
Compartmentalized Polymeric Nanoparticles Deliver Vancomycin in a pH-Responsive Manner

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of VCM-Loaded PLA and PLGA NPs
2.3. Drug Quantification
2.4. Determination of Residual PVA in the Preparation of PLGA and PLA Particles
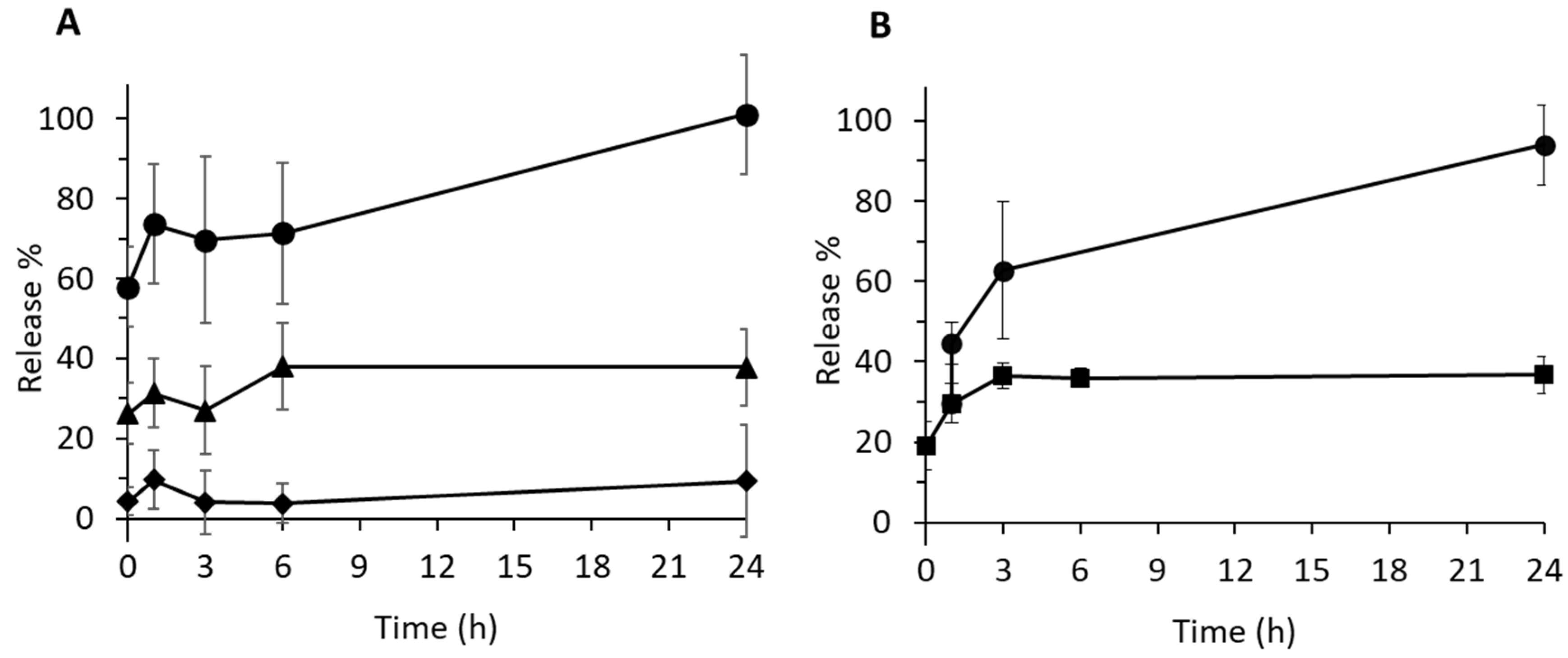
2.5. Release Studies
2.6. Nanoparticle Characterization
2.6.1. NP Size Measurement by Dynamic Light Scattering (DLS) and Zeta Potential Measurements
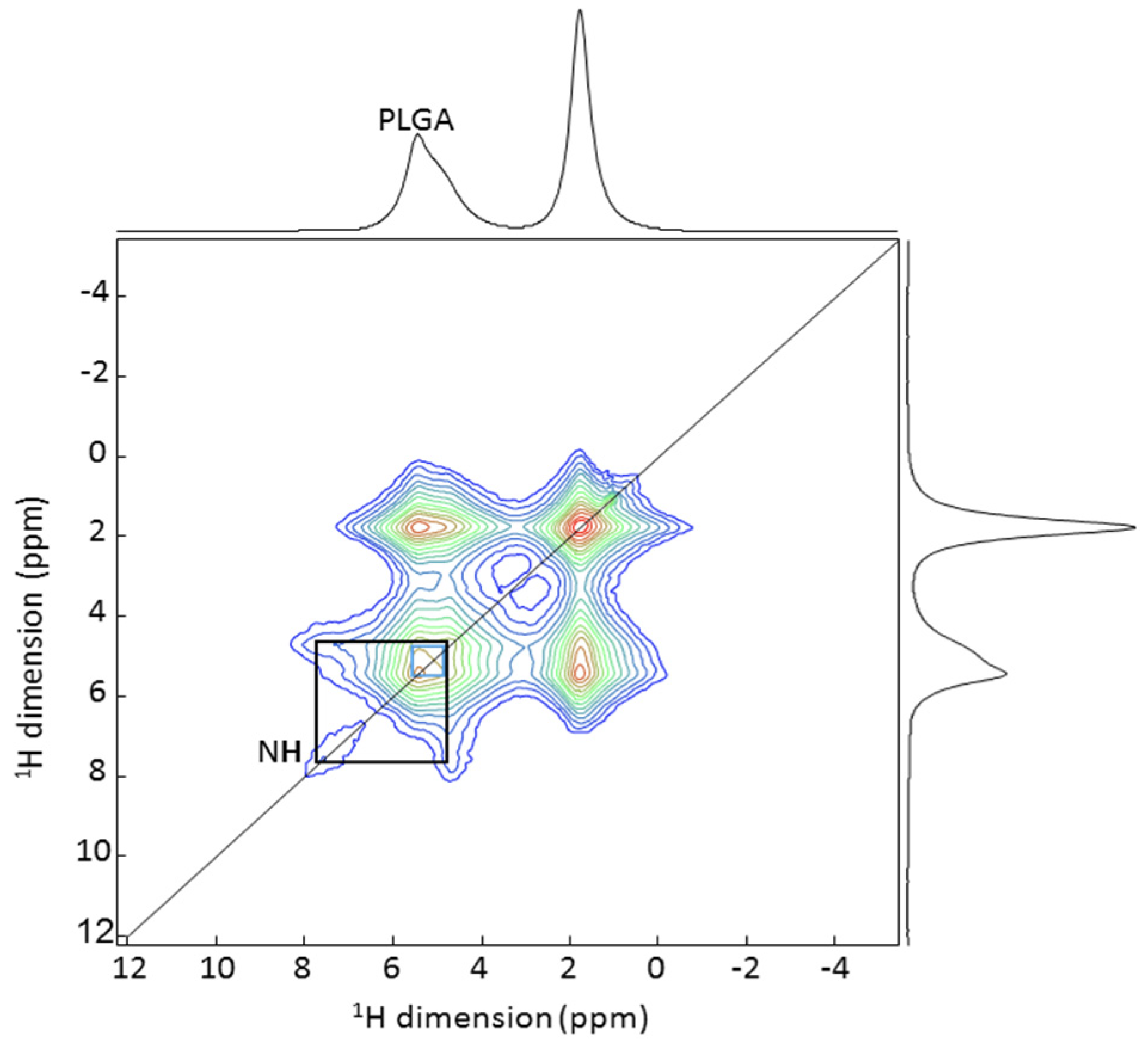
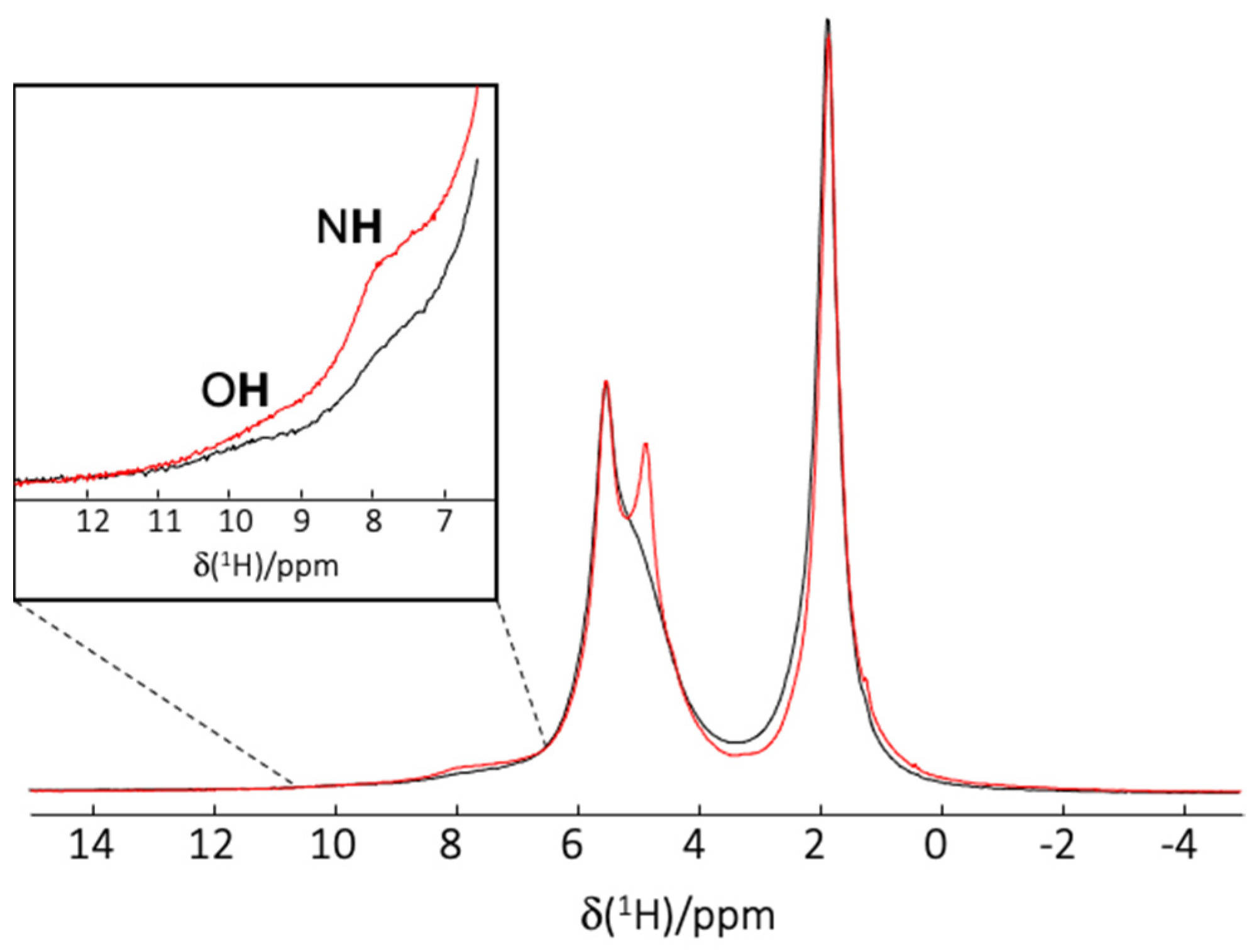
2.6.2. Solid State NMR Spectroscopy
2.6.3. Size-Exclusion Chromatography (SEC)
2.6.4. TEM and Cryo-TEM Investigations
2.6.5. Scanning Electron Microscopy (SEM)-EDX
2.6.6. Scanning TEM-EDX
2.6.7. Fourier Transform IR (FTIR) Spectroscopy
2.6.8. AFM-IR
3. Results and Discussion
3.1. NP Preparation
3.2. Drug Loading and Mechanism
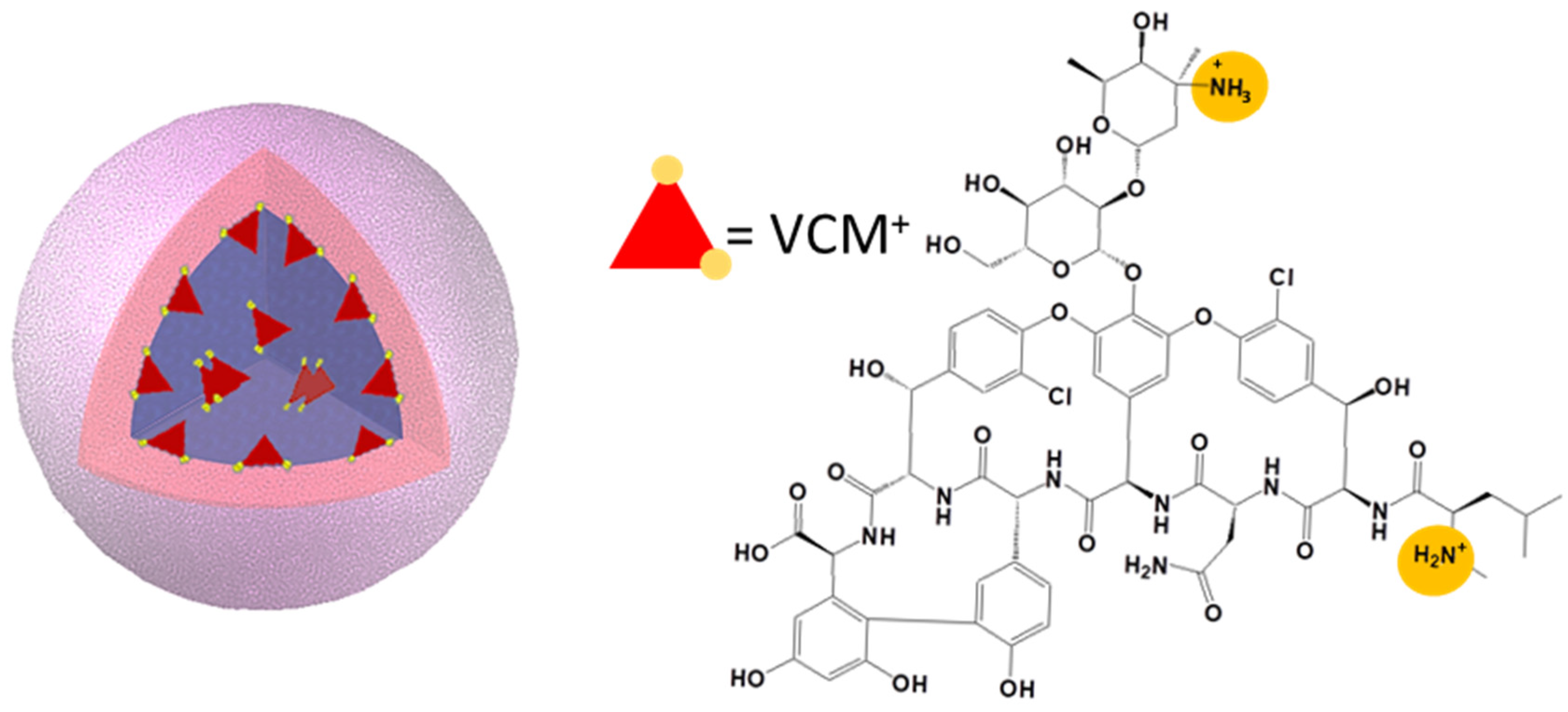
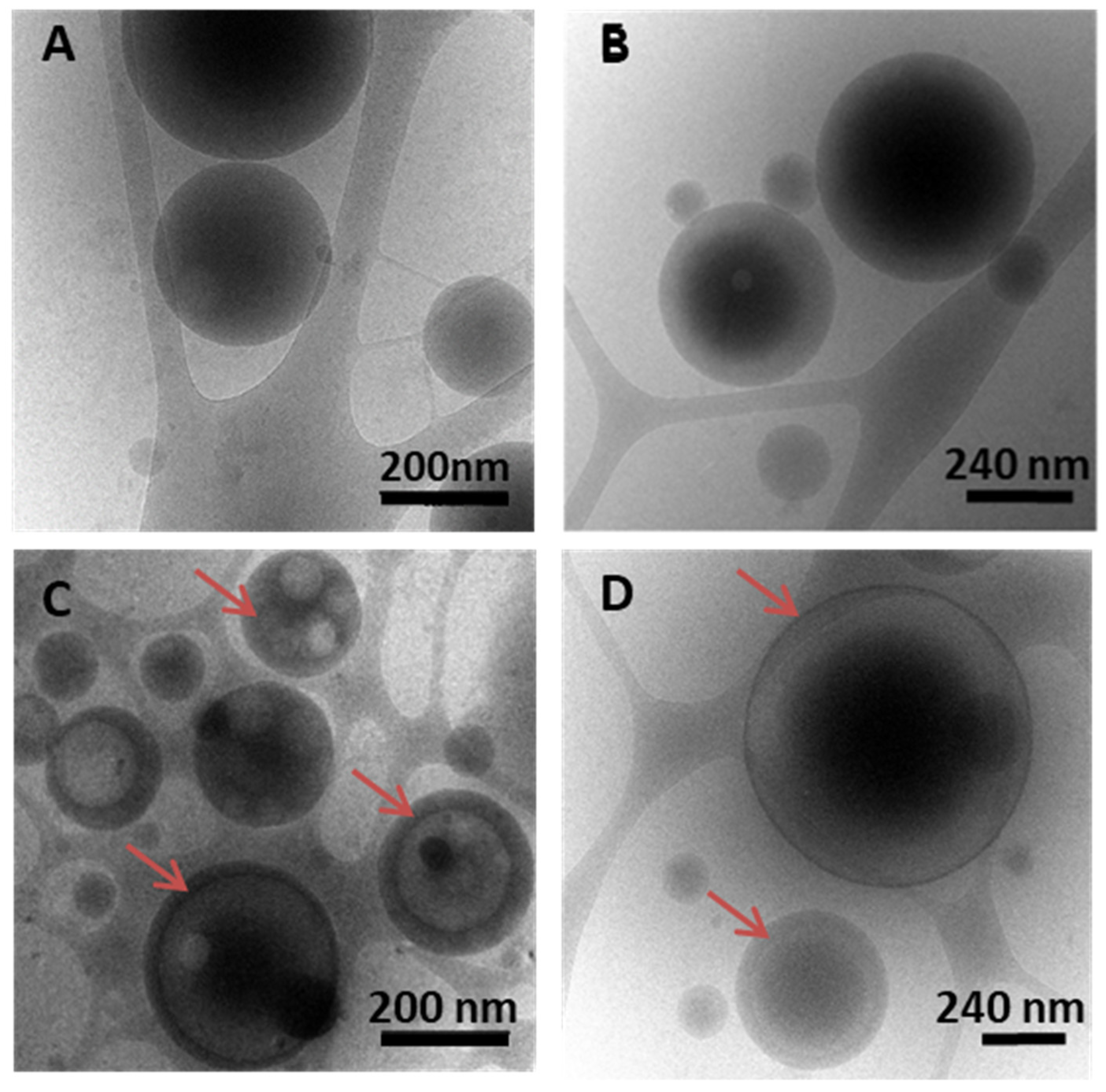
3.3. NPs’ Inner Morphology
3.4. Individual NP Characterization: Localization of VCM
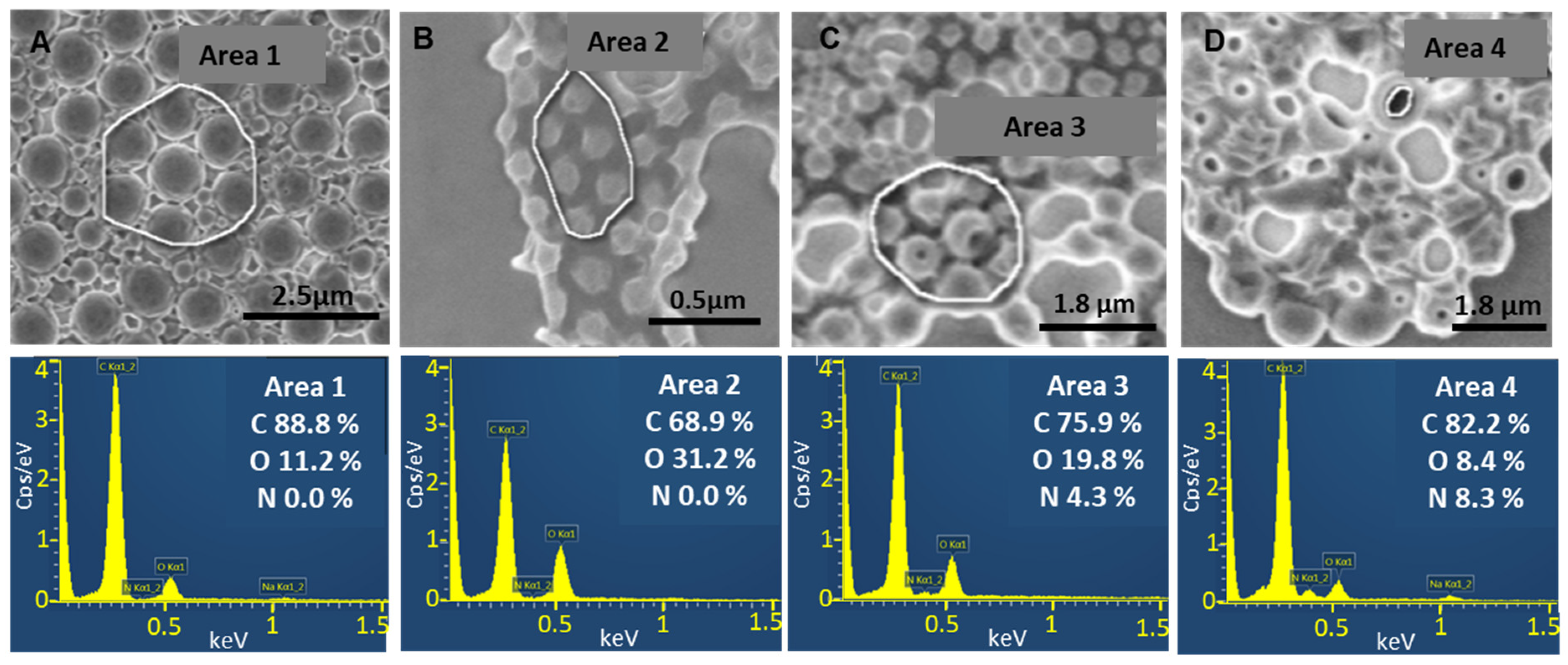
3.4.1. SEM-EDX
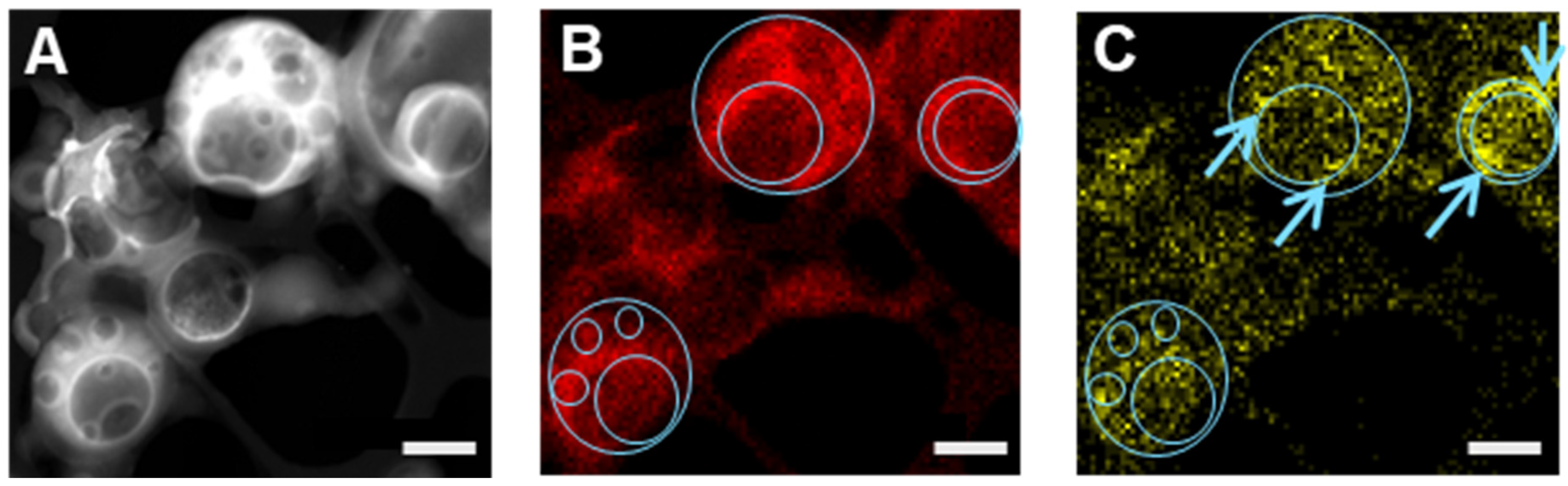
3.4.2. STEM-EDX
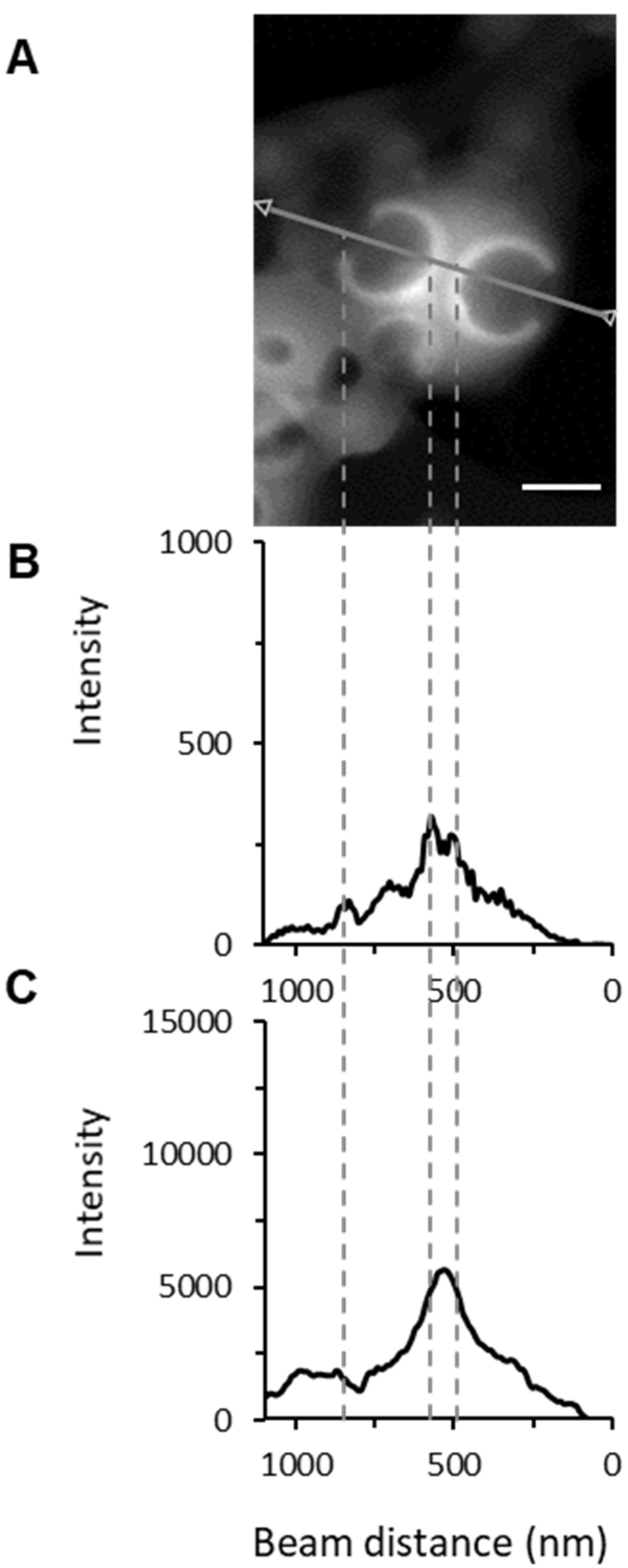
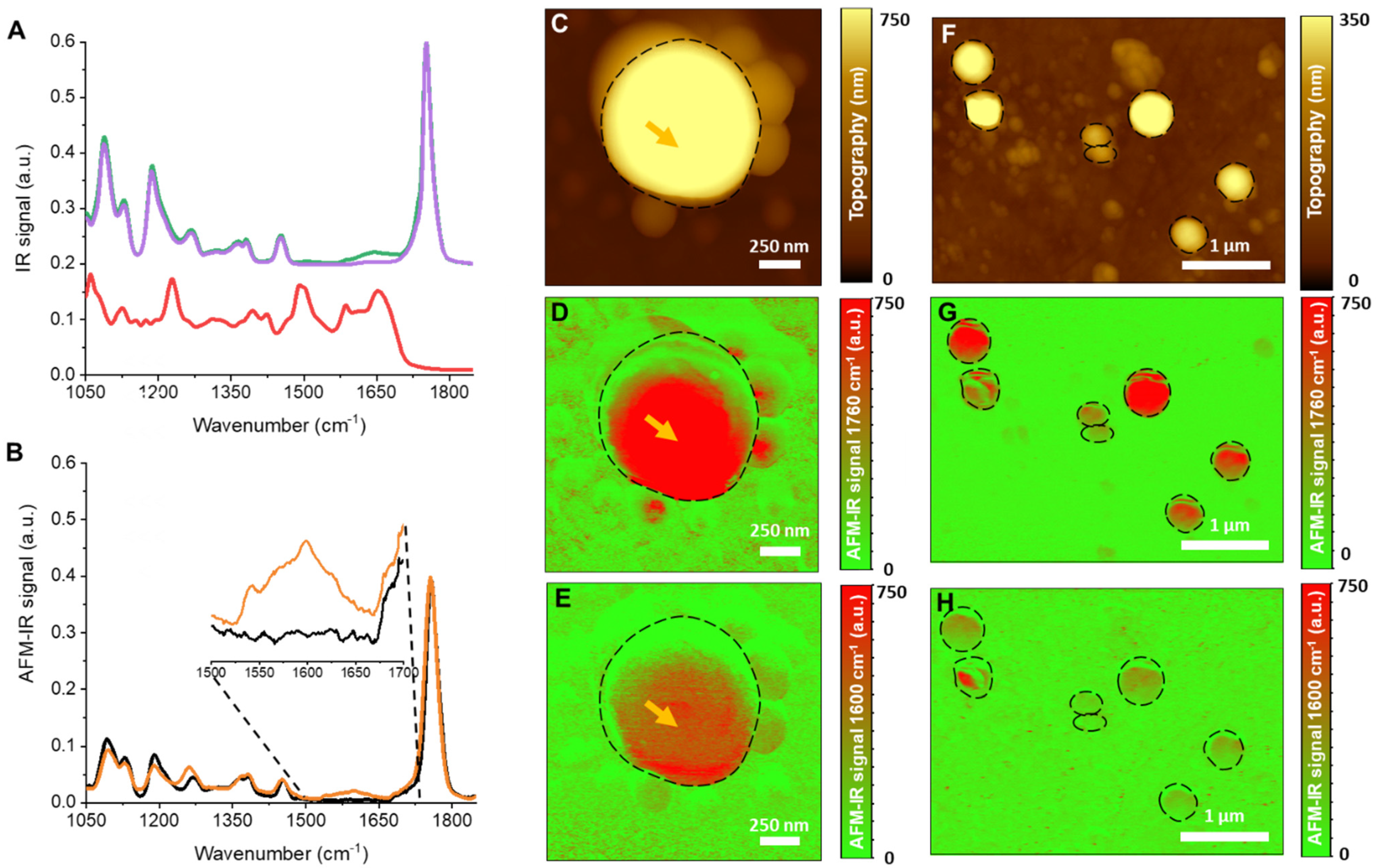
3.4.3. AFM-IR
3.5. pH-Controlled Drug Release and Related Mechanism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Hiroshi Morisaki, J.; et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Guzman Prieto, A.M.; van Schaik, W.; Rogers, M.R.C.; Coque, T.M.; Baquero, F.; Corander, J.; Willems, R.J.L. Global emergence and dissemination of enterococci as nosocomial pathogens: Attack of the clones? Front. Microbiol. 2016, 7, 788. [Google Scholar] [CrossRef] [Green Version]
- Daneman, N.; McGeer, A.; Low, D.E.; Tyrrell, G.; Simor, A.E.; McArthur, M.; Schwartz, B.; Jessamine, P.; Croxford, R.; Green, K.A. Hospital-acquired invasive group A streptococcal infections in Ontario, Canada, 1992-2000. Clin. Infect. Dis. 2005, 41, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, I.; Sanges, M.R.; Folgore, A.; Carratelli, C.R. Apoptosis of human keratinocytes after bacterial invasion. FEMS Immunol. Med. Microbiol. 2000, 27, 235–240. [Google Scholar] [CrossRef]
- Fowler, T.; Wann, E.R.; Joh, D.; Johansson, S.; Foster, T.J.; Höök, M. Cellular invasion by Staphylococcus aureus involves a fibronectin bridge between the bacterial fibronectic-binding MSCRAMMs and host cell β1 integrins. Eur. J. Cell Biol. 2000, 79, 672–679. [Google Scholar] [CrossRef]
