Brockstedt, U.; Kristensson, K. Lectin-binding pattern of neuroepithelial and respiratory epithelial cells in the mouse nasal cavity. Histochem. J. 1989, 21, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Yan, Z.; Hu, K.; Pang, Z.; Cheng, X.; Guo, L.; Zhang, Q.; Jiang, X.; Fang, L.; Lai, R. Odorranalectin-conjugated nanoparticles: Preparation, brain delivery and pharmacodynamic study on Parkinson’s disease following intranasal administration. J. Control. Release 2011, 151, 131–138. [Google Scholar] [CrossRef]
- Lou, H.; Jing, X.; Wei, X.; Shi, H.; Ren, D.; Zhang, X. Naringenin protects against 6-OHDA-induced neurotoxicity via activation of the Nrf2/ARE signaling pathway. Neuropharmacology 2014, 79, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Md, S.; Alhakamy, N.A.; Aldawsari, H.M.; Asfour, H.Z. Neuroprotective and antioxidant effect of naringenin-loaded nanoparticles for nose-to-brain delivery. Brain Sci. 2019, 9, 275. [Google Scholar] [CrossRef] [Green Version]
- Pangeni, R.; Sharma, S.; Mustafa, G.; Ali, J.; Baboota, S. Vitamin E loaded resveratrol nanoemulsion for brain targeting for the treatment of Parkinson’s disease by reducing oxidative stress. Nanotechnology 2014, 25, 485102. [Google Scholar] [CrossRef]
- de Oliveira Junior, E.R.; Truzzi, E.; Ferraro, L.; Fogagnolo, M.; Pavan, B.; Beggiato, S.; Rustichelli, C.; Maretti, E.; Lima, E.M.; Leo, E.; et al. Nasal administration of nanoencapsulated geraniol/ursodeoxycholic acid conjugate: Towards a new approach for the management of Parkinson’s disease. J. Control. Release 2020, 321, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Bender, T.S.; Migliore, M.M.; Campbell, R.B.; John Gatley, S.; Waszczak, B.L. Intranasal administration of glial-derived neurotrophic factor (GDNF) rapidly and significantly increases whole-brain GDNF level in rats. Neuroscience 2015, 303, 569–576. [Google Scholar] [CrossRef]
- Hernando, S.; Herran, E.; Figueiro-Silva, J.; Pedraz, J.L.; Igartua, M.; Carro, E.; Hernandez, R.M. Intranasal administration of TAT-conjugated lipid nanocarriers loading GDNF for Parkinson’s disease. Mol. Neurobiol. 2018, 55, 145–155. [Google Scholar] [CrossRef]
- Zhao, Y.Z.; Li, X.; Lu, C.T.; Lin, M.; Chen, L.J.; Xiang, Q.; Zhang, M.; Jin, R.R.; Jiang, X.; Shen, X.T.; et al. Gelatin nanostructured lipid carriers-mediated intranasal delivery of basic fibroblast growth factor enhances functional recovery in hemiparkinsonian rats. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Kumar, S.; Ali, J.; Baboota, S. Intranasal delivery of tetrabenazine nanoemulsion via olfactory region for better treatment of hyperkinetic movement associated with Huntington’s disease: Pharmacokinetic and brain delivery study. Chem. Phys. Lipids 2020, 230, 104917. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, R.; Singh, D.; Prakash, A.; Mishra, N. Development, characterization and nasal delivery of rosmarinic acid-loaded solid lipid nanoparticles for the effective management of Huntingtons disease. Drug Deliv. 2015, 22, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Passoni, A.; Favagrossa, M.; Colombo, L.; Bagnati, R.; Gobbi, M.; Diomede, L.; Birolini, G.; Di Paolo, E.; Valenza, M.; Cattaneo, E.; et al. Efficacy of cholesterol nose-to-brain delivery for brain targeting in Huntington’s disease. ACS Chem. Neurosci. 2020, 11, 367–372. [Google Scholar] [CrossRef]
- Sava, V.; Fihurka, O.; Khvorova, A.; Sanchez-Ramos, J. Enriched chitosan nanoparticles loaded with siRNA are effective in lowering Huntington’s disease gene expression following intranasal administration. Nanomed. Nanotechnol. Biol. Med. 2020, 24, 102119. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riccardi, C.; Napolitano, F.; Montesarchio, D.; Sampaolo, S.; Melone, M.A.B. Nanoparticle-Guided Brain Drug Delivery: Expanding the Therapeutic Approach to Neurodegenerative Diseases. Pharmaceutics 2021, 13, 1897. https://doi.org/10.3390/pharmaceutics13111897
Riccardi C, Napolitano F, Montesarchio D, Sampaolo S, Melone MAB. Nanoparticle-Guided Brain Drug Delivery: Expanding the Therapeutic Approach to Neurodegenerative Diseases. Pharmaceutics. 2021; 13(11):1897. https://doi.org/10.3390/pharmaceutics13111897
Chicago/Turabian StyleRiccardi, Claudia, Filomena Napolitano, Daniela Montesarchio, Simone Sampaolo, and Mariarosa Anna Beatrice Melone. 2021. "Nanoparticle-Guided Brain Drug Delivery: Expanding the Therapeutic Approach to Neurodegenerative Diseases" Pharmaceutics 13, no. 11: 1897. https://doi.org/10.3390/pharmaceutics13111897
APA StyleRiccardi, C., Napolitano, F., Montesarchio, D., Sampaolo, S., & Melone, M. A. B. (2021). Nanoparticle-Guided Brain Drug Delivery: Expanding the Therapeutic Approach to Neurodegenerative Diseases. Pharmaceutics, 13(11), 1897. https://doi.org/10.3390/pharmaceutics13111897