
Isolation, Structure Determination of Sesquiterpenes from Neurolaena lobata and Their Antiproliferative, Cell Cycle Arrest-Inducing and Anti-Invasive Properties against Human Cervical Tumor Cells

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Experimental Procedures
2.2. Plant Material
2.3. Extraction and Isolation
2.4. Physical Characteristics of New Compounds
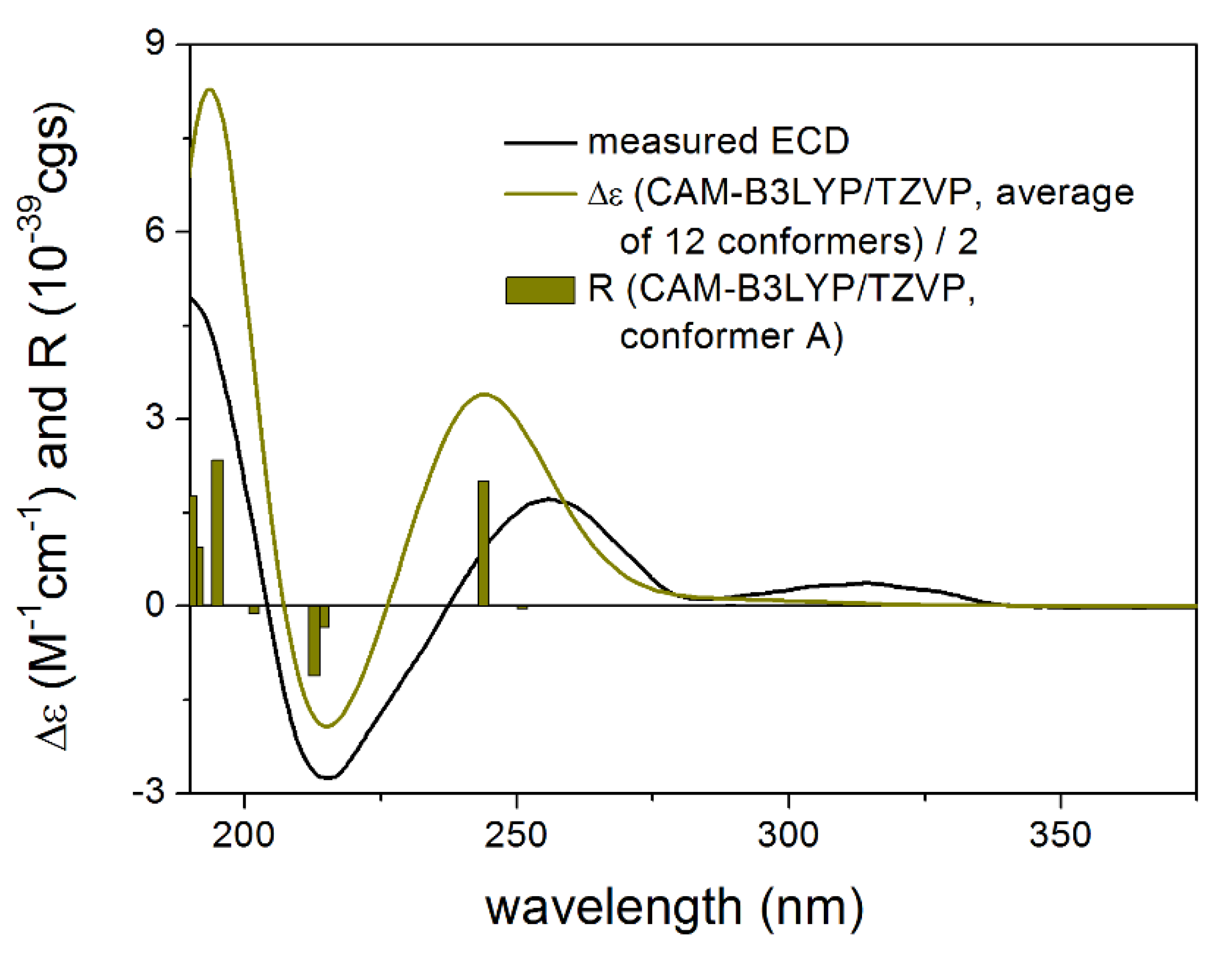
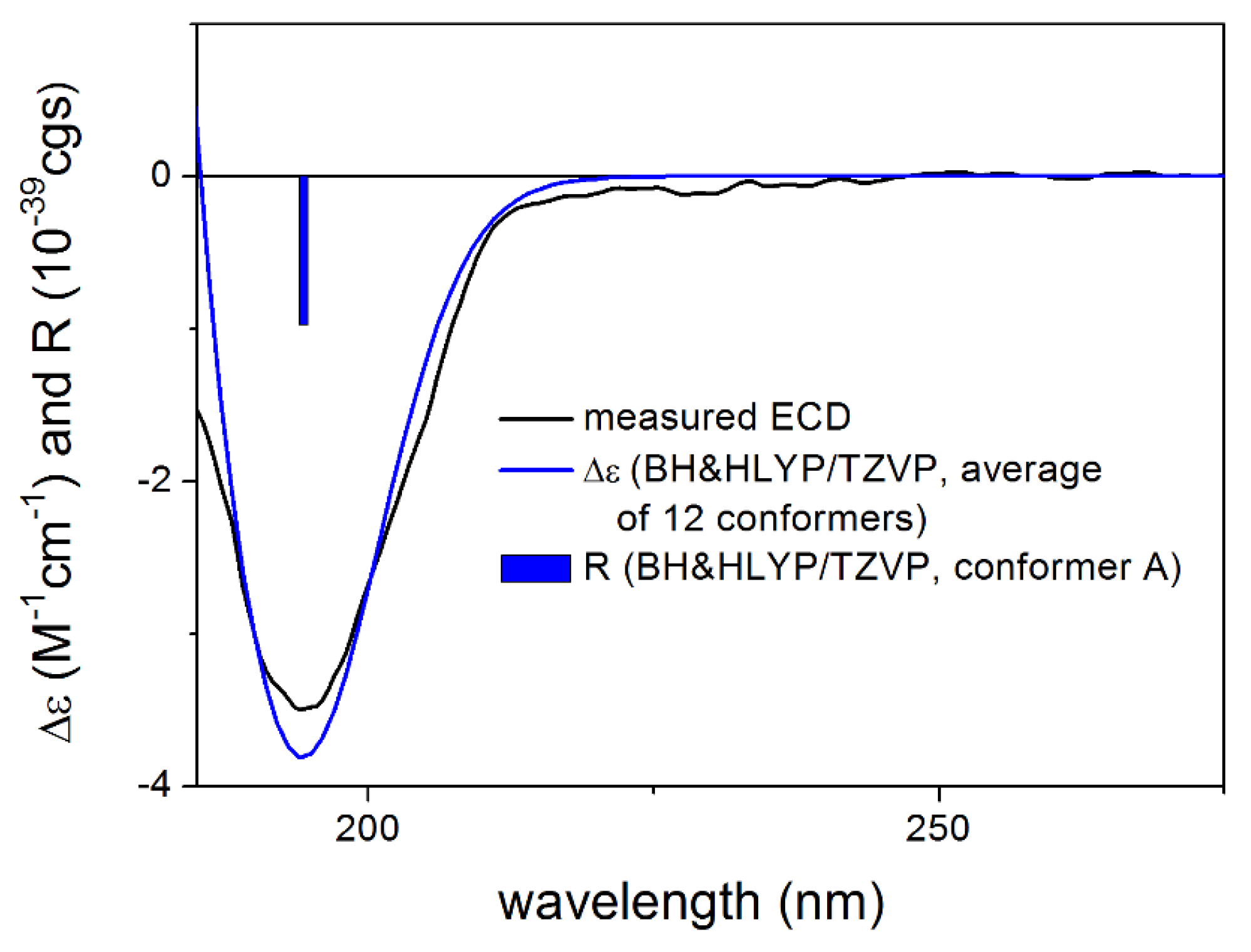
2.5. Computational Section
2.6. Antiproliferative MTT Assay
2.7. Cell Cycle Analysis by Flow Cytometry
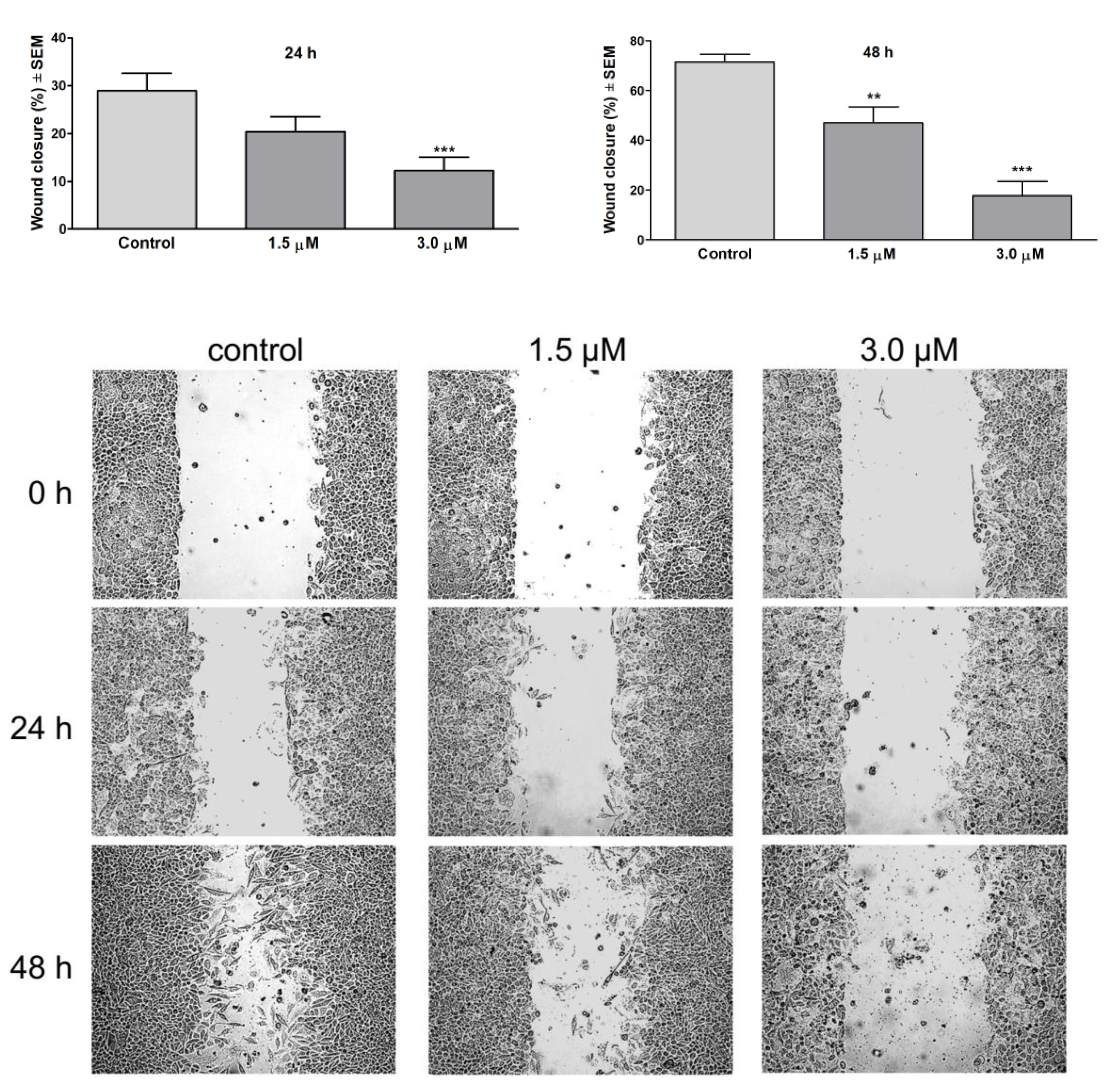
2.8. Wound Healing Assay
2.9. Statistical Analysis
3. Results and Discussion
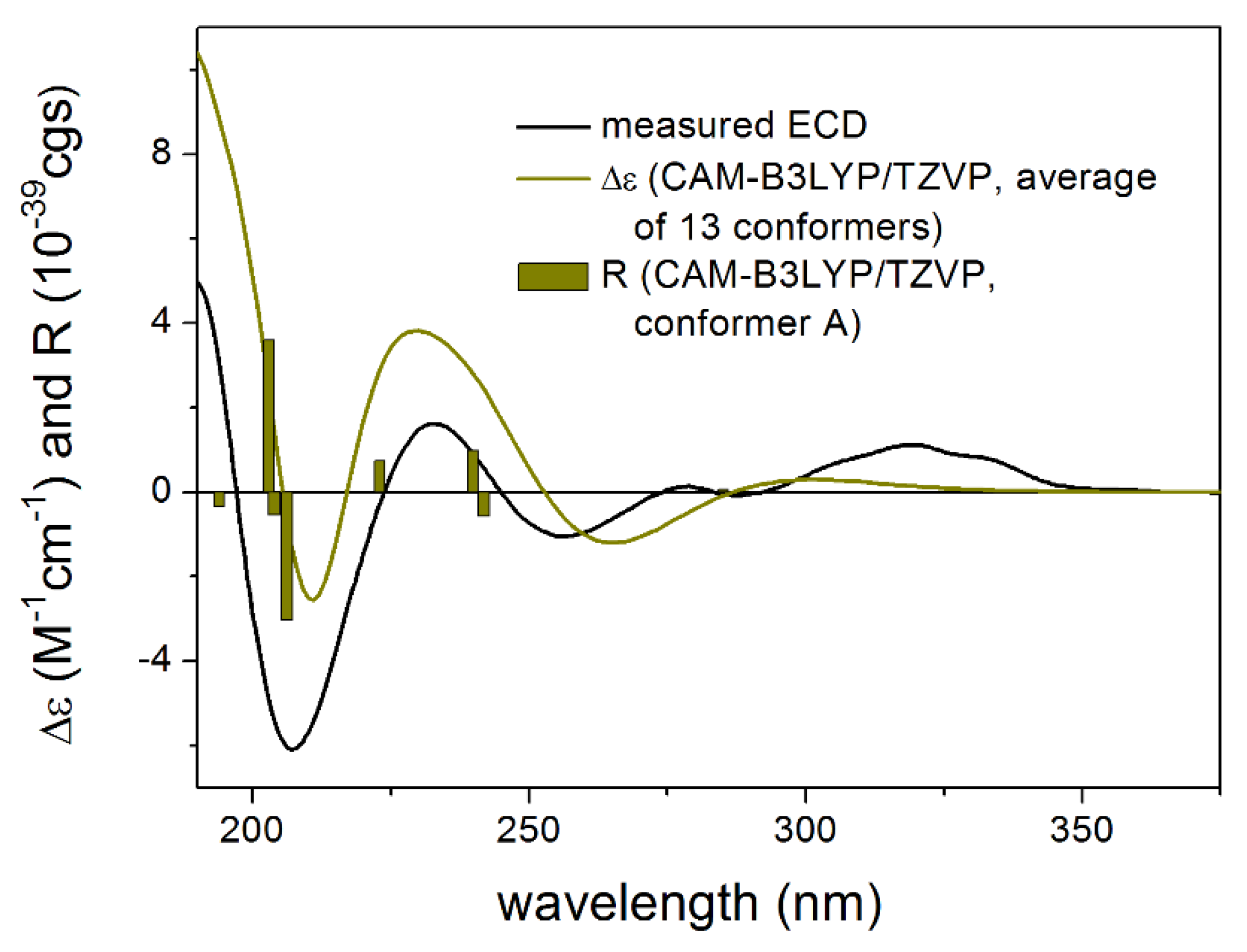
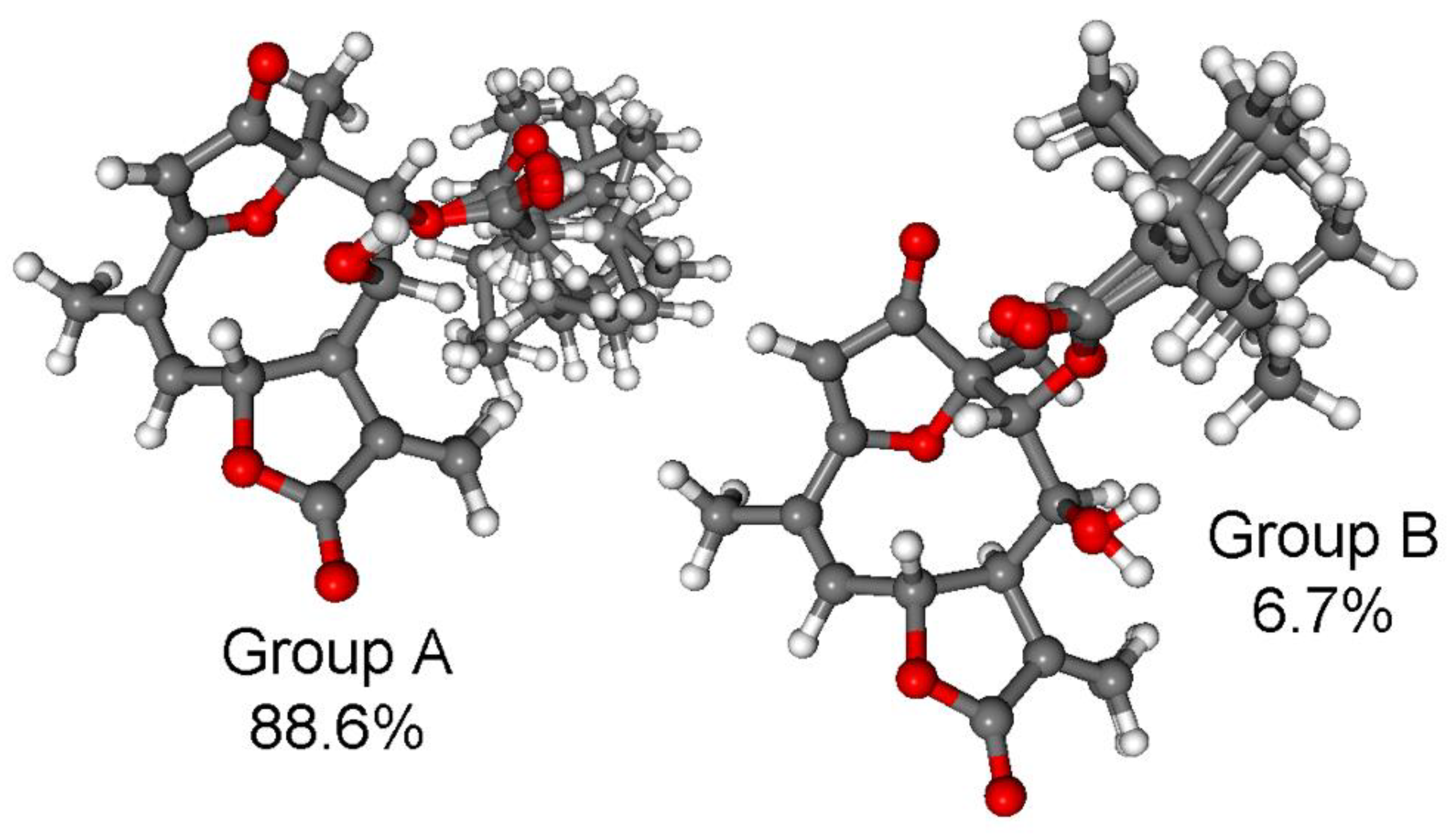
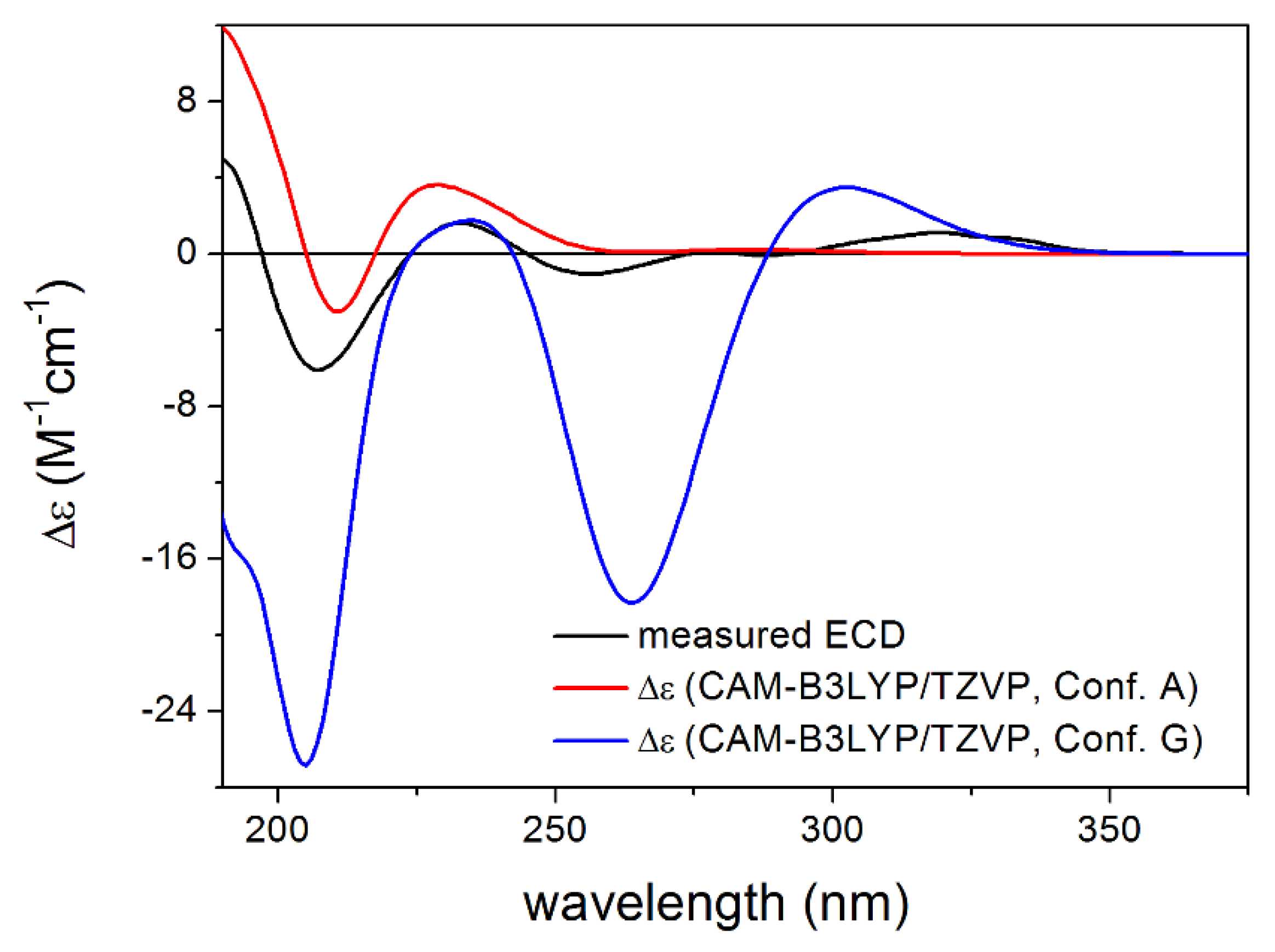
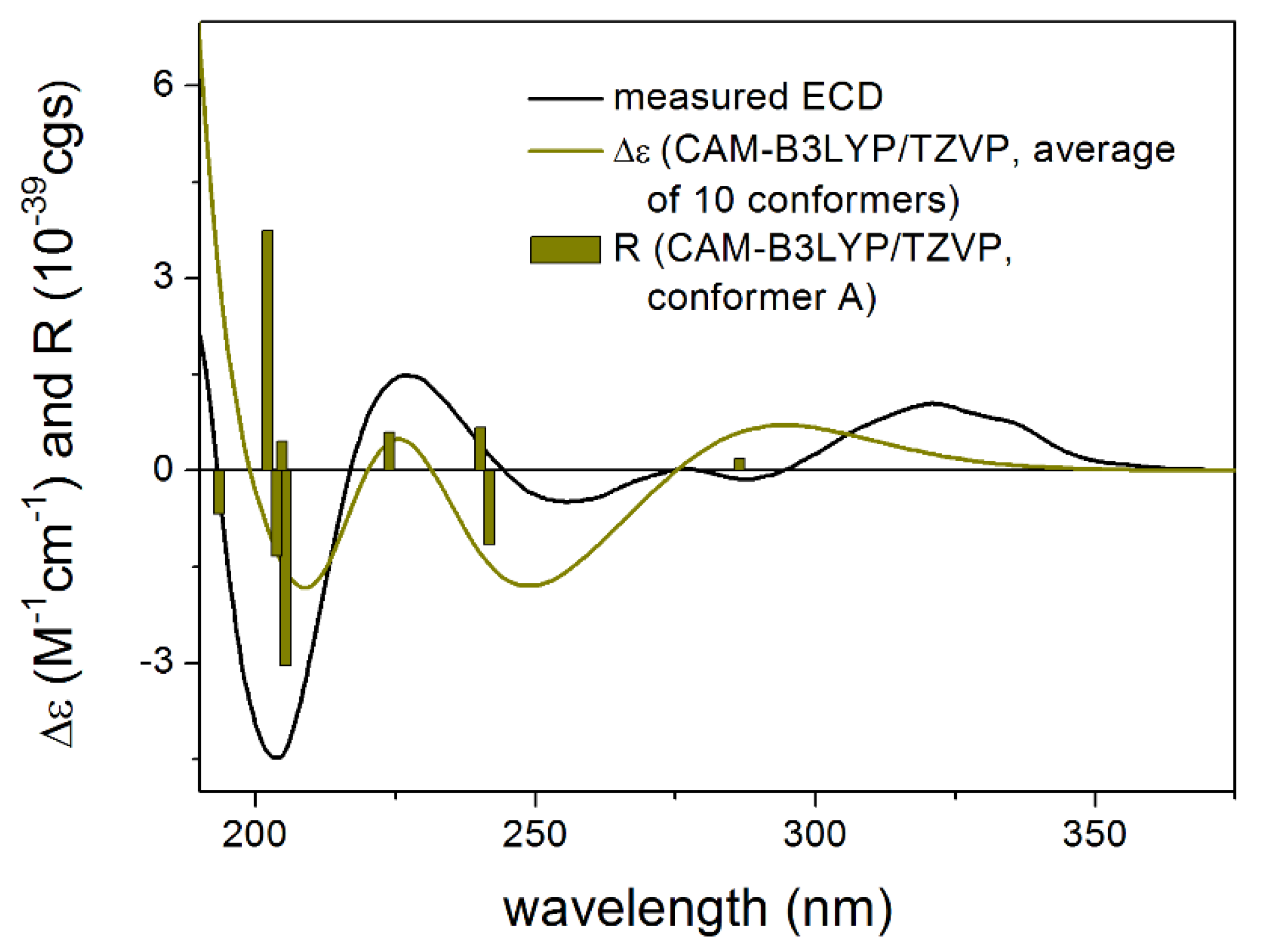
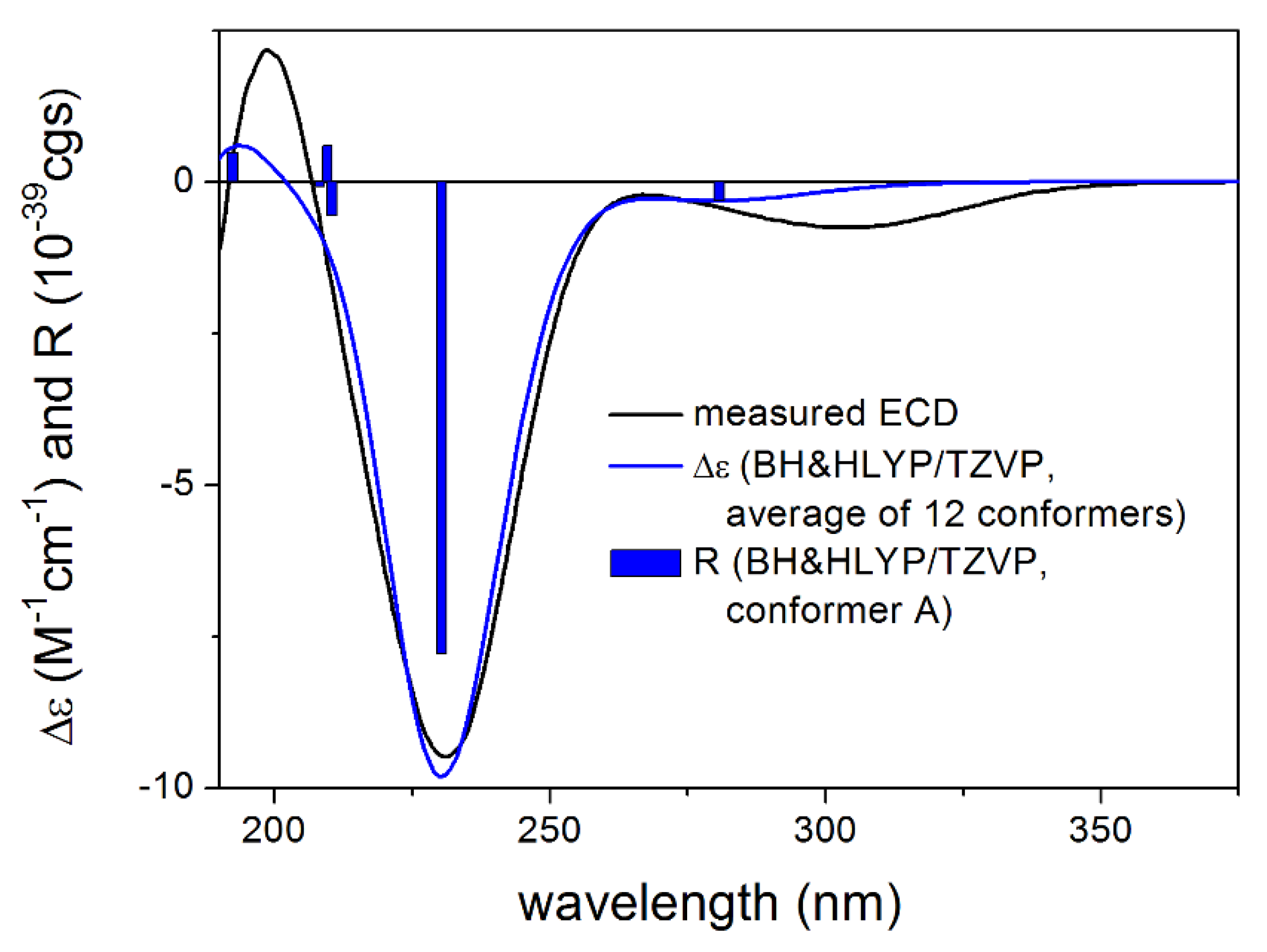
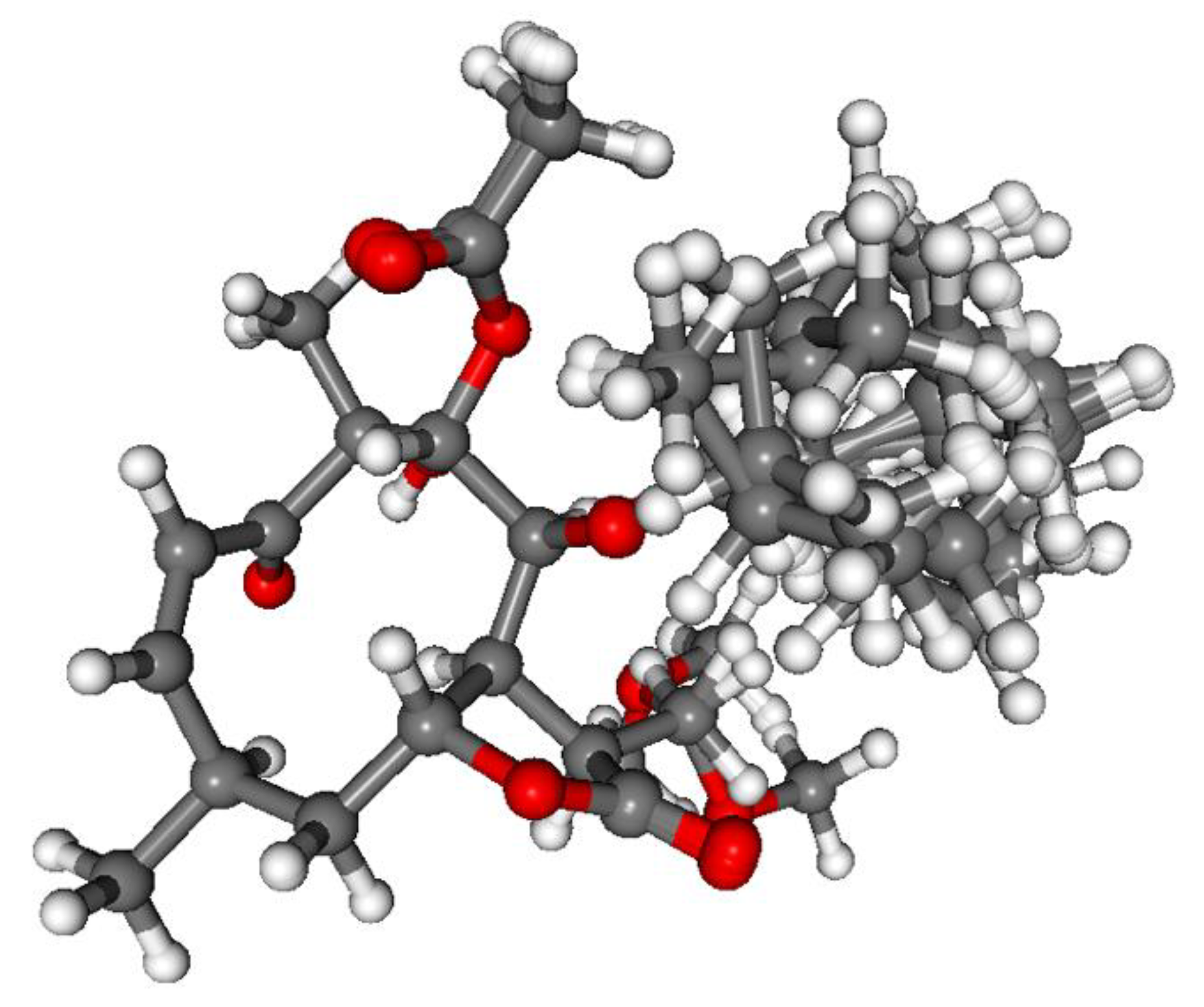
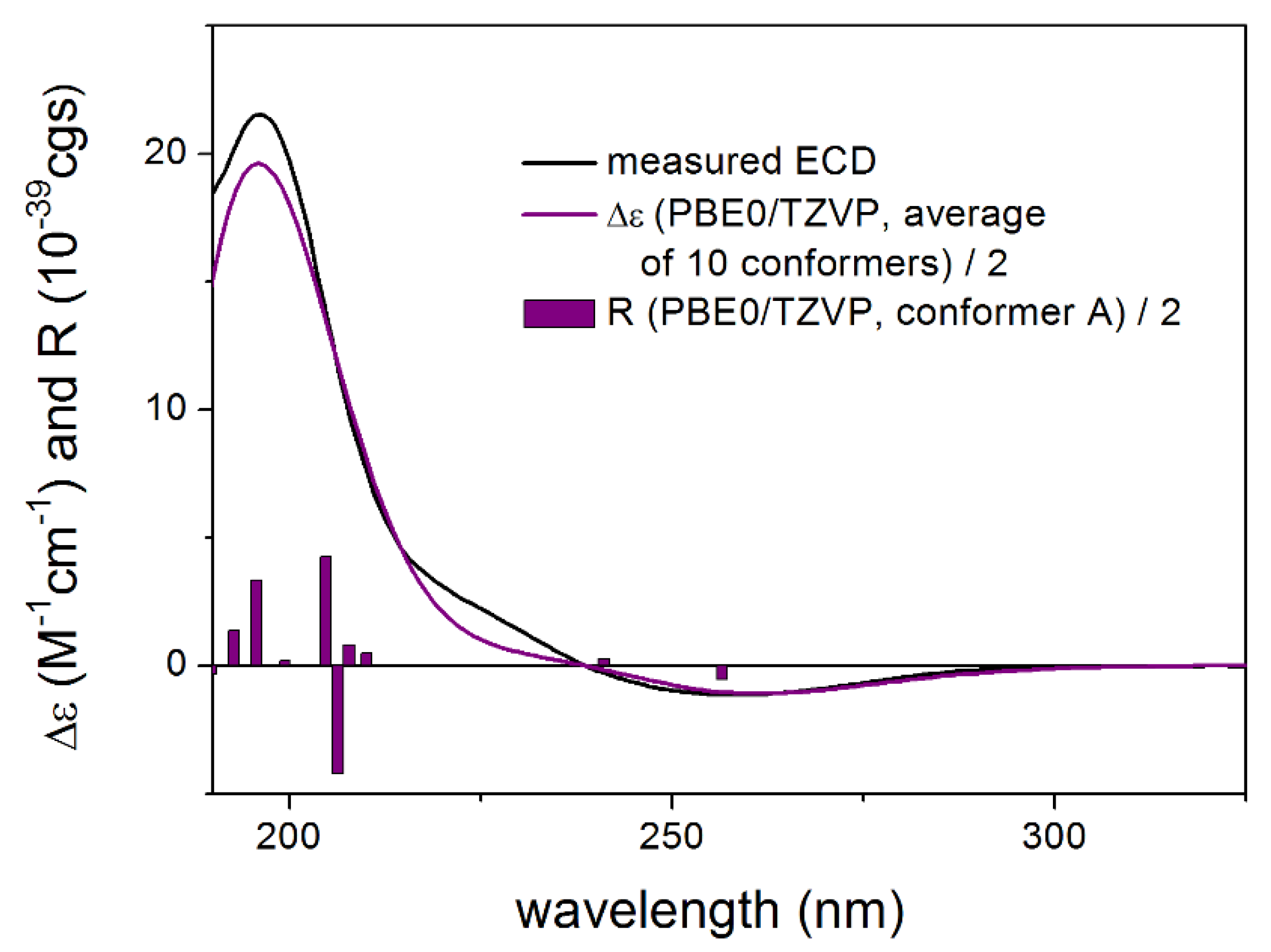
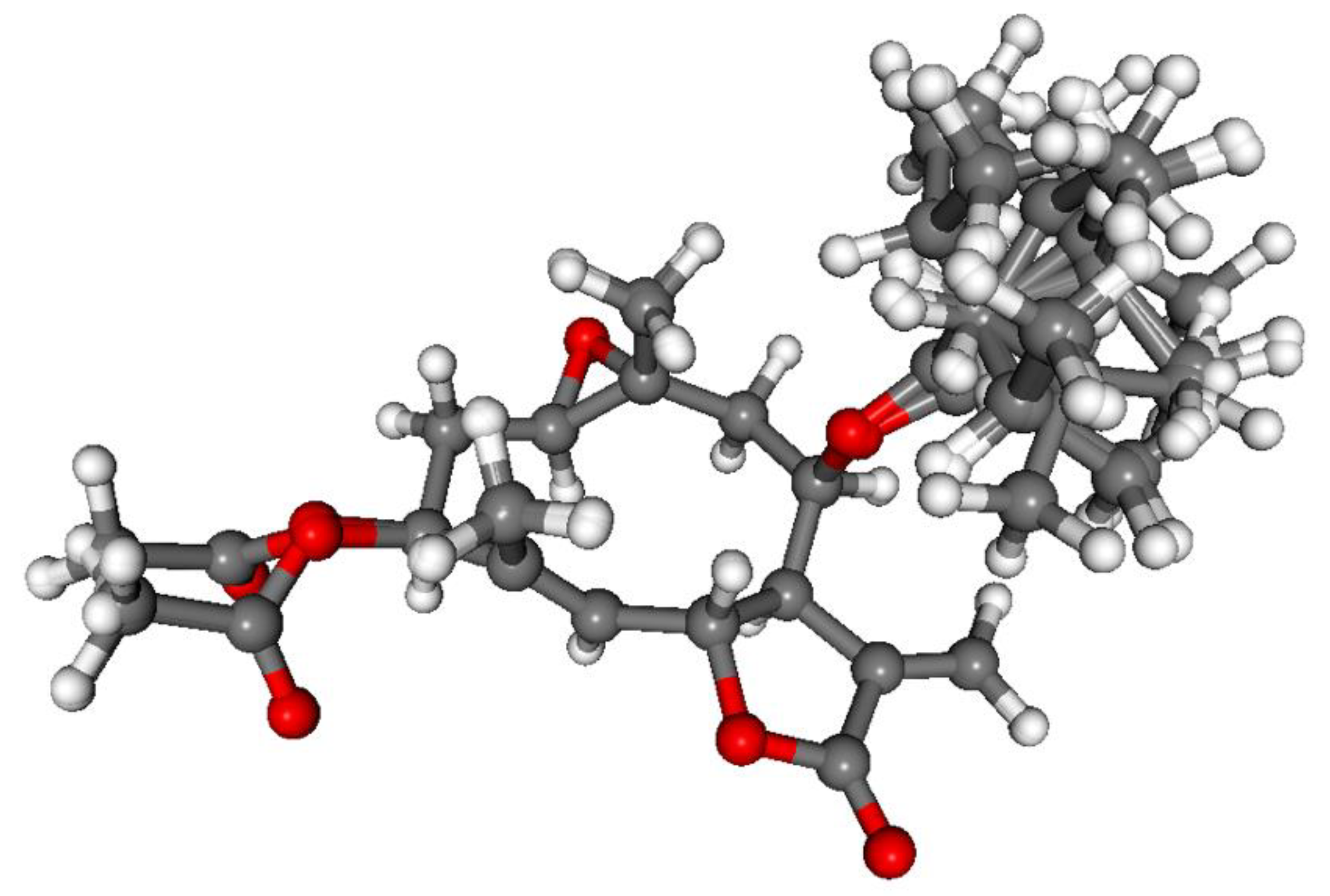
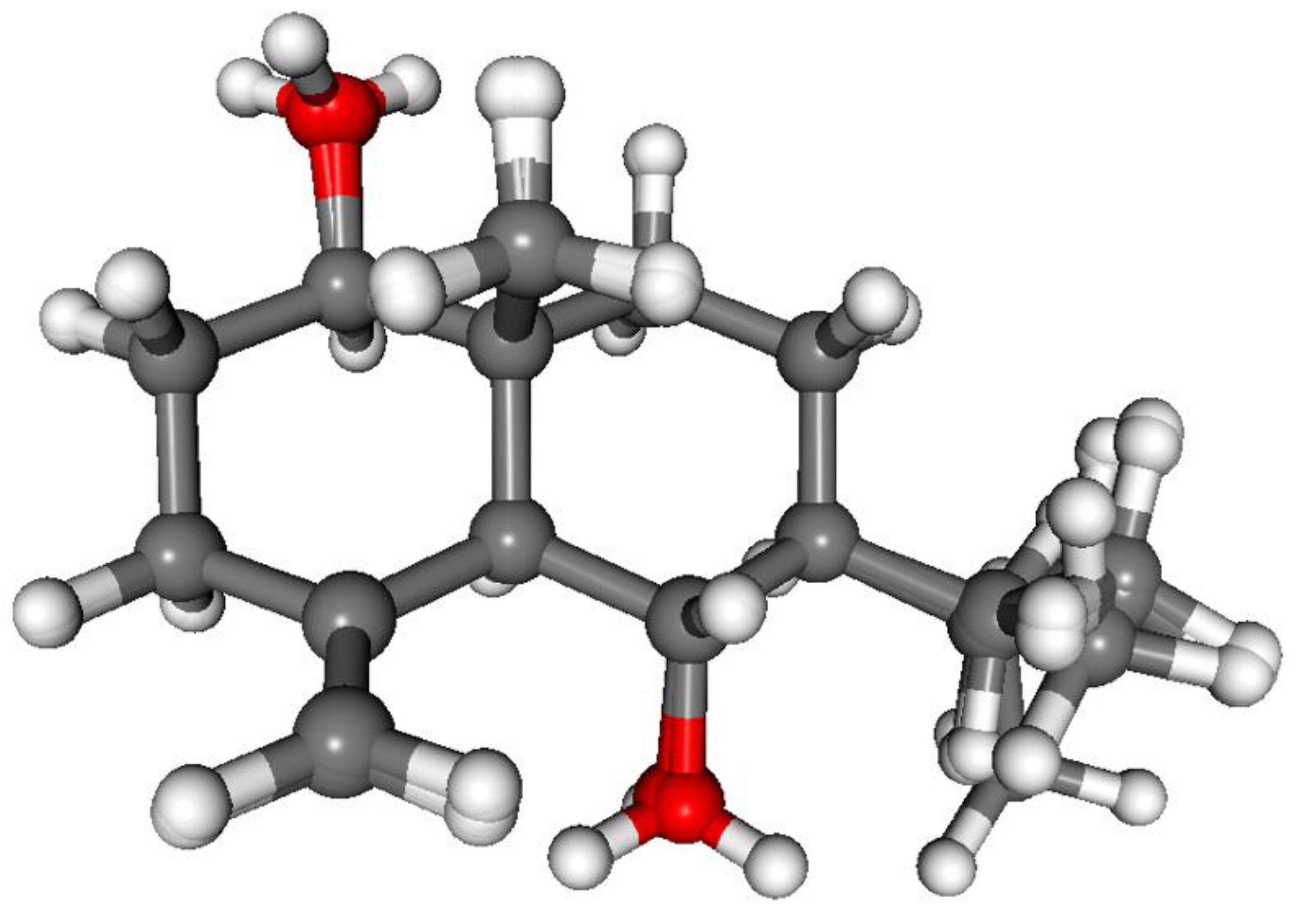
3.1. Structure Elucidation of the Isolated Compounds
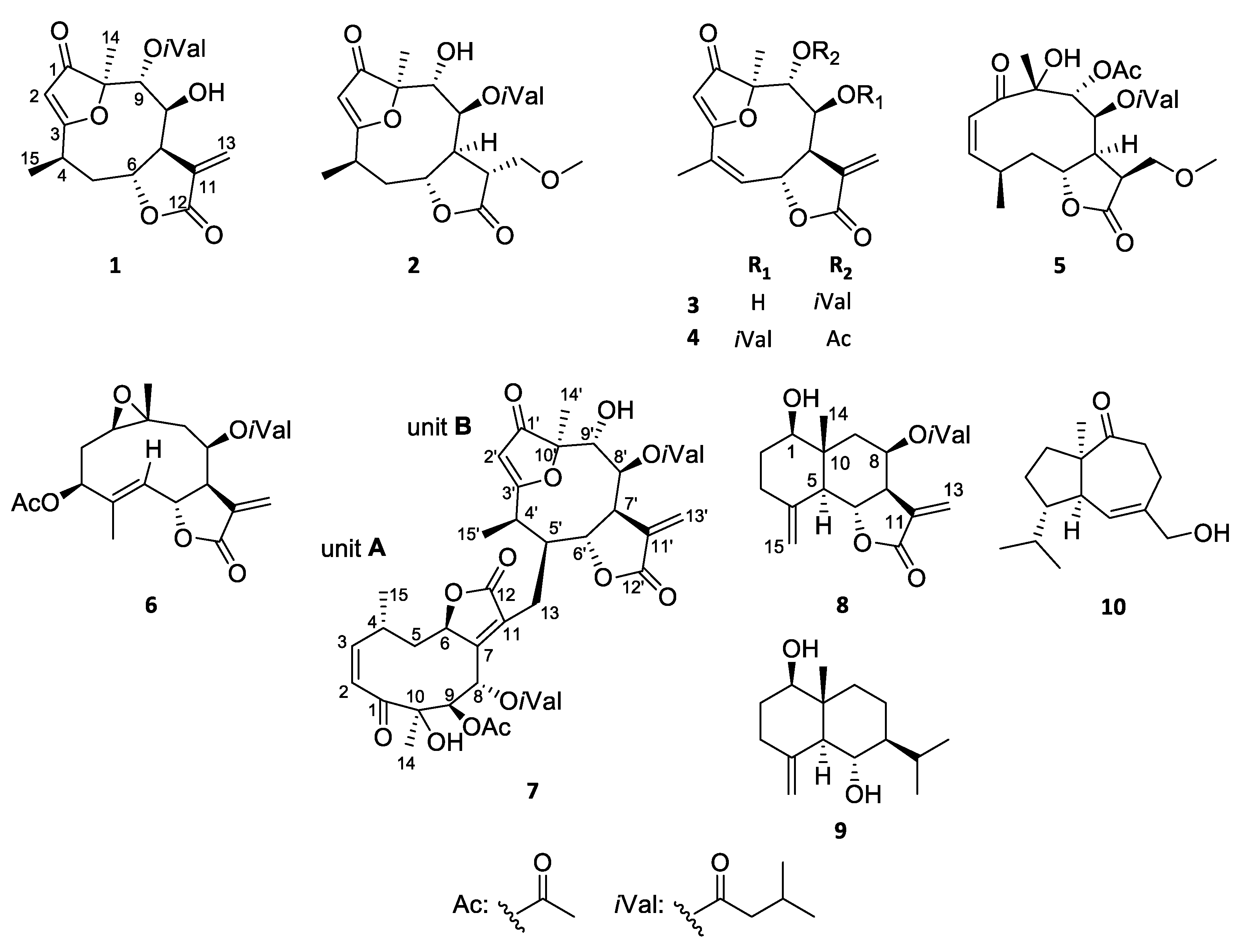
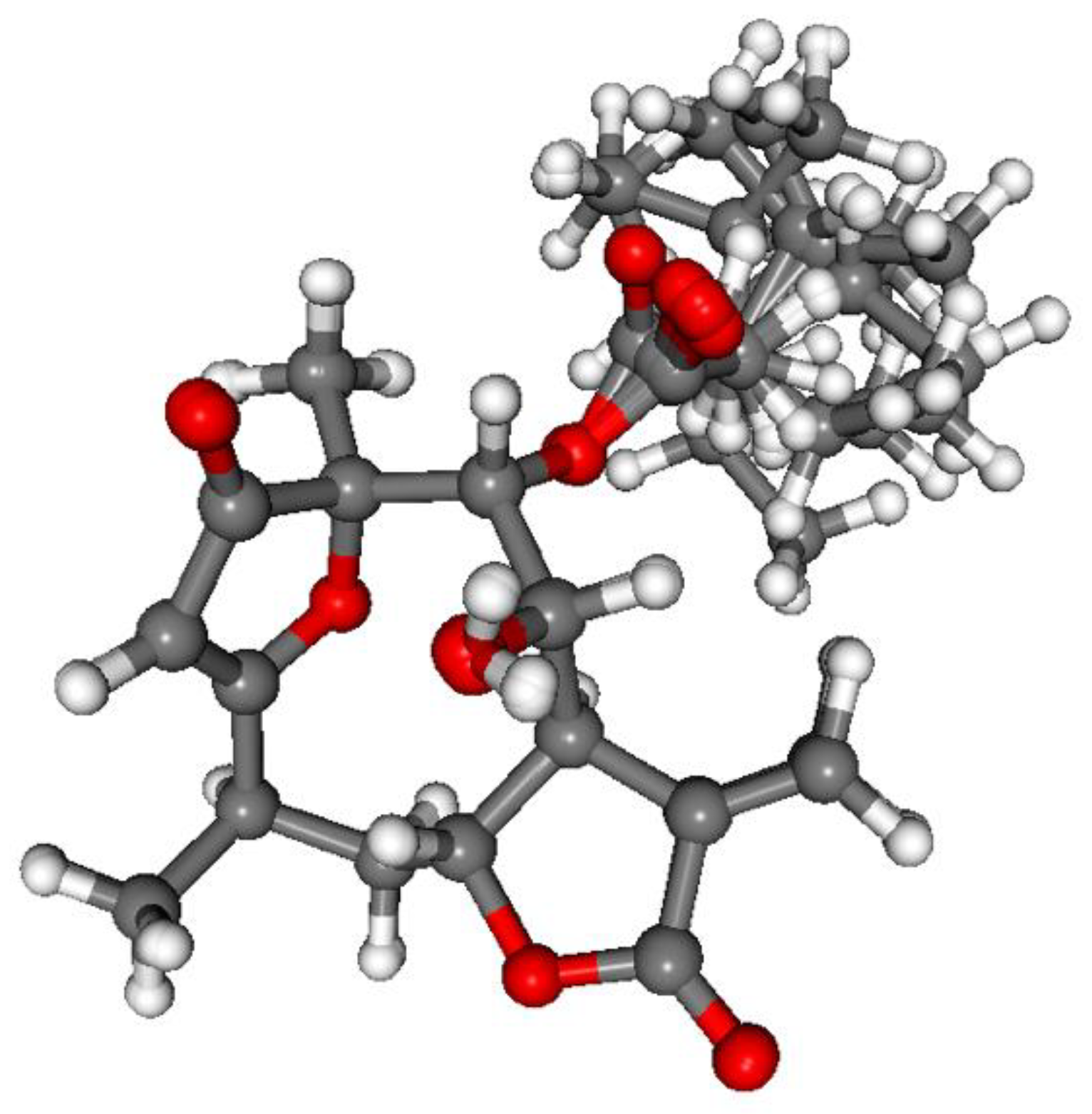
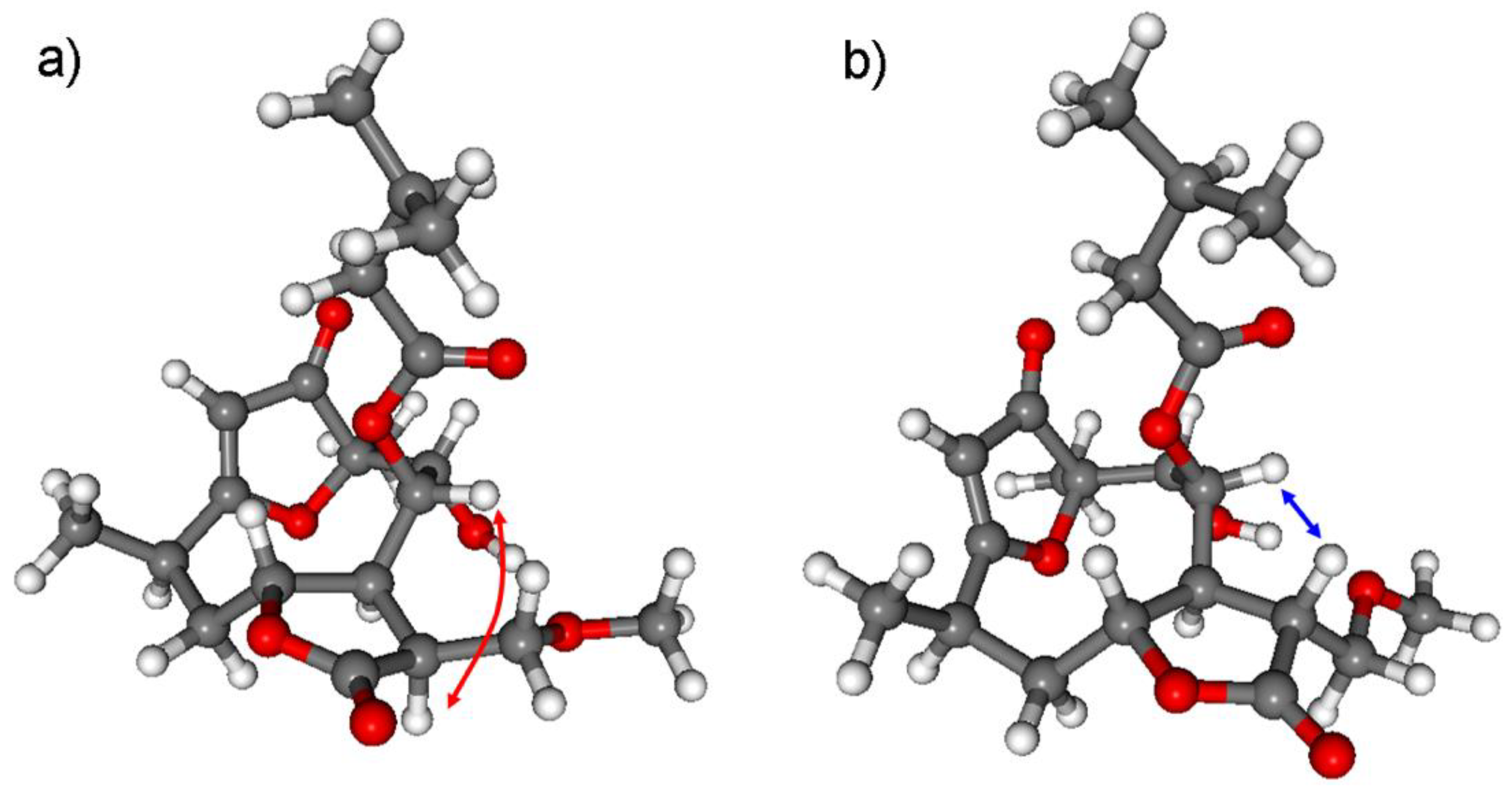
3.1.1. Lobatolide A (1)
3.1.2. Lobatolide B (2)
3.1.3. Lobatolide C (3)