- Hess, D.J.; Henry-Stanley, M.J.; Erickson, E.A.; Wells, C.L. Intracellular survival of Staphylococcus aureus within cultured enterocytes. J. Surg. Res. 2003, 114, 42–49. [Google Scholar] [CrossRef]
- Nair, S.P.; Bischoff, M.; Senn, M.M.; Berger-Bächi, B. The σB regulon influences internalization of Staphylococcus aureus by osteoblasts. Infect. Immun. 2003, 71, 4167–4170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pumerantz, A.; Muppidi, K.; Agnihotri, S.; Guerra, C.; Venketaraman, V.; Wang, J.; Betageri, G. Preparation of liposomal vancomycin and intracellular killing of meticillin-resistant Staphylococcus aureus (MRSA). Int. J. Antimicrob. Agents 2011, 37, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J. Immune evasion by staphylococci. Nat. Rev. Microbiol. 2005, 3, 948–958. [Google Scholar] [CrossRef]
- Garzoni, C.; Kelley, W.L. Staphylococcus aureus: New evidence for intracellular persistence. Trends Microbiol. 2009, 17, 59–65. [Google Scholar] [CrossRef]
- Barcia-Macay, M.; Seral, C.; Mingeot-Leclercq, M.P.; Tulkens, P.M.; Van Bambeke, F. Pharmacodynamic evaluation of the intracellular activities of antibiotics against Staphylococcus aureus in a model of THP-1 macrophages. Antimicrob. Agents Chemother. 2006, 50, 841–851. [Google Scholar] [CrossRef] [Green Version]
- Prasad, Y.; Puthli, S.P.; Eaimtrakarn, S.; Ishida, M.; Yoshikawa, Y.; Shibata, N.; Takada, K. Enhanced intestinal absorption of vancomycin with Labrasol and D-α-tocopheryl PEG 1000 succinate in rats. Int. J. Pharm. 2003, 250, 181–190. [Google Scholar] [CrossRef]
- Patel, S.; Preuss, C.V.; Bernice, F. Vancomycin; StatPearls Publishing: St. Petersburg, FL, USA, 2021. [Google Scholar]
- Roszell, S.; Jones, C. Intravenous Administration Issues. J. Infus. Nurs. 2010, 33, 112–118. [Google Scholar] [CrossRef] [Green Version]
- Rybak, M.; Lomaestro, B.; Rotschafer, J.C.; Moellering, R.; Craig, W.; Billeter, M.; Dalovisio, J.R.; Levine, D.P.; Reilly, C. Therapeutic monitoring of vancomycin in adult patients: A consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am. J. Health Pharm. 2009, 66, 82–98. [Google Scholar] [CrossRef]
