Nano Co-Crystal Embedded Stimuli-Responsive Hydrogels: A Potential Approach to Treat HIV/AIDS



Abstract
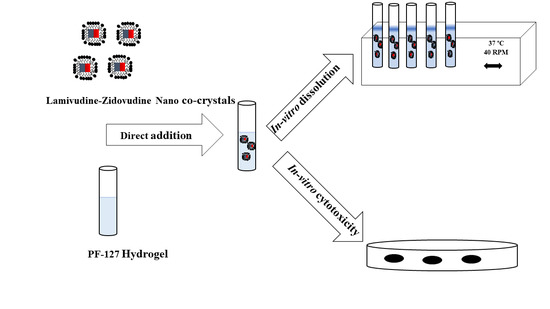
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Micro Co-Crystal (MCC) and Nano Co-Crystal (NCC) Synthesis
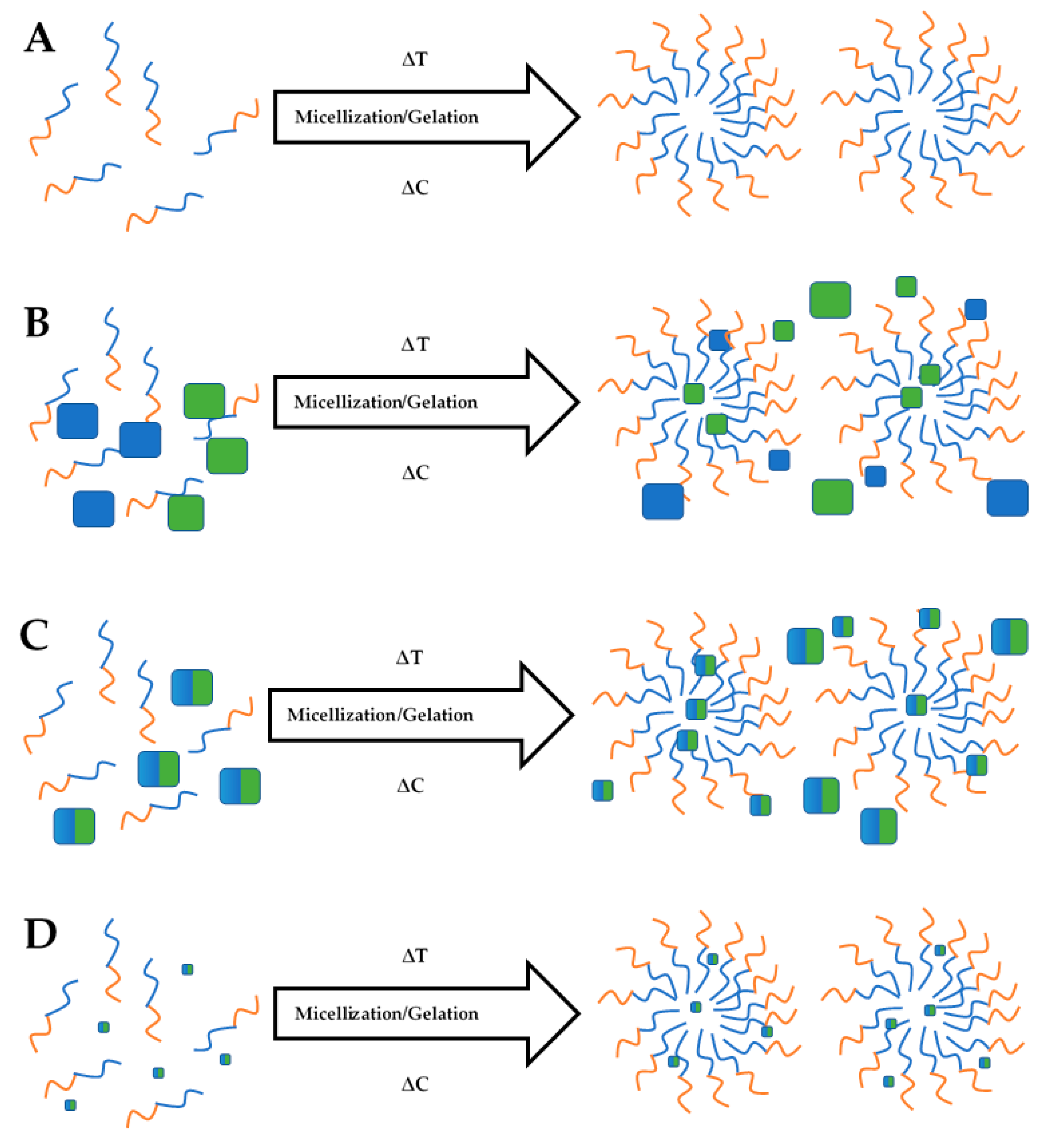
2.2.2. Stimuli-Responsive Gel Carrier Synthesis
Thermosensitive Gel Carriers
Ion-Sensitive Gels
Dual pH and Thermal Responsive Gels
Dual Ion and Thermal Responsive Gels
2.2.3. Stimuli-Responsive Gel Screening
Sol–Gel Transition Time
Erosion Time
Scanning Electron Microscopy
2.2.4. NCC Characterization
2.2.5. High-Performance Liquid Chromatography
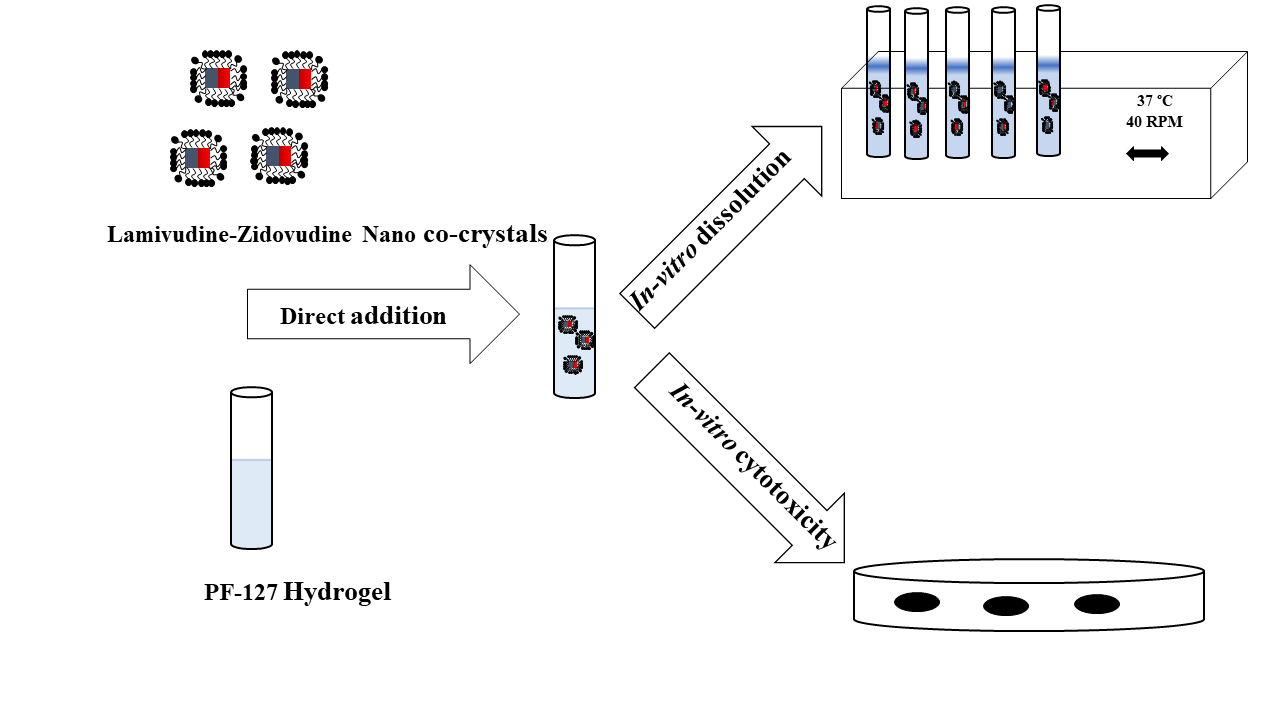
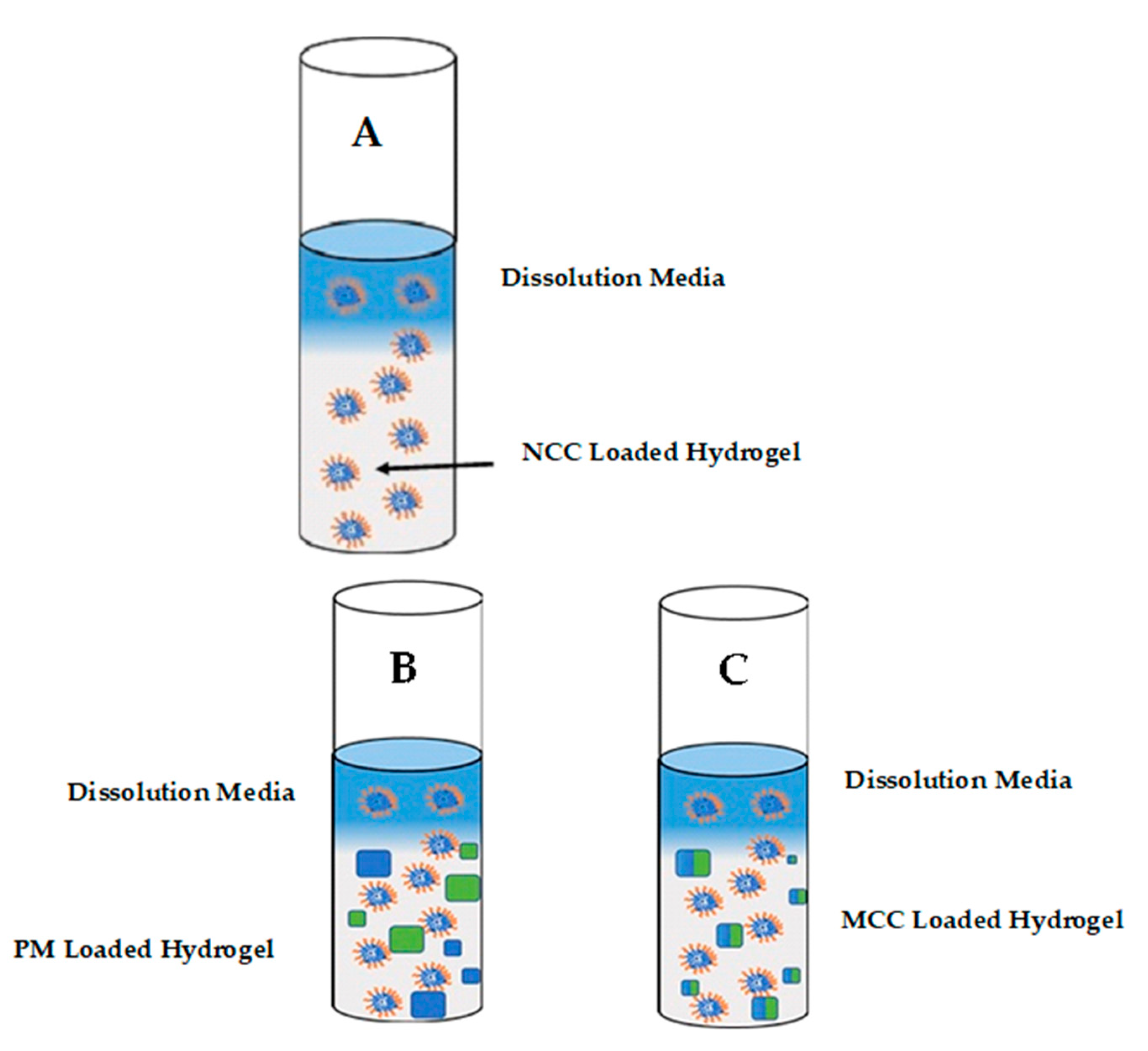
2.2.6. In Vitro Release
2.2.7. Determination of Best Fit Model
2.2.8. Cytotoxicity Studies
3. Results and Discussion
3.1. Stimuli-Responsive Carrier Screening
PF-127 25% w/w Gel Characterization
3.2. Nano Co-Crystal (NCC) Characterization
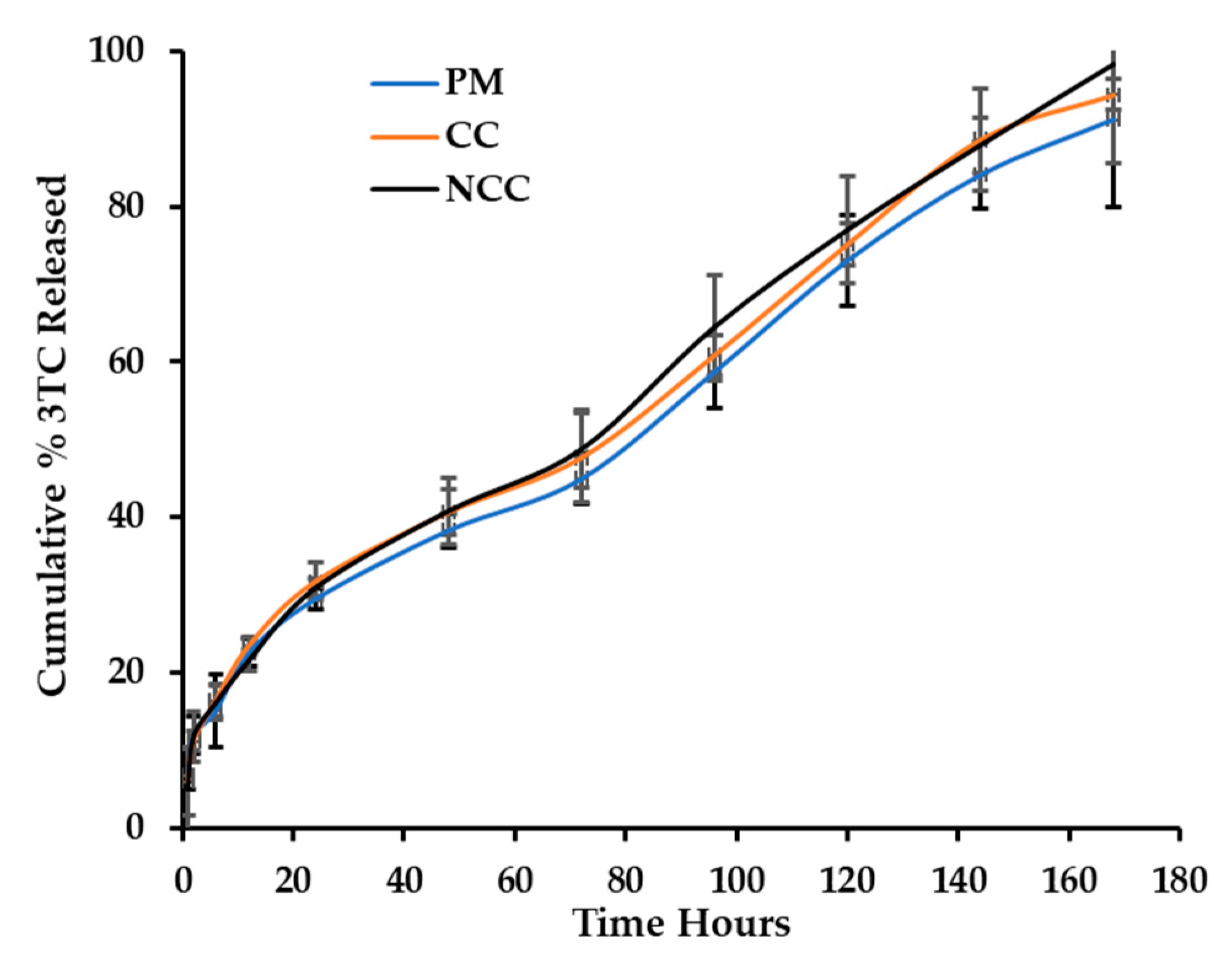
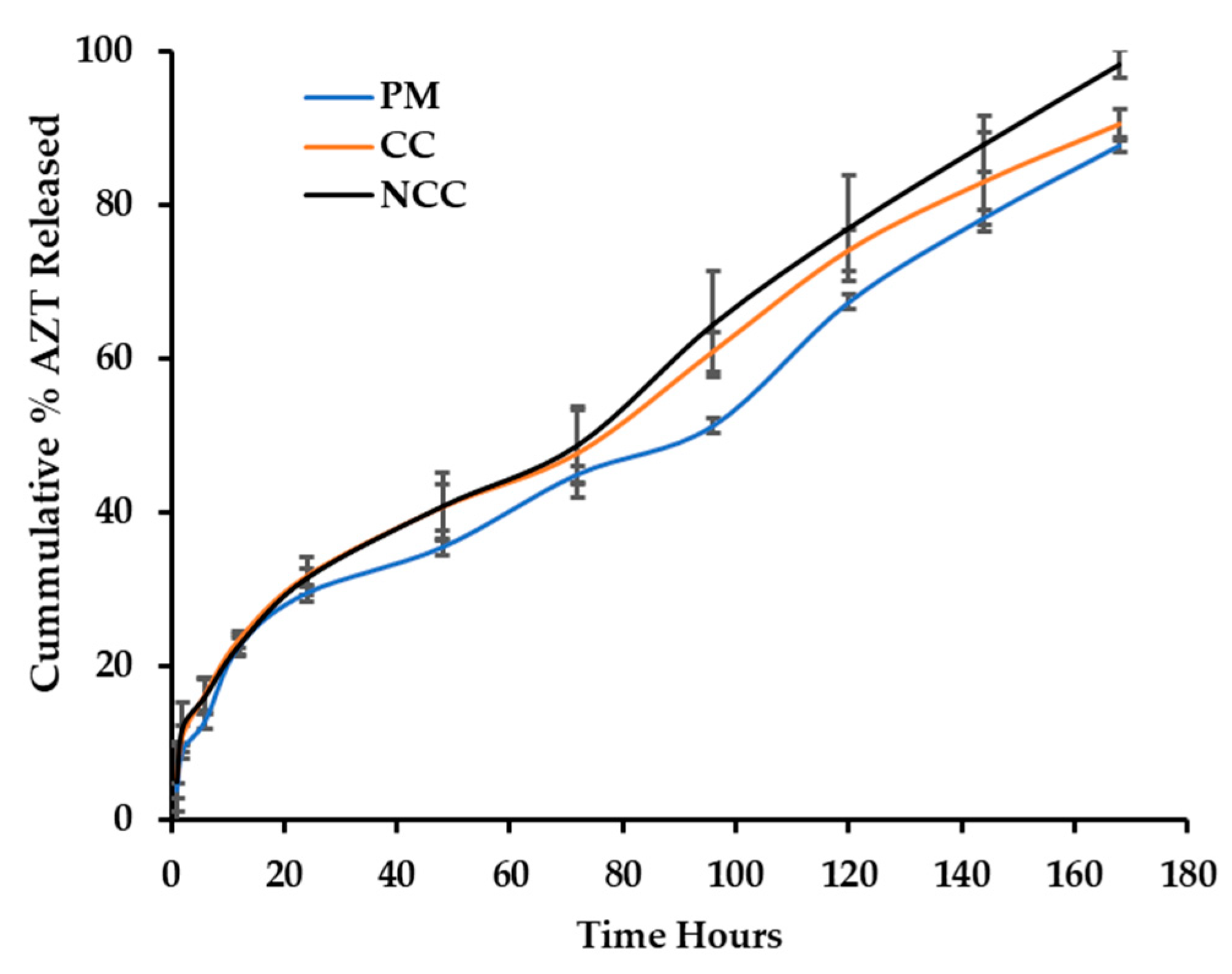
3.3. In Vitro Release of 3TC and AZT
3.4. Best Fit Model
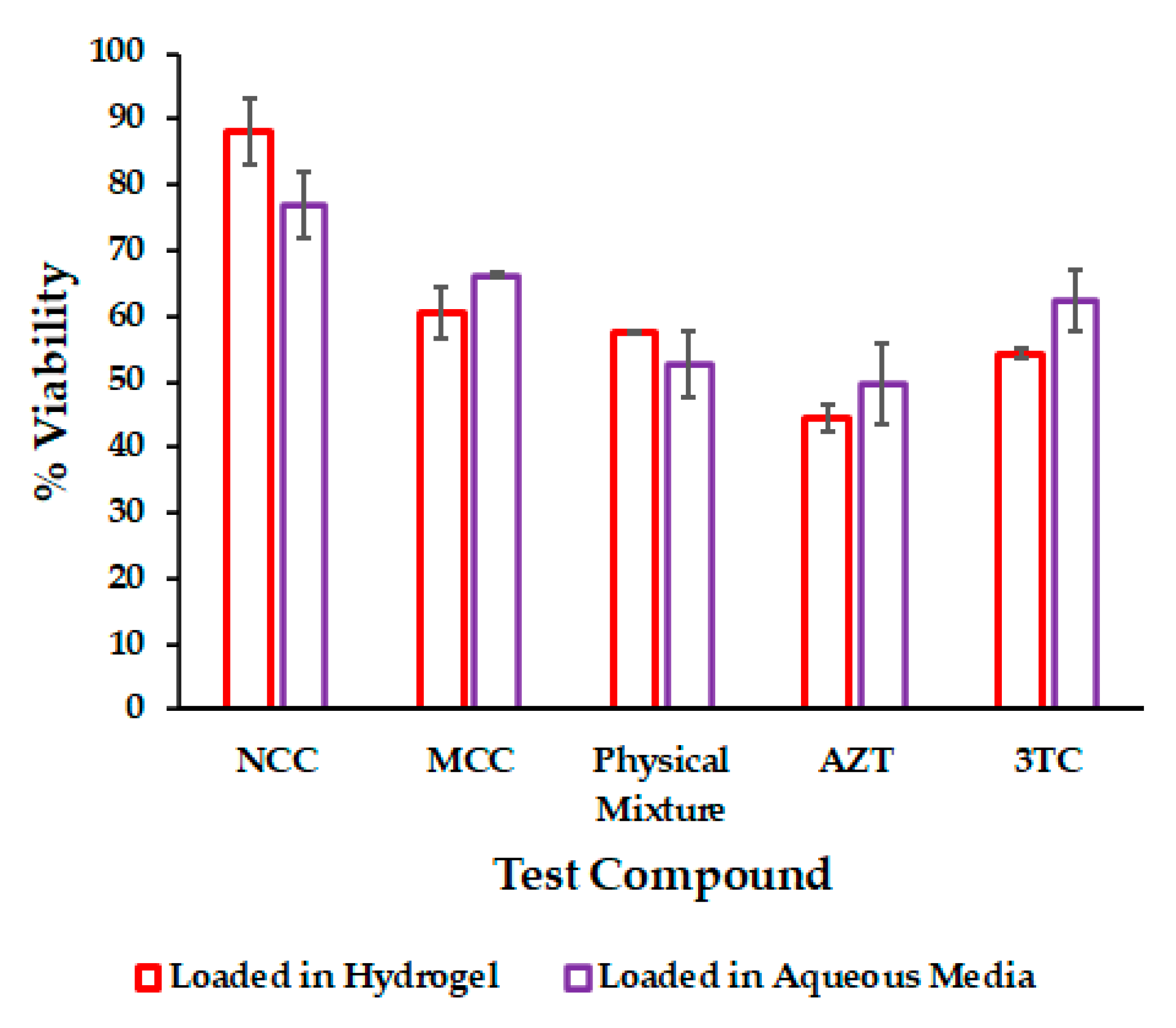
3.5. In Vitro Cytotoxicity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lohse, N.; Obel, N. Update of survival for persons with HIV infection in Denmark. Ann. Intern. Med. 2016, 165, 749–750. [Google Scholar] [CrossRef] [PubMed]
- Samji, H.; Cescon, A.; Hogg, R.S.; Modur, S.P.; Althoff, K.N.; Buchacz, K.; Burchell, A.N.; Cohen, M.; Gebo, K.A.; Gill, M.J.; et al. Closing the gap: Increases in life expectancy among treated HIV-positive individuals in the United States and Canada. PLoS ONE 2013, 8, e81355. [Google Scholar] [CrossRef] [PubMed]
- Cihlar, T.; Ray, A.S. Nucleoside and nucleotide HIV reverse transcriptase inhibitors: 25 years after zidovudine. Antivir. Res. 2010, 85, 39–58. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.M.; Faulds, D. Lamivudine. Drugs 1997, 53, 657–680. [Google Scholar] [CrossRef] [PubMed]
- Mandelbrot, L.; Landreau-Mascaro, A.; Rekacewicz, C.; Berrebi, A.; Bénifla, J.L.; Burgard, M.; Lachassine, E.; Barret, B.; Chaix, M.L.; Bongain, A.; et al. Lamivudine-zidovudine combination for prevention of maternal-infant transmission of HIV-1. JAMA 2001, 285, 2083–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Department of Health and Human Services-Panel on Antiretroviral Guidelines for Adults and Adolescents—A Working Group of the Office of AIDS Research Advisory Council (OARAC). Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV; OARAC: Redwood City, CA, USA, 2018. [Google Scholar]
- Rathbun, R.; Lockhart, S.; Stephens, J. Current HIV Treatment Guidelines—An Overview. Curr. Pharm. Des. 2006, 12, 1045–1063. [Google Scholar] [CrossRef]
- Schalm, S.W. Clinical implications of lamivudine resistance by HBV. Lancet 1997, 349, 3–4. [Google Scholar] [CrossRef]
- Kopp, S. International Pharmacopoeia Monograph on Zidovudine Revised Draft for Comments; World Health Organisation: Geneva, Switzerland, 2005. [Google Scholar]
- Kopp, S. Working Document QAS International Pharmacopoeia Monograph on Lamivudine Draft for Comment; World Health Organisation: Geneva, Switzerland, 2005. [Google Scholar]
- World Health Organization International Agency for Research on Cancer. Some Antiviral and Antineoplastic Drugs, and Other Pharmaceutical Agents; WHO: Lyon, France, 2000; Volume 76. [Google Scholar]