3.1.4. Lobatin C (4)
3.1.5. Lobatolide D (5)
3.1.6. Lobatolide E (6)
3.1.7. Lobatolide F (7)
3.1.8. Lobatolide G (8)
3.2. Antiproliferative Activity of Isolated Compounds In Vitro
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chadwick, M.; Trewin, H.; Gawthrop, F.; Wagstaff, C. Sesquiterpenoids lactones: Benefits to plants and people. Int. J. Mol. Sci. 2013, 14, 12780–12805. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, E.; Towers, G.N.H.; Mitchell, J.C. Biological activities of sesquiterpene lactones. Phytochemistry 1976, 15, 1573–1580. [Google Scholar] [CrossRef]
- Seaman, F.C. Sesquiterpene lactones as taxonomic characters in the Asteraceae. Bot. Rev. 1982, 48, 121–595. [Google Scholar] [CrossRef]
- Ren, Y.; Yu, J.; Kinghorn, A.D. Development of anticancer agents from plant-derived sesquiterpene lactones. Curr. Med. Chem. 2016, 23, 2397–2420. [Google Scholar] [CrossRef] [PubMed]
- Zhumakayeva, A.; Rakhimov, K.; Sirota, V.; Arystan, L.; Madiyarov, A.; Adekenov, S. Long-term results of combination therapy for locally advanced breast cancer. Georgian Med. News 2018, 282, 30–35. [Google Scholar]
- Babaei, G.; Aliarab, A.; Abroon, S.; Rasmi, Y.; Gholizadeh, S.; Aziz, G. Application of sesquiterpene lactone: A new promising way for cancer therapy based on anticancer activity. Biomed Pharm. 2018, 106, 239–246. [Google Scholar] [CrossRef]