- Sun, H.; Maderazo, E.G.; Krusell, A.R. Serum protein-binding characteristics of vancomycin. Antimicrob. Agents Chemother. 1993, 37, 1132–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levison, M.E.; Levison, J.H. Pharmacokinetics and Pharmacodynamics of Antibacterial Agents. Infect. Dis. Clin. N. Am. 2009, 23, 791–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrell, T.C.; Collignon, P.J. A prospective study of adverse reactions associated with vancomycin therapy. J. Antimicrob. Chemother. 1985, 16, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Mohamed, M.F.; Seleem, M.N.; Yeo, Y. Particle engineering for intracellular delivery of vancomycin to methicillin-resistant Staphylococcus aureus (MRSA)-infected macrophages. J. Control. Release 2017, 267, 133–143. [Google Scholar] [CrossRef]
- Kollef, M.H. Limitations of vancomycin in the management of resistant staphylococcal infections. Clin. Infect. Dis. 2007, 45, 191–195. [Google Scholar] [CrossRef]
- Ladavière, C.; Gref, R. Toward an optimized treatment of intracellular bacterial infections: Input of nanoparticulate drug delivery systems. Nanomedicine 2015, 10, 3033–3055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, Y.; Grazon, C.; Clavier, G.; Rieger, J.; Audibert, J.F.; Sclavi, B.; Méallet-Renault, R. Rapid and accurate detection of Escherichia coli growth by fluorescent pH-sensitive organic nanoparticles for high-throughput screening applications. Biosens. Bioelectron. 2016, 75, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Radovic-Moreno, A.F.; Lu, T.K.; Puscasu, V.A.; Yoon, C.J.; Langer, R.; Farokhzad, O.C. Surface charge-switching polymeric nanoparticles for bacterial cell wall-targeted delivery of antibiotics. ACS Nano 2012, 6, 4279–4287. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Lillard, J.W. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, M.; Zafar, N.; Fessi, H.; Elaissari, A. Double emulsion solvent evaporation techniques used for drug encapsulation. Int. J. Pharm. 2015, 496, 173–190. [Google Scholar] [CrossRef] [PubMed]
- Sande, L.; Sanchez, M.; Montes, J.; Wolf, A.J.; Morgan, M.A.; Omri, A.; Liu, G.Y. Liposomal encapsulation of vancomycin improves killing of methicillin-resistant Staphylococcus aureus in a murine infection model. J. Antimicrob. Chemother. 2012, 67, 2191–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, T.; Shang, B.C.; Tang, H.; Zhou, T.H.; Xu, G.L.; Li, H.L.; Chen, Q.H.; Xu, Y.Q. Nano-hydroxyapatite/chitosan/konjac glucomannan scaffolds loaded with cationic liposomal vancomycin: Preparation, in vitro release and activity against staphylococcus aureus biofilms. J. Biomater. Sci. Polym. Ed. 2011, 22, 1669–1681. [Google Scholar] [CrossRef]