- Coleman, N.J.; Craig, D.Q.M. Modulated temperature differential scanning calorimetry: A novel approach to pharmaceutical thermal analysis. Int. J. Pharm. 1996, 135, 13–29. [Google Scholar] [CrossRef]
- Shamsipur, M.; Pourmortazavi, S.M.; Beigi, A.A.M.; Heydari, R.; Khatibi, M. Thermal Stability and Decomposition Kinetic Studies of Acyclovir and Zidovudine Drug Compounds. AAPS PharmSciTech 2013, 14, 287–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.V.; Nath, L.K. Evaluation of compatibility of lamivudine with tablet excipients and a novel synthesized polymer. J. Mater. Environ. Sci. 2011, 2, 243–250. [Google Scholar] [CrossRef]
- Villard, A.L.; Coussot, G.; Lefebvre, I.; Augustijns, P.; Aubertin, A.M.; Gosselin, G.; Peyrottes, S.; Périgaud, C. Phenyl phosphotriester derivatives of AZT: Variations upon the SATE moiety. Bioorganic Med. Chem. 2008, 16, 7321–7329. [Google Scholar] [CrossRef]
- Kumar, P.; Lakshmi, Y.S.; Bhaskar, C.; Golla, K.; Kondapi, A.K. Improved safety, bioavailability and pharmacokinetics of Zidovudine through lactoferrin nanoparticles during oral administration in rats. PLoS ONE 2015, 10, e0140399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauch, S.; Jantratid, E.; Dressman, J.B.; Junginger, H.E.; Kopp, S.; Midha, K.K.; Shah, V.P.; Stavchansky, S.; Barends, D.M. Biowaiver Monographs for Immediate Release Solid Oral Dosage Forms: Lamivudine. J. Pharm. Sci. 2011, 100, 2054–2063. [Google Scholar] [CrossRef] [PubMed]
- Jozwiakowski, M.J.; Nguyen, N.A.T.; Sisco, J.M.; Spancake, C.W. Solubility behavior of lamivudine crystal forms in recrystallization solvents. J. Pharm. Sci. 1996, 85, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Kashuba, A.D.; Dyer, J.R.; Kramer, L.M.; Raasch, R.H.; Eron, J.J.; Cohen, M.S. Antiretroviral-drug concentrations in semen: Implications for sexual transmission of human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 1999, 43, 1817–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, N.H.S.; Patnala, S.; Löbenberg, R.; Kanfer, I. In Vitro Dissolution of Generic Immediate-Release Solid Oral Dosage Forms Containing BCS Class I Drugs: Comparative Assessment of Metronidazole, Zidovudine, and Amoxicillin versus Relevant Comparator Pharmaceutical Products in South Africa and India. AAPS PharmSciTech 2014, 15, 1076–1086. [Google Scholar] [CrossRef] [Green Version]
- Kasim, N.A.; Whitehouse, M.; Ramachandran, C.; Bermejo, M.; Lennernäs, H.; Hussain, A.S.; Junginger, H.E.; Stavchansky, S.A.; Midha, K.K.; Shah, V.P.; et al. Molecular Properties of WHO Essential Drugs and Provisional Biopharmaceutical Classification. Mol. Pharm. 2004, 1, 85–96. [Google Scholar] [CrossRef]
- Lindenberg, M.; Kopp, S.; Dressman, J.B. Classification of orally administered drugs on the World Health Organization Model list of Essential Medicines according to the biopharmaceutics classification system. Eur. J. Pharm. Biopharm. 2004, 58, 265–278. [Google Scholar] [CrossRef]
- Anderson, F.D.; Archer, D.F.; Harman, S.M.; Leonard, R.J.; Wilborn, W.H. Tissue Response to Bioerodible, Subcutaneous Drug Implants: A Possible Determinant of Drug Absorption Kinetics. Pharm. Res. 1993, 10, 369–380. [Google Scholar] [CrossRef]
- Anderson, J.M.; Niven, H.; Pelagalli, J.; Olanoff, L.S.; Jones, R.D. The role of the fibrous capsule in the function of implanted drug-polymer sustained release systems. J. Biomed. Mater. Res. 1981, 15, 889–902. [Google Scholar] [CrossRef]