- Giron, L.M.; Freire, V.; Alonz, A.; Caceres, A. Ethnobotanical survey of the medicinal flora used by the Caribs of Guatemala. J. Ethnopharmacol. 1991, 34, 173–187. [Google Scholar] [CrossRef]
- Hartwell, J.L. Plants used against cancer. A survey. J. Nat. Prod. 1968, 31, 71–170. [Google Scholar]
- Passreiter, C.M.; Wendisch, D.; Gondol, D. Sesquiterpene lactones from Neurolaena Lobata. Phytochemistry 1995, 39, 133–137. [Google Scholar] [CrossRef]
- Borges-del-Castillo, J.; Manresa-Ferrero, M.T.; Rodríguez-Luis, F.; Vázquez-Bueno, P. Panama flora II. New sesquiterpene lactones from Neurolaena lobata. J. Nat. Prod. 1982, 45, 762–765. [Google Scholar] [CrossRef]
- Manchand, P.S.; Blount, J.F. Stereostructures of neurolenins A and B, novel germacranolide sesquiterpenes from Neurolaena lobata (L.) R.Br. J. Org. Chem. 1978, 43, 4352–4354. [Google Scholar] [CrossRef]
- Lajter, I.; Vasas, A.; Béni, Z.; Forgo, P.; Binder, M.; Bochkov, V.; Zupkó, I.; Krupitza, G.; Frisch, R.; Kopp, B.; et al. Sesquiterpenes from Neurolaena lobata and their antiproliferative and anti-inflammatory activities. J. Nat. Prod. 2014, 77, 576–582. [Google Scholar] [CrossRef]