- Shahverdi, A.R.; Fakhimi, A.; Shahverdi, H.R.; Minaian, S. Synthesis and effect of silver nanoparticles on the antibacterial activity of different antibiotics against Staphylococcus aureus and Escherichia coli. Nanomed. Nanotechnol. Biol. Med. 2007, 3, 168–171. [Google Scholar] [CrossRef]
- Hur, Y.E.; Park, Y. Vancomycin-Functionalized gold and silver nanoparticles as an antibacterial nanoplatform against methicillin-resistant staphylococcus aureus. J. Nanosci. Nanotechnol. 2016, 16, 6393–6399. [Google Scholar] [CrossRef]
- Kaur, A.; Preet, S.; Kumar, V.; Kumar, R.; Kumar, R. Synergetic effect of vancomycin loaded silver nanoparticles for enhanced antibacterial activity. Colloids Surf. B Biointerfaces 2019, 176, 62–69. [Google Scholar] [CrossRef]
- Esmaeillou, M.; Zarrini, G.; Rezaee, M.A.; Mojarrad, J.S.; Bahadori, A. Vancomycin capped with silver nanoparticles as an antibacterial agent against multi-drug resistance bacteria. Adv. Pharm. Bull. 2017, 7, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Gu, H.; Ho, P.L.; Tong, E.; Wang, L.; Xu, B. Presenting vancomycin on nanoparticles to enhance antimicrobial activities. Nano Lett. 2003, 3, 1261–1263. [Google Scholar] [CrossRef]
- Singla, R.; Guliani, A.; Kumari, A.; Yadav, S.K. Metallic Nanoparticles, Toxicity Issues and Applications in Medicine. In Nanoscale Materials in Targeted Drug Delivery, Theragnosis and Tissue Regeneration; Springer: Singapore, 2016; pp. 41–80. ISBN 9789811008184. [Google Scholar]
- Koo, O.M.; Rubinstein, I.; Onyuksel, H. Role of nanotechnology in targeted drug delivery and imaging: A concise review. Nanomedicine Nanotechnology, Biol. Med. 2005, 1, 193–212. [Google Scholar] [CrossRef]
- Sayin, B.; Çalifi, S. Influence of Accelarated Storage Conditions on The Stability of Vancomycin Loaded Poly(D,L-Lactide-Co- Glycolide) Microspheres. Fabad J. Pharm. Sci. 2005, 29, 111–116. [Google Scholar]
- Özalp, Y.; Özdemir, N.; Kocagöz, S.; Hasirci, V. Controlled release of vancomycin from biodegradable microcapsules. J. Microencapsul. 2001, 18, 89–110. [Google Scholar] [CrossRef] [PubMed]