- Higgins, D.M.; Basaraba, R.J.; Hohnbaum, A.C.; Lee, E.J.; Grainger, D.W.; Gonzalez-Juarrero, M. Localized immunosuppressive environment in the foreign body response to implanted biomaterials. Am. J. Pathol. 2009, 175, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Daugherty, A.L.; Cleland, J.L.; Duenas, E.M.; Mrsny, R.J. Pharmacological modulation of the tissue response to implanted polylactic-co-glycolic acid microspheres. Eur. J. Pharm. Biopharm. 1997, 44, 89–102. [Google Scholar] [CrossRef]
- Wright, J.C.; Burgess, D.J. Advances in Delivery Science and Technology: Long Acting Injections and Implants; Rathbone, M., Ed.; Springer: New York, NY, USA, 2009; Volume 92, ISBN 9781461405535. [Google Scholar]
- Medlicott, N.J.; Waldron, N.A.; Foster, T.P. Sustained release veterinary parenteral products. Adv. Drug Deliv. Rev. 2004, 56, 1345–1365. [Google Scholar] [CrossRef] [PubMed]
- Medlicott, N.J.; Tucker, I.G. Pulsatile release from subcutaneous implants. Adv. Drug Deliv. Rev. 1999, 38, 139–149. [Google Scholar] [CrossRef]
- Qiu, Y.; Park, K. Environment-sensitive hydrogels for drug delivery. Adv. Drug Deliv. Rev. 2012, 64, 49–60. [Google Scholar] [CrossRef]
- Traitel, T.; Goldbart, R.; Kost, J. Smart polymers for responsive drug-delivery systems. J. Biomater. Sci. Polym. Ed. 2008, 19, 755–767. [Google Scholar] [CrossRef] [Green Version]
- Marieb, E.N.; Hoehn, K.N. Human Anatomy and Physiology, 10th ed.; Pearson Education: San Francisco, CA, USA, 2016; Volume 51. [Google Scholar]
- Larsen, C.; Larsen, S.W.; Jensen, H.; Yaghmur, A.; Østergaard, J. Role of in vitro release models in formulation development and quality control of parenteral depots. Expert Opin. Drug Deliv. 2009, 6, 1283–1295. [Google Scholar] [CrossRef]
- Sun, Y.; Jensen, H.; Petersen, N.J.; Larsen, S.W.; Østergaard, J. Concomitant monitoring of implant formation and drug release of in situ forming poly (lactide-co-glycolide acid) implants in a hydrogel matrix mimicking the subcutis using UV–vis imaging. J. Pharm. Biomed. Anal. 2018, 150, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Veyries, M.L.; Couarraze, G.; Geiger, S.; Agnely, F.; Massias, L.; Kunzli, B.; Faurisson, F.; Rouveix, B. Controlled release of vancomycin from Poloxamer 407 gels. Int. J. Pharm. 1999, 192, 183–193. [Google Scholar] [CrossRef]
- Haglund, B.O.; Joshi, R.; Himmelstein, K.J. An in situ gelling system for parenteral delivery. J. Control. Release 1996, 41, 229–235. [Google Scholar] [CrossRef]
- Matschke, C.; Isele, U.; Van Hoogevest, P.; Fahr, A. Sustained-release injectables formed in situ and their potential use for veterinary products. J. Control. Release 2002, 85, 1–15. [Google Scholar] [CrossRef]
- Ding, K.; Yang, Z.; Zhang, Y.L.; Xu, J.Z. Injectable thermosensitive chitosan/b-glycerophosphate/collagen hydrogel maintains the plasticity of skeletal muscle satellite cells and supports their in vivo viability. Cell Biol. Int. 2013, 37, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Ruel-Gariépy, E.; Leroux, J.-C.C. In situ-forming hydrogels—Review of temperature-sensitive systems. Eur. J. Pharm. Biopharm. 2004, 58, 409–426. [Google Scholar] [CrossRef]
- Witika, B.A.; Makoni, P.A.; Matafwali, S.K.; Chabalenge, B.; Mwila, C.; Kalungia, A.C.; Nkanga, C.I.; Bapolisi, A.M.; Walker, R.B. Biocompatibility of Biomaterials for Nanoencapsulation: Current Approaches. Nanomaterials 2020, 10, 1649. [Google Scholar] [CrossRef]