- De Las Heras, B.; Slowing, K.; Benedi, J.; Carretero, E.; Ortega, T.; Toledo, C.; Bermejo, P.; Iglesias, I.; Abad, M.J.; Gomez-Serranillos, P.; et al. Antiinflammatory and antioxidant activity of plants used in traditional medicine in Ecuador. J. Ethnopharmacol. 1998, 61, 161–166. [Google Scholar] [CrossRef]
- Walshe-Roussel, B.; Choueiri, C.; Saleem, A.; Asim, M.; Caal, F.; Cal, V.; Rojas, M.O.; Pesek, T.; Durst, T.; Arnason, J.T. Potent anti-inflammatory activity of sesquiterpene lactones from Neurolaena lobata (L.) R. Br. ex Cass., a Q’eqchi’ Maya traditional medicine. Phytochemistry 2013, 92, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Gracioso, J.S.; Hiruma-Lima, C.A.; Souza Brito, A.R.M. Antiulcerogenic effect of a hydroalcoholic extract and its organic fractions of Neurolaena lobata (L.) R.Br. Phytomedicine 2000, 7, 283–289. [Google Scholar] [CrossRef]
- Gracioso, J.S.; Paulo, M.Q.; Hiruma-Lima, C.A.; Souza Brito, A.R. Antinociceptive effect in mice of a hydroalcoholic extract of Neurolaena lobata (L.) R. Br. and its organic fractions. J. Pharm. Pharmacol. 1998, 50, 1425–1429. [Google Scholar] [CrossRef] [PubMed]
- Fujimaki, Y.; Kamachi, T.; Yanagi, T.; Caceres, A.; Maki, J.; Aoki, Y. Macrofilaricidal and microfilaricidal effects of Neurolaena lobata, a Guatemalan medicinal plant, on Brugia pahangi. J. Helminthol. 2005, 79, 23–28. [Google Scholar] [CrossRef]