- Ritsema, J.A.S.; Herschberg, E.M.A.; Borgos, S.E.; Løvmo, C.; Schmid, R.; te Welscher, Y.M.; Storm, G.; van Nostrum, C.F. Relationship between polarities of antibiotic and polymer matrix on nanoparticle formulations based on aliphatic polyesters. Int. J. Pharm. 2018, 548, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Posadowska, U.; Brzychczy-Wloch, M.; Pamula, E. Injectable gellan gum-based nanoparticles-loaded system for the local delivery of vancomycin in osteomyelitis treatment. J. Mater. Sci. Mater. Med. 2016, 27, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakeri-Milani, P.; Loveymi, B.D.; Jelvehgari, M.; Valizadeh, H. The characteristics and improved intestinal permeability of vancomycin PLGA-nanoparticles as colloidal drug delivery system. Colloids Surf. B Biointerfaces 2013, 103, 174–181. [Google Scholar] [CrossRef]
- Huang, W.F.; Tsui, C.P.; Tang, C.Y.; Yang, M.; Gu, L. Surface charge switchable and pH-responsive chitosan/polymer core-shell composite nanoparticles for drug delivery application. Compos. Part B Eng. 2017, 121, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Zambaux, M.F.; Bonneaux, F.; Gref, R.; Maincent, P.; Dellacherie, E.; Alonso, M.J.; Labrude, P.; Vigneron, C. Influence of experimental parameters on the characteristics of poly(lactic acid) nanoparticles prepared by a double emulsion method. J. Control. Release 1998, 50, 31–40. [Google Scholar] [CrossRef]
- Massiot, D.; Fayon, F.; Capron, M.; King, I.; Le Calvé, S.; Alonso, B.; Durand, J.O.; Bujoli, B.; Gan, Z.; Hoatson, G. Modelling one- and two-dimensional solid-state NMR spectra. Magn. Reson. Chem. 2002, 40, 70–76. [Google Scholar] [CrossRef]
- Mathurin, J.; Pancani, E.; Deniset-Besseau, A.; Kjoller, K.; Prater, C.B.; Gref, R.; Dazzi, A. How to unravel the chemical structure and component localization of individual drug-loaded polymeric nanoparticles by using tapping AFM-IR. Analyst 2018, 143, 5940–5949. [Google Scholar] [CrossRef]
- Xing, B.; Jiang, T.; Wu, X.; Liew, R.; Zhou, J.; Zhang, D.; Yeow, E.K.L. Molecular interactions between glycopeptide vancomycin and bacterial cell wall peptide analogues. Chem. A Eur. J. 2011, 17, 14170–14177. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; O’Mara, M.L.; Zuegg, J.; Cooper, M.A.; Mark, A.E. Vancomycin: Ligand recognition, dimerization and super-complex formation. FEBS J. 2013, 280, 1294–1307. [Google Scholar] [CrossRef]