- Nie, S.; Hsiao, W.W.; Pan, W.; Yang, Z. Thermoreversible pluronic® F127-based hydrogel containing liposomes for the controlled delivery of paclitaxel: In vitro drug release, cell cytotoxicity, and uptake studies. Int. J. Nanomedicine 2011, 6, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Priya James, H.; John, R.; Alex, A.; Anoop, K.R. Smart polymers for the controlled delivery of drugs—A concise overview. Acta Pharm. Sin. B 2014, 4, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Sacco, P.; Furlani, F.; de Marzo, G.; Marsich, E.; Paoletti, S.; Donati, I. Concepts for Developing Physical Gels of Chitosan and of Chitosan Derivatives. Gels 2018, 4, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, F.; Morin, A.; Monsan, P. Microbial polysaccharides with actual potential industrial applications. Biotechnol. Adv. 1986, 4, 245–259. [Google Scholar] [CrossRef]
- Totosaus, A.; Pérez-Chabela, M.L. Textural properties and microstructure of low-fat and sodium-reduced meat batters formulated with gellan gum and dicationic salts. Food Sci. Technol. 2009, 42, 563–569. [Google Scholar] [CrossRef]
- Banerjee, S.; Bhattacharya, S. Compressive textural attributes, opacity and syneresis of gels prepared from gellan, agar and their mixtures. J. Food Eng. 2011, 102, 287–292. [Google Scholar] [CrossRef]
- Rehm, B.H.A. Bacterial polymers: Biosynthesis, modifications and applications. Nat. Rev. Microbiol. 2010, 8, 578. [Google Scholar] [CrossRef] [PubMed]
- Cox, P.J.; Sujja, A.J.; Munday, D.L.; Khan, K.A. Development and evaluation of a multiple unit oral sustained release dosage form for S (+)-ibuprofen;preparation and release kinetics. Int. J. Pharm. 2000, 193, 73–84. [Google Scholar] [CrossRef]
- Toti, U.S.; Soppimath, K.S.; Mallikarjuna, N.N.; Aminabhavi, T.M. Acrylamide-grafted-acacia gum polymer matrix tablets as erosion-controlled drug delivery systems. J. Appl. Polym. Sci. 2004, 93, 2245–2253. [Google Scholar] [CrossRef]
- Vendruscolo, C.W.; Andreazza, I.F.; Ganter, J.L.M.S.; Ferrero, C.; Bresolin, T.M.B. Xanthan and galactomannan (from M. scabrella) matrix tablets for oral controlled delivery of theophylline. Int. J. Pharm. 2005, 296, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rasul, A.; Iqbal, M.; Murtaza, G.; Waqas, M.K.; Hanif, M.; Khan, S.A.; Bhatti, N.S. Design, development and in-vitro evaluation of metoprolol tartrate tablets containing xanthan-tragacanth. Acta Pol. Pharm. Drug Res. 2010, 67, 517–522. [Google Scholar]
- Mundargi, R.C.; Shelke, N.B.; Babu, V.R.; Patel, P.; Rangaswamy, V.; Aminabhavi, T.M. Novel thermo-responsive semi-interpenetrating network microspheres of gellan gum-poly(N-isopropylacrylamide) for controlled release of atenolol. J. Appl. Polym. Sci. 2010, 116, 1832–1841. [Google Scholar] [CrossRef]
- Zoratto, N.; Matricardi, P. Semi-IPNs and IPN-based hydrogels. In Biomaterials; Pal, K., Banerjee, I., Eds.; Woodhead Publishing: Cambridge, UK, 2018; pp. 91–124. ISBN 978-0-08-102179-8. [Google Scholar]
- Witika, B.A.; Smith, V.J.; Walker, R.B. Quality by design optimization of cold sonochemical synthesis of zidovudine-lamivudine nanosuspensions. Pharmaceutics 2020, 12, 367. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, P.M.; Azim, Y.; Thakur, T.S.; Desiraju, G.R. Co-Crystals of the Anti-HIV Drugs Lamivudine and Zidovudine. Cryst. Growth Des. 2009, 9, 951–957. [Google Scholar] [CrossRef]