- Chinchilla, M.; Valerio, I.; Sanchez, R.; Mora, V.; Bagnarello, V.; Martinez, L.; Gonzalez, A.; Vanegas, J.C.; Apestegui, A. In vitro antimalarial activity of extracts of some plants from a biological reserve in Costa Rica. Rev. Biol. Trop. 2012, 60, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Bedoya, L.M.; Alvarez, A.; Bermejo, M.; Gonzalez, N.; Beltran, M.; Sanchez-Palomino, S.; Cruz, S.M.; Gaitan, I.; del Olmo, E.; Escarcena, R.; et al. Guatemalan plants extracts as virucides against HIV-1 infection. Phytomedicine 2008, 15, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Unger, C.; Popescu, R.; Giessrigl, B.; Laimer, D.; Heider, S.; Seelinger, M.; Diaz, R.; Wallnofer, B.; Egger, G.; Hassler, M.; et al. The dichloromethane extract of the ethnomedicinal plant Neurolaena lobata inhibits NPM/ALK expression which is causal for anaplastic large cell lymphomagenesis. Int. J. Oncol. 2013, 42, 338–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unger, C.; Kiss, I.; Vasas, A.; Lajter, I.; Kramer, N.; Atanasov, A.G.; Nguyen, C.H.; Chatuphonprasert, W.; Brenner, S.; Krieger, S.; et al. The germacranolide sesquiterpene lactone neurolenin B of the medicinal plant Neurolaena lobata (L.) R.Br. ex Cass inhibits NPM/ALK-driven