- Loll, P.J.; Bevivino, A.E.; Korty, B.D.; Axelsen, P.H. Simultaneous recognition of a carboxylate-containing ligand and an intramolecular surrogate ligand in the crystal structure of an asymmetric vancomycin dimer. J. Am. Chem. Soc. 1997, 119, 1516–1522. [Google Scholar] [CrossRef]
- Loll, P.J.; Miller, R.; Weeks, C.M.; Axelsen, P.H. A ligand-mediated dimerization mode for vancomycin. Chem. Biol. 1998, 5, 293–298. [Google Scholar] [CrossRef] [Green Version]
- Giammarco, J.; Mochalin, V.N.; Haeckel, J.; Gogotsi, Y. The adsorption of tetracycline and vancomycin onto nanodiamond with controlled release. J. Colloid Interface Sci. 2016, 468, 253–261. [Google Scholar] [CrossRef]
- Prosapio, V.; De Marco, I.; Reverchon, E. PVP/corticosteroid microspheres produced by supercritical antisolvent coprecipitation. Chem. Eng. J. 2016, 292, 264–275. [Google Scholar] [CrossRef]
- Quellec, P.; Gref, R.; Perrin, L.; Dellacherie, E.; Sommer, F.; Verbavatz, J.M.; Alonso, M.J. Protein encapsulation within polyethylene glycol-coated nanospheres. I. Physicochemical characterization. J. Biomed. Mater. Res. 1998, 42, 45–54. [Google Scholar] [CrossRef]
- Lotfipour, F.; Abdollahi, S.; Jelvehgari, M.; Valizadeh, H.; Hassan, M.; Milani, M. Study of antimicrobial effects of vancomycin loaded PLGA nanoparticles against enterococcus clinical isolates. Drug Res. (Stuttg) 2014, 64, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Kentish, S.; Wooster, T.J.; Ashokkumar, M.; Balachandran, S.; Mawson, R.; Simons, L. The use of ultrasonics for nanoemulsion preparation. Innov. Food Sci. Emerg. Technol. 2008, 9, 170–175. [Google Scholar] [CrossRef]
- Mahdi Jafari, S.; He, Y.; Bhandari, B. Nano-emulsion production by sonication and microfluidization—A comparison. Int. J. Food Prop. 2006, 9, 475–485. [Google Scholar] [CrossRef]
- Takács-Novák, K.; Noszál, B.; Tókés-Kövesdi, M.; Szász, G. Acid-base properties and proton-speciation of vancomycin. Int. J. Pharm. 1993, 89, 261–263. [Google Scholar] [CrossRef]
- Johnson, J.L.H.; Yalkowsky, S.H. Reformulation of a new vancomycin analog: An example of the importance of buffer species and strength. AAPS PharmSciTech 2006, 7, E1–E5. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.W.; Mitragotri, S. Polymer particles that switch shape in response to a stimulus. Proc. Natl. Acad. Sci. USA 2010, 107, 11205–11210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shestakova, P.; Martineau, C.; Mavrodinova, V.; Popova, M. Solid state NMR characterization of zeolite beta based drug formulations containing Ag and sulfadiazine. RSC Adv. 2015, 5, 81957–81964. [Google Scholar] [CrossRef]