- Lupin Limited. LAMIVUDINE: ZIDOVUDINE: WATER 1: 1: 1 COCRYSTAL. Patent WO 2009/116055 A1, 24 September 2009. [Google Scholar]
- Witika, B.A.; Smith, V.J.; Walker, R.B. A comparative study of the effect of different stabilizers on the critical quality attributes of self-assembling nano co-crystals. Pharmaceutics 2020, 12, 182. [Google Scholar] [CrossRef] [Green Version]
- Schmolka, I.R. Artificial skin I. Preparation and properties of pluronic F127 gels for treatment of burns. J. Biomed. Mater. Res. 1972, 6, 571–582. [Google Scholar] [CrossRef]
- Meng, Y.C.; Hong, L.B.; Jin, J.Q. A Study on the Gelation Properties and Rheological Behavior of Gellan Gum. Appl. Mech. Mater. 2013, 284–287, 20–24. [Google Scholar] [CrossRef]
- Varshosaz, J.; Tabbakhian, M.; Salmani, Z. Designing of a Thermosensitive Chitosan/Poloxamer In Situ Gel for Ocular Delivery of Ciprofloxacin. Open Drug Deliv. J. 2008, 2, 61–70. [Google Scholar] [CrossRef]
- Marques, M.R.C.; Loebenberg, R.; Almukainz, M. Simulated Biological Fluids with Possible Application in Dissolution Testing. Dissolution Technol. 2011, 101, 1212–1220. [Google Scholar] [CrossRef]
- Oyane, A.; Kim, H.-M.; Furuya, T.; Kokubo, T.; Miyazaki, T.; Nakamura, T. Preparation and assessment of revised simulated body fluids. J. Biomed. Mater. Res. 2002, 65, 401–413. [Google Scholar] [CrossRef]
- Tamer, Y.; Yildirim, H. Biodegradable and stimuli sensitive amphiphilic graft copolymers and their sol-gel phase transition behavior. Polym. Adv. Technol. 2015, 26, 399–407. [Google Scholar] [CrossRef]
- Saindane, N.S.; Pagar, K.P.; Vavia, P.R. Nanosuspension based in situ gelling nasal spray of carvedilol: Development, in vitro and in vivo characterization. AAPS PharmSciTech 2013, 14, 189–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malemane, S. Development and Assessment of a Smart Thermosetting Intranasal Hydrogel for Lamotrigine. Master’s Thesis, Rhodes University, Makhanda, South Africa, 2018. [Google Scholar]
- Witika, B.A. Formulation Development, Manufacture and Evaluation of a Lamivudine-Zidovudine Nano Co-Crystal Thermo-Responsive Suspension. Ph.D. Thesis, Rhodes University, Makhanda, South Africa, 2020. [Google Scholar]
- Chaibva, F.A.; Walker, R.B. The comparison of in vitro release methods for the evaluation of oxytocin release from pluronic® F127 parenteral formulations. Dissolution Technol. 2007, 14, 15–25. [Google Scholar] [CrossRef]
- Chaibva, F.A. Development and Assessment of an Oxytocin Parenteral Dosage Form Prepared Using Pluronic® F127. Master’s Thesis, Rhodes University, Makhanda, South Africa, 2006. [Google Scholar]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foldbjerg, R.; Wang, J.; Beer, C.; Thorsen, K.; Sutherland, D.S.; Autrup, H. Biological effects induced by BSA-stabilized silica nanoparticles in mammalian cell lines. Chem. Biol. Interact. 2013, 204, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, D.R.; Carmen Morán, M.; Mitjans, M.; Martínez, V.; Pérez, L.; Pilar Vinardell, M. New cationic nanovesicular systems containing lysine-based surfactants for topical administration: Toxicity assessment using representative skin cell lines. Eur. J. Pharm. Biopharm. 2013, 83, 33–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bácskay, I.; Nemes, D.; Fenyvesi, F.; Váradi, J.; Vasvári, G.; Fehér, P.; Vecsernyés, M.; Ujhelyi, Z. Role of Cytotoxicity Experiments in Pharmaceutical Development. In Cytotoxicity; IntechOpen: Rijeka, Croatia, 2012; pp. 131–145. [Google Scholar]