cell expansion and NF-κB-driven tumour intravasation. Phytomedicine 2015, 22, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Kiss, I.; Unger, C.; Huu, C.N.; Atanasov, A.G.; Kramer, N.; Chatruphonprasert, W.; Brenner, S.; McKinnon, R.; Peschel, A.; Vasas, A.; et al. Lobatin B inhibits NPM/ALK and NF-κB attenuating anaplastic-large-cell-lymphomagenesis and lymphendothelial tumour intravasation. Cancer Lett. 2015, 356, 994–1006. [Google Scholar] [CrossRef]
- Francois, G.; Passreiter, C.M.; Woerdenbag, H.J.; van Looveren, M. Antiplasmodial activities and cytotoxic effects of aqueous extracts and sesquiterpene lactones from Neurolaena lobata. Planta Med. 1996, 62, 126–129. [Google Scholar] [CrossRef] [PubMed]
- MacroModel. Schrödinger, LLC. 2015. Available online: http://www.schrodinger.com/MacroModel (accessed on 30 September 2021).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E. 01; Gaussian: Wallingford, CT, USA, 2013. [Google Scholar]
- Stephens, P.J.; Harada, N. ECD cotton effect approximated by the Gaussian curve and other methods. Chirality 2010, 22, 229–233. [Google Scholar] [CrossRef] [PubMed]
- CHESHIRE CCAT, the Chemical Shift Repository for Computed NMR Scaling Factors. Available online: http://cheshirenmr.info/index.htm (accessed on 30 September 2021).