- Knox, J.R.; Pratt, R.F. Different modes of vancomycin and D-alanyl-D-alanine peptidase binding to cell wall peptide and a possible role for the vancomycin resistance protein. Antimicrob. Agents Chemother. 1990, 34, 1342–1347. [Google Scholar] [CrossRef] [Green Version]
- Dazzi, A.; Prater, C.B. AFM-IR: Technology and applications in nanoscale infrared spectroscopy and chemical imaging. Chem. Rev. 2017, 117, 5146–5173. [Google Scholar] [CrossRef]
- Lu, F.; Jin, M.; Belkin, M.A. Tip-enhanced infrared nanospectroscopy via molecular expansion force detection. Nat. Photonics 2014, 8, 307–312. [Google Scholar] [CrossRef]
- Pancani, E.; Mathurin, J.; Bilent, S.; Bernet-Camard, M.F.; Dazzi, A.; Deniset-Besseau, A.; Gref, R. High-Resolution Label-Free Detection of Biocompatible Polymeric Nanoparticles in Cells. Part. Part. Syst. Charact. 2018, 35. [Google Scholar] [CrossRef]
- Yuniarto, K.; Aris Purwanto, Y.; Purwanto, S.; Welt, B.A.; Karia Purwadaria, H.; Candra Sunarti, T. Infrared and Raman studies on polylactide acid and polyethylene glycol-400 blend. AIP Conf. Proc. 2016, 1725, 20101. [Google Scholar] [CrossRef] [Green Version]
- Salter, C.J.; Mitchell, R.C.; Drake, A.F. Infrared spectroscopic studies of vancomycin and its interactions with N-acetyl-D-Ala-D-Ala and N,N′-diacetyl-L-Lys-D-Ala-D-Ala. J. Chem. Soc. Perkin Trans. 2 1995, 2203–2211. [Google Scholar] [CrossRef]
- Phillips-Jones, M.K.; Lithgo, R.; DInu, V.; Gillis, R.B.; Harding, J.E.; Adams, G.G.; Harding, S.E. Full hydrodynamic reversibility of the weak dimerization of vancomycin and elucidation of its interaction with VanS monomers at clinical concentration. Sci. Rep. 2017, 7, 12697. [Google Scholar] [CrossRef] [PubMed]
- Tu, F.; Lee, D. Controlling the stability and size of double-emulsion-templated poly(lactic- co -glycolic) acid microcapsules. Langmuir 2012, 28, 9944–9952. [Google Scholar] [CrossRef] [PubMed]
- Gorrepati, E.A.; Wongthahan, P.; Raha, S.; Fogler, H.S. Silica precipitation in acidic solutions: Mechanism, pH effect, and salt effect. Langmuir 2010, 26, 10467–10474. [Google Scholar] [CrossRef]
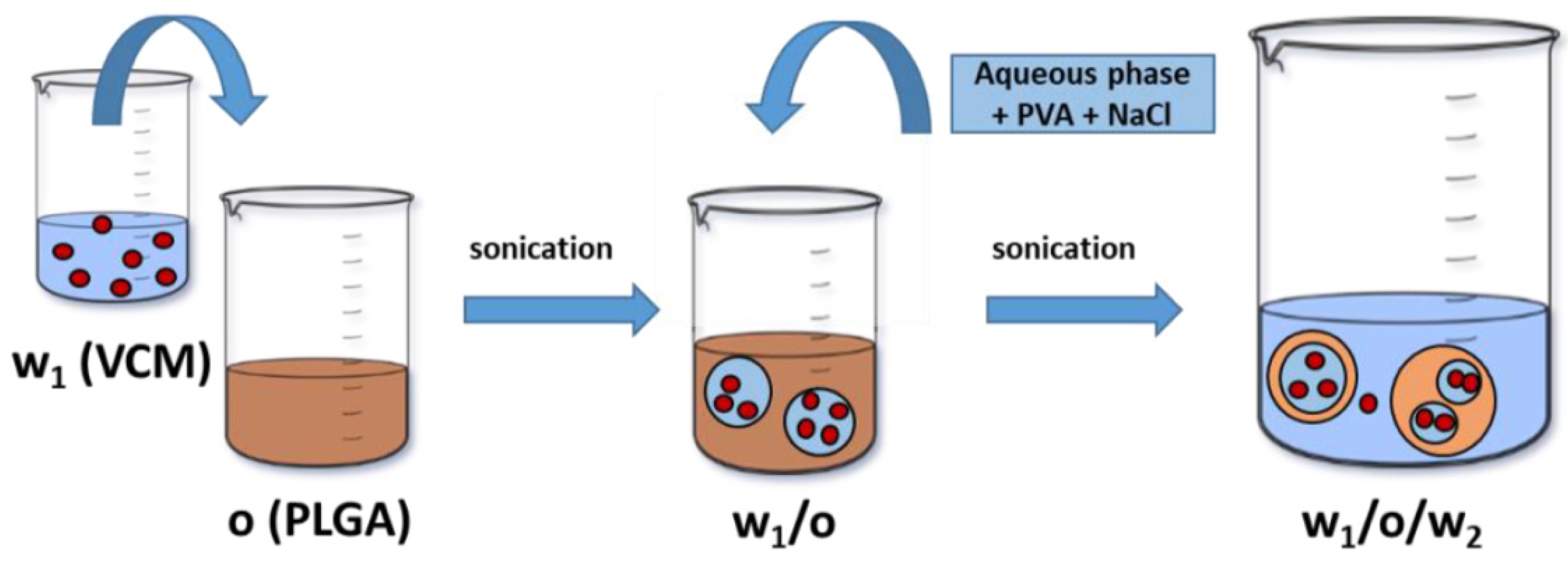
 ) is solubilized in w1.