- Nogueira, D.; Mitjans, M.; Rolim, C.; Vinardell, M. Mechanisms Underlying Cytotoxicity Induced by Engineered Nanomaterials: A Review of in Vitro Studies. Nanomaterials 2014, 4, 454–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, S.-W.; Sikorski, J.A.; Weitzmann, M.N.; Beck, G.R. Bio-active engineered 50 nm silica nanoparticles with bone anabolic activity: Therapeutic index, effective concentration, and cytotoxicity profile in vitro. Toxicol. Vitr. 2014, 28, 354–364. [Google Scholar] [CrossRef] [Green Version]
- Sosnik, A.; Cohn, D. Reverse thermo-responsive poly(ethylene oxide) and poly(propylene oxide) multiblock copolymers. Biomaterials 2005, 26, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Gioffredi, E.; Boffito, M.; Calzone, S.; Giannitelli, S.M.; Rainer, A.; Trombetta, M.; Mozetic, P.; Chiono, V. Pluronic F127 Hydrogel Characterization and Biofabrication in Cellularized Constructs for Tissue Engineering Applications. Procedia CIRP 2016, 49, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Malli, S.; Bories, C.; Pradines, B.; Loiseau, P.M.; Ponchel, G.; Bouchemal, K. In situ forming pluronic® F127/chitosan hydrogel limits metronidazole transmucosal absorption. Eur. J. Pharm. Biopharm. 2017, 112, 143–147. [Google Scholar] [CrossRef]
- Vadnere, M.; Amidon, G.; Lindenbaum, S.; Haslam, J.L. Thermodynamic studies on the gel-sol transition of some pluronic polyols. Int. J. Pharm. 1984, 22, 207–218. [Google Scholar] [CrossRef]
- Groseclose, M.R.; Castellino, S. Intramuscular and subcutaneous drug depot characterization of a long-acting cabotegravir nanoformulation by MALDI IMS. Int. J. Mass Spectrom. 2019, 437, 92–98. [Google Scholar] [CrossRef]
- Liu, T.; Yu, X.; Yin, H.; Möschwitzer, J.P. Advanced modification of drug nanocrystals by using novel fabrication and downstream approaches for tailor-made drug delivery. Drug Deliv. 2019, 26, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Cidade, M.T.; Ramos, D.J.; Santos, J.; Carrelo, H.; Calero, N.; Borges, J.P. Injectable hydrogels based on pluronic/water systems filled with alginate microparticles for biomedical applications. Materials (Basel) 2019, 12, 1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higuchi, W.I. Analysis of data on the medicament release from ointments. J. Pharm. Sci. 1962, 51, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T. Rate of Release of Medicaments from Ointment Bases Containing Drugs in Suspension. J. Pharm. Sci. 1961, 50, 874–875. [Google Scholar] [CrossRef] [PubMed]
- Hixson, A.W.; Crowell, J.H. Dependence of Reaction Velocity upon surface and Agitation. Ind. Eng. Chem. 1931, 23, 923–931. [Google Scholar] [CrossRef]
- Siepmann, J.; Siepmann, F. Modeling of diffusion controlled drug delivery. J. Control. Release 2012, 161, 351–362. [Google Scholar] [CrossRef]
- Anderson, B.C. Development of Novel Polymeric Materials for Gene Therapy and pH-Sensitive Drug Delivery: Modeling, Synthesis, Characterization, and Analysis. Ph.D. Thesis, Iowa State University, Ames, IA, USA, 2002. [Google Scholar]
- Anderson, B.C.; Pandit, N.K.; Mallapragada, S.K. Understanding drug release from poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide) gels. J. Control. Release 2001, 70, 157–167. [Google Scholar] [CrossRef]
- Bruschi, M.L. Mathematical models of drug release. In Strategies to Modify the Drug Release from Pharmaceutical Systems; Woodhead Publishing: Cambridge, UK, 2015; pp. 63–86. ISBN 9780081000922. [Google Scholar]