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef] [PubMed]
- Varetto, U. MOLEKEL 5.4; Swiss National Supercomputing Centre: Manno, Switzerland, 2009. [Google Scholar]
- Kulmány, Á.E.; Frank, É.; Papp, D.; Szekeres, A.; Szebeni, G.J.; Zupkó, I. Biological evaluation of antiproliferative and anti-invasive properties of an androstadiene derivative on human cervical cancer cell lines. J. Steroid Biochem. Mol. Biol. 2021, 214, 105990. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Reutelingsperger, C. Flow cytometry of apoptotic cell death. J. Immunol. Methods 2000, 243, 167–190. [Google Scholar] [CrossRef]
- Latif, D.A.; Gonda, T.; Vágvölgyi, M.; Kúsz, N.; Kulmány, Á.; Ocsovszki, I.; Zomborszki, P.Z.; Zupkó, I.; Hunyadi, A. Synthesis and in vitro antitumor activity of naringenin oxime and oxime ether derivatives. Int. J. Mol. Sci. 2019, 20, 2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrens, J.; Kameritsch, P.; Wallner, S.; Pohl, U.; Pogoda, K. The carboxyl tail of Cx43 augments p38 mediated cell migration in a gap junction-independent manner. Eur. J. Cell Biol. 2010, 89, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Superchi, S.; Scafato, P.; Gorecki, M.; Pescitelli, G. Absolute configuration determination by quantum mechanical calculation of chiroptical spectra: Basics and applications to fungal metabolites. Curr. Med. Chem. 2018, 25, 287–320. [Google Scholar] [CrossRef] [PubMed]
- Maándi, A.; Kurtaán, T. Applications of OR/ECD/VCD to the structure elucidation of natural products. Nat. Prod. Rep. 2019, 36, 889–918. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Li, W.S.; Yan, R.J.; Yu, Y.; Shi, Z.; Mándi, A.; Shen, L.; Kurtán, T.; Wu, J. Determination of the absolute configuration of super-carbon-chain compounds by a combined chemical, spectroscopic, and computational approach: Gibbosols A and B. Angew. Chem. Int. Ed. 2020, 59, 13028–13036. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- Qiu, S.; de Gussem, E.; Tehrani, K.A.; Sergeyev, S.; Bultinck, P.; Herrebout, W. Stereochemistry of the tadalafil diastereoisomers: A critical assessment of Vibrational Circular Dichroism, Electronic Circular Dichroism, and Optical Rotatory Dispersion. J. Med. Chem. 2013, 56, 8903–8914. [Google Scholar] [CrossRef]
- Dračínský, M.; Buděšínský, M.; Warżajtis, B.; Rychlewska, U. Solution and solid-state effects on NMR chemical shifts in sesquiterpene lactones: NMR, X-ray, and theoretical methods. J. Phys. Chem. A 2012, 116, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Li, W.S.; Mándi, A.; Liu, J.J.; Shen, L.; Kurtán, T.; Wu, J. Xylomolones A–D from the Thai mangrove Xylocarpus moluccensis: Assignment of absolute stereostructures and unveiling a convergent strategy for limonoid biosynthesis. J. Org. Chem. 2019, 84, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.F.; Lan, L.F.; Taglialatela-Scafati, O.; Guo, Y.W. Sartrolides A–G and bissartrolide, new cembranolides from the South China Sea soft coral Sarcophyton trocheliophorum Marenzeller. Tetrahedron 2013, 69, 7381–7386. [Google Scholar] [CrossRef]
- Liang, L.F.; Kurtán, T.; Mándi, A.; Yao, L.G.; Li, J.; Lan, L.F.; Guo, Y.W. Structural, stereochemical, and bioactive studies of cembranoids from Chinese soft coral Sarcophyton trocheliophorum. Tetrahedron 2018, 74, 1933–1941. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.G.; Goodman, J.M. Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: The DP4 probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef] [PubMed]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Cai, F.Y.; Lauro, G.; Tang, H.; Su, L.; Wang, H.L.; Li, H.H.; Mándi, A.; Kurtaán, T.; Riccio, R.; et al. Immunomodulatory biscembranoids and assignment of their relative and absolute configurations: Data set modulation in the Density Functional Theory/Nuclear Magnetic Resonance approach. J. Nat. Prod. 2019, 82, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- Passreiter, C.M.; Stöber, S.; Ortega, A. Furanoheliangolides from leaves of Neurolaena macrocephala. Z. Naturforsch. 2000, 55c, 1026–1029. [Google Scholar] [CrossRef] [PubMed]
- Bohlmann, F.; Jakupovic, J.; Ahmed, M.; Grenz, M.; Suding, H.; Robinson, H.; Kino, R.M. Germacranolides and diterpenes from Viguiera species. Phytochemistry 1981, 20, 113–116. [Google Scholar] [CrossRef]
- Scotta, R.; Zdero, C.; Bohlmann, F. Germacranolides, guaianolides and eudesmanolides from Greenmaniella resinosa. Phytochemistry 1987, 26, 1999–2006. [Google Scholar] [CrossRef]
- Lee, K.H.; Min, Y.D.; Choi, S.Z.; Kwon, H.C.; Cho, O.R.; Lee, K.C.; Lee, K.R. A new sesquiterpene lactone from Artemisia rubripes Nakai. Arch. Pharm. Res. 2004, 27, 1016–1019. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, M.; Inoue, A.; Hayashi, Y.; Sastrapradja, S.; Kosela, S.; Iwashita, T. Structure of aphanamol I and II. J. Org. Chem. 1984, 49, 3660–3662. [Google Scholar] [CrossRef]
- Hansson, T.; Wickberg, B. A short enantiospecific route to isodaucane sesquiterpenes from limonene. On the absolute configuration of (+)-aphanamol I and II. J. Org. Chem. 1992, 57, 5370–5376. [Google Scholar] [CrossRef]
- Krishnaswamy, N.R. Chemistry of Natural Products: A Unified Approach; Universities Press: Hyderabad, India, 1999; pp. 66–67. [Google Scholar]
- Schröpfer, A.; Kammerer, U.; Kapp, M.; Dietl, J.; Feix, S.; Anacker, J. Expression pattern of matrix metalloproteinases in human gynecological cancer cell lines. BMC Cancer 2010, 10, 553. [Google Scholar] [CrossRef] [Green Version]
- Molnár, J.; Szebeni, G.J.; Csupor-Löffler, B.; Hajdú, Z.; Szekeres, T.; Saiko, P.; Ocsovszki, I.; Puskás, L.G.; Hohmann, J.; Zupkó, I. Investigation of the antiproliferative properties of natural sesquiterpenes from Artemisia asiatica and Onopordum acanthium on HL-60 cells in vitro. Int. J. Mol. Sci. 2016, 17, 83. [Google Scholar] [CrossRef] [Green Version]
- Jeyamohan, S.; Moorthy, R.K.; Kannan, M.K.; Velanganni Arockiam, A.J. Parthenolide induces apoptosis and autophagy through the suppression of PI3K/Akt signaling pathway in cervical cancer. Biotech. Lett. 2016, 38, 1251–1260. [Google Scholar] [CrossRef]
- Abu-Izneid, T.; Abdur Rauf, A.; Shariati, M.A.; Khalil, A.A.; Imran, M.; Rebezov, M.; Uddin, S.; Mahomoodally, M.F.; Rengasamy, K.R.R. Sesquiterpenes and their derivatives-natural anticancer compounds: An update. Pharmacol. Res. 2020, 161, 105165. [Google Scholar] [CrossRef]
- Li, H.; Li, M.; Wang, G.; Shao, F.; Chen, W.; Xia, C.; Wang, S.; Li, Y.; Zhou, G.; Liu, Z. EM23, a natural sesquiterpene lactone from Elephantopus mollis, induces apoptosis in human myeloid leukemia cells through thioredoxin- and reactive oxygen species–mediated signalling pathways. Front. Pharmacol. 2016, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Berdan, A.C.; Ho, R.; Lehtola, H.S.; To, M.; Hu, X.; Huffman, T.R.; Petri, Y.; Altobelli, C.R.; Demeulenaere, S.G.; Olzmann, J.A.; et al. Parthenolide covalently targets and inhibits focal adhesion kinase in breast cancer cells. Cell. Chem. Biol. 2019, 26, 1027–1035. [Google Scholar] [CrossRef]
- Zhang, L.; Qian, H.; Sha, M.; Luan, Z.; Lin, M.; Yuan, D.; Li, X.; Huang, J.; Ye, L. Downregulation of HOTAIR ex-pression mediated antimetastatic effect of artesunate on cervical cancer by inhibiting COX-2 expression. PLoS ONE 2016, 11, e0164838. [Google Scholar] [CrossRef] [Green Version]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 2.93 dd (11.3, 2.3) | |||||
| 2 | 5.60 s | 5.52 s | 5.64 s | 5.65 s | 6.58 d (11.7) | 2.35 m, 1.64 m |
| 3 | 5.97 t (11.7) | 5.50 dd (11.4, 5.7) | ||||
| 4 | 3.01 m | 3.01 m | 3.09 m | |||
| 5 | 2.59 ddd (14.2, 9.3, 4.8) (α) 2.05 d (14.2) (β) | 2.63 ddd (14.2, 9.6, 7.0) 2.03 m | 6.00 dd (4.2, 1.8) | 5.99 d (3.7, 1.8) | 1.79 dt (5.1, 12.3) 1.63 dt (5.1, 12.1) | 5.66 d (9.6) |
| 6 | 4.78 dd (9.3, 4.7) | 4.29 dd (9.6, 6.9) | 5.71 m | 5.24 m | 4.56 dd (12.1, 5.3) | 5.22 t (9.4) |
| 7 | 3.30 m | 3.16 dd (8.9, 6.9) | 3.57 dd (7.6, 4.0) | 3.86 dd (4.6, 1.6) | 3.04 ddd (10.3, 8.1, 5.3) | 3.21 br d (9.4) |