) is solubilized in w1.
) is solubilized in w1.
) is solubilized in w1.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Mw Range (kDa) | End Group | Lactic Unit (%) | Inherent Viscosity Range (dL/g) | DL (wt% + SD) | EE (wt% + SD) | Mean Diameter *,† (nm) | PDI | Zeta Potential (mV + SD) |
|---|---|---|---|---|---|---|---|---|---|
| PLA8C | 6–10 | Carboxyl | 100 | 0.05–0.20 | 7 ± 2 | 16 ± 5 | 319 | 0.18 ± 0.05 | −1.6 ± 0.1 |
| PLA15C | 10–20 | Carboxyl | 100 | 0.15–0.30 | <1 | <1 | 322 | 0.25 ± 0.07 | −1.7 ± 0.1 |
| PLGA13C | 5–20 | Carboxyl | 75 | 0.08–0.21 | 5 ± 1 | 11 ± 2 | 312 | 0.13 ± 0.01 | −1.9 ± 0.1 |
| PLGA15C | 10–20 | Carboxyl | 50 | 0.15–0.25 | 14 ± 4 | 36 ± 2 | 325 | 0.19 ± 0.02 | −1.9 ± 0.1 |
| PLGA23C | 15–30 | Carboxyl | 45 | 0.15–0.30 | 9 ± 1 | 24 ± 1 | 323 | 0.18 ± 0.03 | −1.3 ± 0.1 |
| PLGA28C | 15–40 | Carboxyl | 50 | 0.25–0.40 | 8 ± 1 | 18 ± 2 | 326 | 0.21 ± 0.03 | −0.7 ± 0.3 |
| PLGA54C | 42–65 | Carboxyl | 50 | 0.40–0.55 | <1 | <1 | 324 | 0.14 ± 0.01 | −1.4 ± 0.3 |
| PLGA61C | 37–84 | Carboxyl | 75 | 0.38–0.64 | <1 | <1 | 350 | 0.16 ± 0.03 | −1.4 ± 0.1 |
| PLGA103C | 76–130 | Carboxyl | 75 | 0.70–0.90 | <1 | <1 | 352 | 0.14 ± 0.01 | −1.3 ± 0.3 |
| PLGA113C | 95–130 | Carboxyl | 50 | 0.65–0.90 | <1 | <1 | 367 | 0.17 ± 0.03 | −1.4 ± 0.1 |
| PLGA15E | 10–20 | Ester | 50 | 0.15–0.25 | <1 | <1 | 323 | 0.26 ± 0.04 | −0.9 ± 0.1 |
| PLGA82E | 72–91 | Ester | 50 | 0.60–0.70 | <1 | <1 | 344 | 0.15 ± 0.04 | −1.4 ± 0.2 |
| pH of 1st Emulsion | pH of 2nd Emulsion | DL (wt% ± SD) | EE (wt% ± SD) | Mean Diameter *,† (nm) | PDI |
|---|---|---|---|---|---|
| 4.0 | 6.3 | 14 ± 4 | 36 ± 2 | 325 | 0.16 ± 0.17 |
| 7.4 | 12 ± 1 | 36 ± 1 | 340 | 0.17 ± 0.04 | |
| 8.5 | 14 ± 4 | 43 ± 9 | 353 | 0.19 ± 0.06 | |
| 7.4 | 6.3 | 22 ± 1 | 47 ± 2 | 318 | 0.15 ± 0.01 |
| 7.4 | 25 ± 3 | 53 ± 7 | 312 | 0.15 ± 0.03 | |
| 8.5 | 23 ± 3 | 50 ± 7 | 319 | 0.19 ± 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ural, M.S.; Menéndez-Miranda, M.; Salzano, G.; Mathurin, J.; Aybeke, E.N.; Deniset-Besseau, A.; Dazzi, A.; Porcino, M.; Martineau-Corcos, C.; Gref, R. Compartmentalized Polymeric Nanoparticles Deliver Vancomycin in a pH-Responsive Manner. Pharmaceutics 2021, 13, 1992. https://doi.org/10.3390/pharmaceutics13121992
Ural MS, Menéndez-Miranda M, Salzano G, Mathurin J, Aybeke EN, Deniset-Besseau A, Dazzi A, Porcino M, Martineau-Corcos C, Gref R. Compartmentalized Polymeric Nanoparticles Deliver Vancomycin in a pH-Responsive Manner. Pharmaceutics. 2021; 13(12):1992. https://doi.org/10.3390/pharmaceutics13121992
Chicago/Turabian StyleUral, Merve Seray, Mario Menéndez-Miranda, Giuseppina Salzano, Jérémie Mathurin, Ece Neslihan Aybeke, Ariane Deniset-Besseau, Alexandre Dazzi, Marianna Porcino, Charlotte Martineau-Corcos, and Ruxandra Gref. 2021. "Compartmentalized Polymeric Nanoparticles Deliver Vancomycin in a pH-Responsive Manner" Pharmaceutics 13, no. 12: 1992. https://doi.org/10.3390/pharmaceutics13121992
APA StyleUral, M. S., Menéndez-Miranda, M., Salzano, G., Mathurin, J., Aybeke, E. N., Deniset-Besseau, A., Dazzi, A., Porcino, M., Martineau-Corcos, C., & Gref, R. (2021). Compartmentalized Polymeric Nanoparticles Deliver Vancomycin in a pH-Responsive Manner. Pharmaceutics, 13(12), 1992. https://doi.org/10.3390/pharmaceutics13121992