- Lin, Z.; Gao, W.; Hu, H.; Ma, K.; He, B.; Dai, W.; Wang, X.; Wang, J.; Zhang, X.; Zhang, Q. Novel thermo-sensitive hydrogel system with paclitaxel nanocrystals: High drug-loading, sustained drug release and extended local retention guaranteeing better efficacy and lower toxicity. J. Control. Release 2014, 174, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, R.; Blanchard, J. Controlled-release delivery system for the α-MSH analog Melanotan-I using poloxamer 407. J. Pharm. Sci. 1996, 85, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.; Croy, S.; Mallapragada, S.; Pandit, N. Experimental investigation and mathematical modeling of Pluronic® F127 gel dissolution: Drug release in stirred systems. J. Control. Release 2000, 67, 191–202. [Google Scholar] [CrossRef]
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Gilbert, J.C.; Hadgraft, J.; Bye, A.; Brookes, L.G. Drug release from Pluronic F-127 gels. Int. J. Pharm. 1986, 32, 223–228. [Google Scholar] [CrossRef]
- Lagarce, F.; Faisant, N.; Desfontis, J.C.; Marescaux, L.; Gautier, F.; Richard, J.; Menei, P.; Benoit, J.P. Baclofen-loaded microspheres in gel suspensions for intrathecal drug delivery: In vitro and in vivo evaluation. Eur. J. Pharm. Biopharm. 2005, 61, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Etxeberria, A.E.; Kornelsen, C.; Nand, A.V.; Ray, S.; Bunt, C.R.; Seyfoddin, A. Development of a Long-Term Drug Delivery System with Levonorgestrel-Loaded Chitosan Microspheres Embedded in Poly(vinyl alcohol) Hydrogel. ACS Appl. Bio Mater. 2019, 2, 2766–2779. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of Solute Release from Porous Hydrophilic Polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Sosnik, A.; Chiappetta, D.A.; Carcaboso, Á.M. Drug delivery systems in HIV pharmacotherapy: What has been done and the challenges standing ahead. J. Control. Release 2009, 138, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Woodrow, K.A. Nanotechnology approaches to eradicating HIV reservoirs. Eur. J. Pharm. Biopharm. 2019, 138, 48–63. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | AZT | 3TC |
|---|---|---|
| Chemical name | 1-[(2R,4S,5S)-4-azido-5-(hydroxymethyl)tetrahydrofuran-2-yl]-5-methyl-pyrimidine-2,4(1H,3H)-dione [9] | (-)-4-amino-1-[(2R,5S)-2-(hydroxymethyl)-1,3-oxathiolan-5-yl]pyrimidin-2(1H)-one [10] |
| Melting point | 106–112 °C (from petroleum ether); 120–122 °C (from water) [9,11,12,13] | 183 °C [14] |
| Molecular weight (g/mol) | 267.2 [9] | 229.3 [10] |
| Chemical formula | C10H13N5O4 [9] | C8H11N3O3S [10] |
| Description | A white or brownish powder [9] | A white or almost white powder [10] |
| pKa and log P | pKa and log P of AZT was reported as 9.68 and 0.06, respectively [15,16] | 3TC is a weak base with a pKa of 4.3 by the protonation of the NH2 group. The log P is −1.46 [17,18,19]. |
| BCS class | III [20] | I/III [21,22] |
| Solubility | Soluble in ethanol; sparingly soluble in water [9,15,16] | Soluble in water, sparingly soluble in methanol and practically insoluble in acetone [10] |
| Formulation | PF-127% w/w | Volume PF-127 mL | GG% w/v | Volume GG mL | Chitosan % w/v |
|---|---|---|---|---|---|
| Thermosensitive gels | |||||
| 1 | 20 | 5.0 | - | - | - |
| 2 | 25 | 5.0 | - | - | - |
| Ion-sensitive gels | |||||
| 3 | - | - | 0.1 | 5.0 | - |
| 4 | - | - | 0.3 | 5.0 | - |
| 5 | - | - | 0.5 | 5.0 | - |