| 8 | 4.13 t (4.7) | 5.07 d (5.4) | 4.04 m | 5.01 dd (5.2, 1.6) | 5.57 d (9.6) | 5.74 d (5.7) |
| 9 | 5.27 d (4.7) | 3.94 d (5.4) | 5.27 d (3.6) | 5.31 d (5.2) | 5.57 d (9.6) | 2.63 dd (15.2, 5.7) 1.40 dd (15.2, 1.7) |
| 11 | 2.89 m | 2.23 br d (8.1) | ||||
| 13a | 6.30 d (3.0) | 3.74 dd (9.4, 4.1) | 6.29 d (3.0) | 6.35 d (3.0) | 3.68 dd (10.2, 5.3) | 6.23 d (3.5) |
| 13b | 5.35 d (3.0) | 3.67 m | 5.35 d (3.0) | 5.48 d (3.0) | 3.34 t (10.2) | 5.65 s |
| 14 | 1.37 s | 1.45 s | 1.41 s | 1.43 s | 1.31 s | 1.21 s |
| 15 | 1.40 d (7.0) | 1.34 d (7.3) | 2.06 s | 2.08 s | 1.15 d (6.3) | 1.87 d (1.3) |
| OMe | 3.40 s | 3.39 s | ||||
| iVal CO 1′ | ||||||
| 2a′ | 2.38 dd (15.1, 7.2) | 2.14 dd (15.3, 7.2) | 2.37 dd (15.2, 7.3) | 2.15 m | 2.00 m | 2.27 m |
| 2b′ | 2.33 dd (15.1, 7.2) | 2.10 m | 2.31 dd (15.2, 7.2) | 2.12 m | 2.00 m | 2.22 m |
| 3′ | 2.19 m | 2.02 m | 2.16 m | 2.01 m | 2.07 m | 2.07 m |
| 4′ | 1.02 d (6.7) | 0.93 d (7.0) | 1.01 d (6.6) | 0.92 d (7.1) | 0.91 d (6.7) | 0.95 d (6.4) |
| 5′ | 1.02 d (6.7) | 0.91 d (6.9) | 1.01 d (6.6) | 0.90 d (7.1) | 0.89 d (6.7) | 0.94 d (6.4) |
| 8-OH | 2.82 d (4.5) | 4.05 s | ||||
| 3-OAc | 2.10 s | |||||
| 9-OAc | 2.23 s | 2.11 s |
| Position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 203.9 | 204.2 | 203.9 | 202.3 | 204.7 | 65.6 |
| 2 | 104.0 | 103.8 | 104.6 | 104.3 | 125.6 | 31.4 |
| 3 | 192.8 | 192.4 | 185.8 | 185.4 | 147.6 | 77.1 |
| 4 | 31.5 | 31.4 | 130.9 | 134.6 | 28.7 | 145.3 |
| 5 | 40.7 | 41.7 | 134.3 | 134.4 | 38.1 | 123.9 |
| 6 | 75.3 | 75.4 | 74.9 | 75.5 | 77.4 | 75.4 |
| 7 | 47.9 | 46.1 | 45.2 | 44.9 | 37.5 | 53.3 |
| 8 | 73.8 | 75.1 | 75.9 | 75.7 | 68.9 | 68.5 |
| 9 | 77.1 | 74.3 | 77.5 | 73.6 | 74.8 | 43.3 |
| 10 | 88.9 | 90.7 | 88.3 | 88.5 | 79.3 | 61.4 |
| 11 | 141.3 | 49.3 | 141.2 | 139.0 | 40.2 | 138.2 |
| 12 | 169.2 | 174.2 | 170.0 | 168.7 | 174.2 | 171.2 |
| 13 | 122.6 | 72.1 | 123.2 | 123.8 | 66.3 | 121.7 |
| 14 | 18.9 | 18.4 | 18.4 | 18.6 | 23.6 | 20.2 |
| 15 | 16.1 | 16.0 | 19.6 | 21.2 | 19.8 | 13.0 |
| OMe | 59.6 | 59.2 | ||||
| iVal CO 1′ | 171.5 | 171.7 | 171.7 | 169.3 | 171.2 | 173.2 |
| 2′ | 43.1 | 43.0 | 43.1 | 42.6 | 42.9 | 44.1 |
| 3′ | 25.7 | 25.6 | 25.6 | 25.3 | 24.7 | 26.7 |
| 4′ | 22.4 | 22.5 | 22.4 | 22.4 | 22.4 | 22.7 |
| 5′ | 22.4 | 22.6 | 22.4 | 22.4 | 22.5 | 22.7 |
| Ac CO | 168.8 | 170.4 | 171.8 | |||
| Ac Me | 21.3 | 20.6 | 20.8 |
| Position | 7 (Unit A) | 7 (Unit B) | 8 | |||
|---|---|---|---|---|---|---|
| δH, Mult. (J in Hz) | δC | δH, Mult. (J in Hz) | δC | δH, Mult. (J in Hz) | δC | |
| 1 | - | 205.0 | - | 203.8 | 3.50 dd (11.4, 4.7) | 78.8 |
| 2 | 6.22 d (11.6) | 123.8 | 5.64 s | 105.4 | 1.81 m, 1.58 m | 31.1 |
| 3 | 6.04 t (11.6) | 147.3 | - | 193.0 | 2.35 m, 2.13 m | 33.5 |
| 4 | 2.14 m | 25.0 | 2.81 m | 36.8 | - | 142.0 |
| 5 | 2.13 m, 1.96 m | 38.9 | 3.54 m | 44.8 | 2.23 d (11.0) | 53.7 |
| 6 | 5.07 d (5.0) | 79.6 | 4.44 t (4.9) | 75.0 | 4.50 t (11.1) | 75.2 |
| 7 | - | 154.2 | 3.68 m | 45.6 | 2.79 dd (11.1, 2.7) | 52.2 |
| 8 | 6.38 d (8.9) | 66.9 | 5.20 br d (5.1) | 76.1 | 5.75 d (2.7) | 65.9 |
| 9 | 5.69 d (8.9) | 73.1 | 4.05 br s | 74.11 | 2.31 dd (15.2, 2.2), 1.56 m | 40.6 |
| 10 | - | 80.7 | - | 90.7 | - | 42.8 |
| 11 | - | 135.7 | - | 138.0 | - | 134.8 |
| 12 | - | 172.2 | - | 168.9 | - | 170.0 |
| 13a | 3.05 dd (14.5, 8.8) | 23.0 | 6.29 d (3.0), | 124.1 | 6.15 d (3.2) | 119.6 |
| 13b | 2.48 dd (14.5, 6.8) | 5.74 d (3.0) | 5.44 d (3.0) | |||
| 14 | 1.32 s | 23.9 | 1.48 s | 19.2 | 5.01 s, 4.94 s | 111.1 |
| 15 | 1.14 d (6.4) | 22.5 | 1.40 d (6.8) | 10.0 | 0.96 s | 13.8 |
| iVal-CO 1′ | s | 172.0 | - | 171.5 | - | 172.3 |
| 2′ | 2.27 dd (15.8, 6.8), 2.15 m (2H) | 43.1 | 2.08 m (2H) | 42.8 | 2.17 br s, 2.16 br s | 43.8 |
| 3′ | 2.02 m | 25.5 | 1.96 m | 25.3 | 2.06 m | 25.6 |
| 4′ | 0.94 d (6.7) | 22.5 | 0.90 d (6.7) | 22.4 | 0.93 d (4.2) | 22.6 |
| 5′ | 0.93 d (6.7) | 22.5 | 0.87 d (6.7) | 22.4 | 0.92 d (4.2) | 22.6 |
| 9-OAc | 170.3 | |||||
| 2.15 s | 20.7 | |||||
| Comp. | Conc. (μM) | Growth Inhibition Values (%) ± SEM at 10 and 30 μM Calculated IC50 Values (μM) [95% CI] | ||||
|---|---|---|---|---|---|---|
| HeLa | C33A | SiHa | NIH/3T3 | MRC-5 | ||
| 1 | 10 | 20.39 ± 0.62 | 90.00 ± 2.54 | 77.93 ± 0.56 | n.t. a | n.t. |
| 30 | 81.78 ± 1.70 | 91.79 ± 0.22 | 92.10 ± 0.73 | |||
| IC50 | 16.78 [15.57–18.09] | 5.89 [2.32–5.52] | 4.86 [4.45–5.31] | |||
| 2 | 10 | <20 b | <20 | <20 | n.t. | n.t. |
| 30 | 21.55 ± 2.42 | 36.60 ± 2.95 | <20 | |||
| 3 | 10 | 91.42 ± 0.34 | 79.15 ± 3.33 | 60.65 ± 1.58 | 92.96 ± 0.56 | 88.13 ± 1.22 |
| 30 | 96.58 ± 1.17 | 94.46 ± 0.21 | 78.77 ± 1.23 | 98.15 ± 0.13 | 97.58 ± 0.10 | |
| IC50 | 4.76 [4.27–5.32] | 6.82 [5.97–7.79] | 7.81 [6.81–8.96] | 4.12 [3.68–4.61] | 6.67 [5.35–8.30] | |
| 4 | 10 | 83.94 ± 1.80 | 97.27 ± 0.18 | 79.77 ± 1.47 | n.t. | n.t. |
| 30 | 90.10 ± 1.03 | 97.19 ± 0.41 | 94.62 ± 0.27 | |||
| IC50 | 5.10 [5.10–6.51] | 2.05 [2.91–2.18] | 2.22 [1.92–2.56] | |||
| 5 | 10 | <20 | 50.50 ± 3.49 | <20 | n.t. | n.t. |
| 30 | 63.86 ± 3.23 | 89.95 ± 1.4 | 65.52 ± 2.52 | |||
| 6 | 10 | <20 | 96.55 ± 0.22 | 84.19 ± 2.32 | 40.67 ± 2.92 | <20 |
| 30 | 91.88 ± 1.23 | 96.72 ± 0.33 | 84.76 ± 1.89 | 96.80 ± 0.25 | 73.05 ± 1.64 | |
| IC50 | 15.69 [12.51–19.69] | 3.53 [2.83–4.39] | 3.24 [2.95–3.55] | 11.17 [10.64–11.74] | 11.94 [10.62–13.43] | |
| 7 | 10 | 91.87 ± 0.72 | 94.66 ± 0.19 | 77.17 ± 1.04 | 90.65 ± 1.20 | <20 |
| 30 | 96.44 ± 0.10 | 95.18 ± 0.31 | 93.50 ± 0.42 | 98.06 ± 0.19 | 97.63 ± 0.27 | |
| IC50 | 6.00 [5.44–6.62] | 6.62 [4.38–10.00] | 6.02 [5.53–6.54] | 5.68 [5.36–6.02] | 15.42 [12.71–18.71] | |
| 9 | 10 | <20 | <20 | <20 | n.t. | n.t. |
| 30 | 37.27 ± 2.65 | 26.69 ± 2.73 | 35.69 ± 1.29 | |||
| 10 | 10 | <20 | <20 | <20 | n.t. | n.t. |
| 30 | 36.49 ± 1.55 | <20 | <20 | |||
| Neu B | 10 | 94.00 ± 0.59 | 98.29 ± 0.03 | 88.73 ± 0.36 | 98.11 ± 0.09 | 94.26 ± 0.23 |
| 30 | 94.81 ± 0.37 | 97.91 ± 0.15 | 93.42 ± 0.33 | 98.02 ± 0.05 | 97.46 ± 0.21 | |
| IC50 | 1.24 [1.07–1.45] | 0.47 [4.59–4.93] | 1.33 [1.14–1.54] | 1.31 [1.22–1.41] | 5.18 [4.69–5.73] | |
| Cispl. | 10 | 32.23 ± 1.16 | 77.17 ± 1.21 | 60.98 ± 0.92 | 73.88 ± 0.46 | 71.24.± 2.89 |
| 30 | 93.70 ± 0.82 | 97.50 ± 0.13 | 88.95 ± 0.53 | 97.10 ± 0.15 | 70.65 ± 1.34 | |
| IC50 | 12.14 [10.18–14.46] | 5.85 [5.37–6.38] | 4.29 [3.72–4.95] | 5.50 [4.46–6.35] | 5.77 [4.30–7.74] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasas, A.; Lajter, I.; Kúsz, N.; Király, S.B.; Kovács, T.; Kurtán, T.; Bózsity, N.; Nagy, N.; Schelz, Z.; Zupkó, I.; et al. Isolation, Structure Determination of Sesquiterpenes from Neurolaena lobata and Their Antiproliferative, Cell Cycle Arrest-Inducing and Anti-Invasive Properties against Human Cervical Tumor Cells. Pharmaceutics 2021, 13, 2088. https://doi.org/10.3390/pharmaceutics13122088
Vasas A, Lajter I, Kúsz N, Király SB, Kovács T, Kurtán T, Bózsity N, Nagy N, Schelz Z, Zupkó I, et al. Isolation, Structure Determination of Sesquiterpenes from Neurolaena lobata and Their Antiproliferative, Cell Cycle Arrest-Inducing and Anti-Invasive Properties against Human Cervical Tumor Cells. Pharmaceutics. 2021; 13(12):2088. https://doi.org/10.3390/pharmaceutics13122088
Chicago/Turabian StyleVasas, Andrea, Ildikó Lajter, Norbert Kúsz, Sándor Balázs Király, Tibor Kovács, Tibor Kurtán, Noémi Bózsity, Nikolett Nagy, Zsuzsanna Schelz, István Zupkó, and et al. 2021. "Isolation, Structure Determination of Sesquiterpenes from Neurolaena lobata and Their Antiproliferative, Cell Cycle Arrest-Inducing and Anti-Invasive Properties against Human Cervical Tumor Cells" Pharmaceutics 13, no. 12: 2088. https://doi.org/10.3390/pharmaceutics13122088
APA StyleVasas, A., Lajter, I., Kúsz, N., Király, S. B., Kovács, T., Kurtán, T., Bózsity, N., Nagy, N., Schelz, Z., Zupkó, I., Krupitza, G., Frisch, R., Mándi, A., & Hohmann, J. (2021). Isolation, Structure Determination of Sesquiterpenes from Neurolaena lobata and Their Antiproliferative, Cell Cycle Arrest-Inducing and Anti-Invasive Properties against Human Cervical Tumor Cells. Pharmaceutics, 13(12), 2088. https://doi.org/10.3390/pharmaceutics13122088