| Dual thermo- and pH-sensitive gels | |||||
| 6 | 20 | 5.0 | - | - | 1.0 |
| 7 | 20 | 5.0 | - | - | 2.0 |
| 8 | 25 | 5.0 | - | - | 1.0 |
| 9 | 25 | 5.0 | - | - | 2.0 |
| Dual thermo- and ion-sensitive gels | |||||
| 10 | 25 | 4.5 | 0.5 | 0.5 | - |
| 11 | 25 | 4.0 | 0.5 | 1.0 | - |
| 12 | 25 | 3.5 | 0.5 | 1.5 | - |
| Reagents | Concentration mg/L |
|---|---|
| Sodium chloride | 7996 |
| Sodium bicarbonate | 350 |
| Potassium chloride | 224 |
| Potassium phosphate dibasic trihydrate | 228 |
| Magnesium chloride hexahydrate | 305 |
| Calcium chloride | 278 |
| Sodium sulphate | 71 |
| Tris(hydroxymethyl) aminomethane | 6057 |
| 1 M Hydrochloric acid | 40 mL |
| Parameter | Setting |
|---|---|
| Flow rate | 0.3 mL/min |
| Injection volume | 10 µL |
| Wavelength | 266 and 271 nm |
| Temperature | 25 °C |
| Mobile-phase composition | 25:75% v/v ACN: H2O |
| Formulation | Sol–Gel Transition Time Min | Erosion Time Days |
|---|---|---|
| Thermosensitive gels | ||
| 1 | 7 | 5 |
| 2 | 5 | 7 |
| Ion-sensitive gels | ||
| * 3 | - | - |
| 4 | 8 | 7 |
| 5 | 6 | 7 |
| Dual thermo- and pH-sensitive gels | ||
| 6 | 6 | 5 |
| 7 | 6 | 4 |
| 8 | 5 | 6 |
| 9 | 5 | 5 |
| Dual thermo- and ion-sensitive gels | ||
| 10 | 5 | 7 |
| 11 | 5 | 7 |
| 12 | 5 | 7 |
| CQA | Value | |
|---|---|---|
| NCC | NCC in Gel | |
| * PS (nm) | 332.9 ± 42.85 | 243.6 ± 26.58 |
| PDI | 0.474 ± 0.040 | 0.495 ± 0.153 |
| * ZP (mV) | −34.6 ± 5.56 | −18.3 ± 4.45 |
| Formulation | Value for Kinetic Model | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Zero-Order F = K0t | First-Order F = 100 × [1 − e−k1 * t] | Higuchi F = kHt0.5 | Korsmeyer–Peppas F = kKPtn | Hixson-Crowell F = 100 × [1 − (1 − kHCt)3] | ||||||||
| AZT | 3TC | AZT | 3TC | AZT | 3TC | AZT | n | 3TC | n | AZT | 3TC | |
| NCC | * 0.9857 | * 0.9859 | 0.7350 | 0.7835 | 0.9801 | 0.9763 | 0.9797 | 0.613 | 0.9773 | 0.598 | 0.8868 | 0.8774 |
| Physical mixture | 0.9761 | 0.9773 | 0.8170 | 0.7522 | 0.9845 | 0.9691 | * 0.9915 | 0.595 | * 0.9783 | 0.513 | 0.8915 | 0.8655 |
| MCC | 0.9735 | 0.9786 | 0.7793 | 0.7832 | 0.9844 | 0.9832 | * 0.9882 | 0.540 | * 0.9861 | 0.557 | 0.8708 | 0.8739 |
| Compound | Viability % | |
|---|---|---|
| Loaded in Hydrogel | In Aqueous Media | |
| NCC | 88.0 ± 5.0 | 76.9 ± 5.0 |
| MCC | 60.5 ± 3.8 | 66.1 ± 0.4 |
| Physical mixture | 57.5 ± 0.2 | 52.6 ± 5.1 |
| AZT | 44.3 ± 2.1 | 49.6 ± 6.2 |
| 3TC | 54.2 ± 0.7 | 62.3 ± 4.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witika, B.A.; Stander, J.-C.; Smith, V.J.; Walker, R.B. Nano Co-Crystal Embedded Stimuli-Responsive Hydrogels: A Potential Approach to Treat HIV/AIDS. Pharmaceutics 2021, 13, 127. https://doi.org/10.3390/pharmaceutics13020127
Witika BA, Stander J-C, Smith VJ, Walker RB. Nano Co-Crystal Embedded Stimuli-Responsive Hydrogels: A Potential Approach to Treat HIV/AIDS. Pharmaceutics. 2021; 13(2):127. https://doi.org/10.3390/pharmaceutics13020127
Chicago/Turabian StyleWitika, Bwalya A., Jessé-Clint Stander, Vincent J. Smith, and Roderick B. Walker. 2021. "Nano Co-Crystal Embedded Stimuli-Responsive Hydrogels: A Potential Approach to Treat HIV/AIDS" Pharmaceutics 13, no. 2: 127. https://doi.org/10.3390/pharmaceutics13020127
APA StyleWitika, B. A., Stander, J. -C., Smith, V. J., & Walker, R. B. (2021). Nano Co-Crystal Embedded Stimuli-Responsive Hydrogels: A Potential Approach to Treat HIV/AIDS. Pharmaceutics, 13(2), 127. https://doi.org/10.3390/pharmaceutics13020127