Understanding Host Immunity and the Gut Microbiota Inspires the New Development of Vaccines and Adjuvants



Abstract
:1. Introduction
2. The Effect of Vaccination on Our Immune Systems
2.1. Tuberculosis and Bacille Calmette Guerin (BCG) Vaccine
2.2. Bacterial Infectious Pneumonia and Streptococcus pneumoniae Vaccine
2.3. Influenza Virus Infection and Vaccine
3. Adjuvants
3.1. Alum
3.2. Complete Freund’s Adjuvant
3.3. Cholera Toxin (CT)

{kind=link}
{kind=link}
| Adjuvants | Components | Activated Immune Responses | Mechanism |
|---|---|---|---|
| Alum | Aluminum hydroxide | ・Th2 response ・IgG1 and IgE production | ・DNA released by dying cells is sensed as DAMPs. ・IgE is elicited by IRF3-Tbk1 axis, while IgG1 is an IRF3-independent pathway [63]. |
| Complete Freund’s adjuvant (CFA) | Heat-killed M. butyricum (peptidoglycan and trehalose-6,6′-dimycolate (TDM)) | ・Th1 and Th17 response ・Cellular immunity | ・TDM-Mincle-Syk-CARD9 and peptidoglycan-Nod1 signaling pathways are involved in inflammatory cytokine production [69,70]. ・Th1: IL-12-STAT4 axis regulates T-bet expression [71,72,73]. ・Th17: IL-1-IL-1R axis regulates RORγt and IRF4 expression [75]. |
| Cholera toxin | Enterotoxin secreted by Vibrio cholerae | ・Th2-associated cytokines, IL-4, IL-5, and IgG1 production ・Upregulating Th1 and Th17 responses | ・Nod2, Ripk2, and IL-1β are required for CT adjuvanticity [83]. ・Symbiotic bacteria (Streptococcus sciuri and Bacillus clausii) and SCFAs contribute to enhance antigen-specific IgG production by CT [83,90]. ・IL-1β dictates CD4+ T cells to differentiation of Tfh cells, which support antibody production from B cells [88]. |
| Pulmonary surfactant-biomimetic liposomes encapsulating 2′, 3′-cyclic guanosine monophosphate adenosine monophosphate (PS-GAMP) | Liposome ・Dipalmitoylated phosphatidylcholine (DPPC) ・Phosphatidylglycerol (DPPG) ・Cholesterol ・Polyethylene glycol (PEG) (Trehalose lyophilization) Content ・2′, 3′-cyclic guanosine monophosphate adenosine monophosphate (cGAMP) | ・Enhance intrasubtype-specific IgG and IgA production. ・Induce high frequency of CD8+ resident memory T (TRM) cells. ・Activating CD8+ Granzyme B+ T cells | ・Surfactant proteins, SP-A and SP-D, bound to PS-GAMP, and subsequently, PS-GAMP is caught by alveolar macrophages (AMs). ・cGAMP is released in cytoplasm and stimulates STING in AMs and type 2 alveolar epithelial cells (AEC II), resulting in secreting IFN-α/β and GM-CSF. ・Secreted cytokines activate DCs, which subsequently induce adaptive immunity, CD8+ T cells activation, and immunoglobulin production [91]. |
3.4. PS-GAMP
4. Trained Immunity
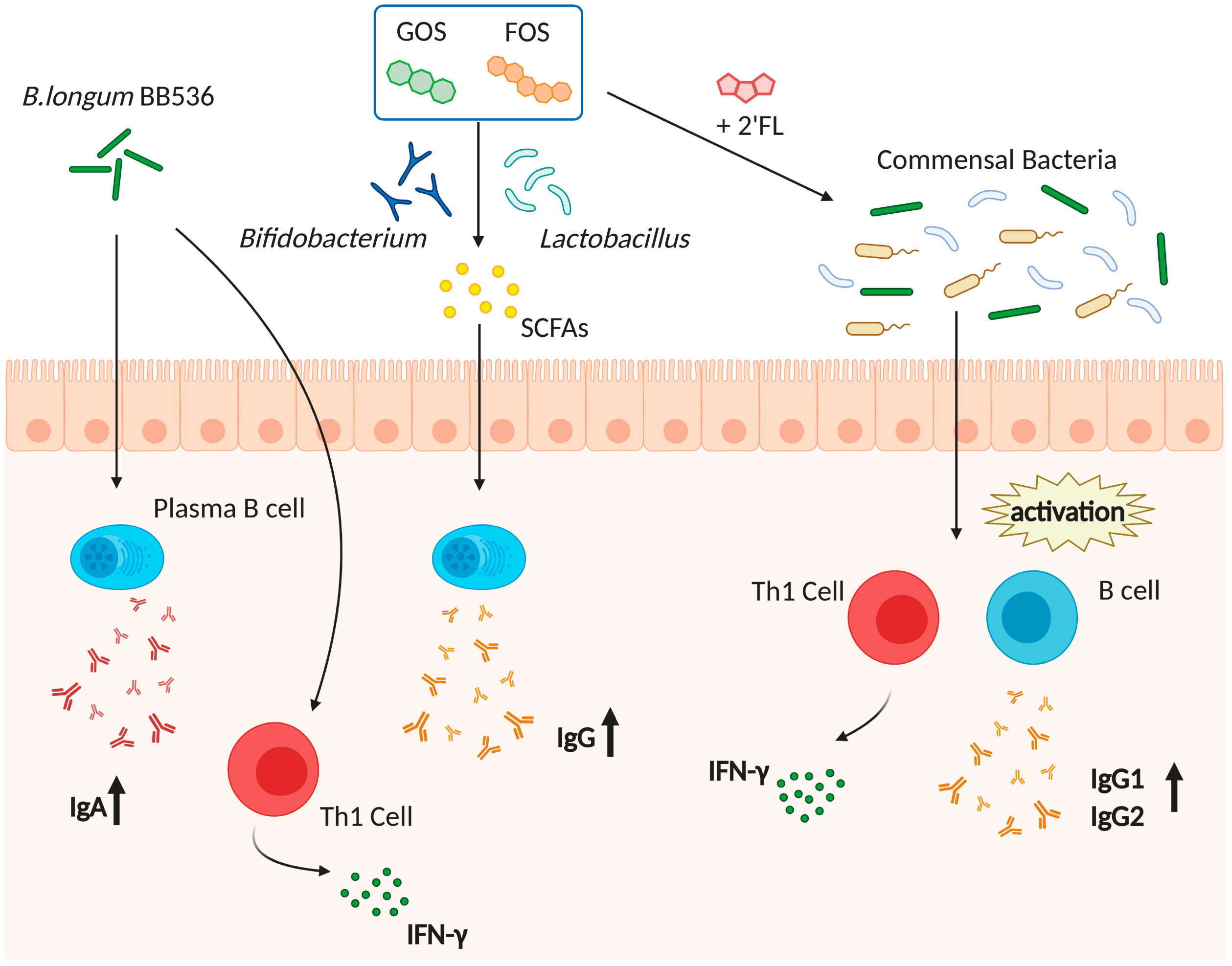
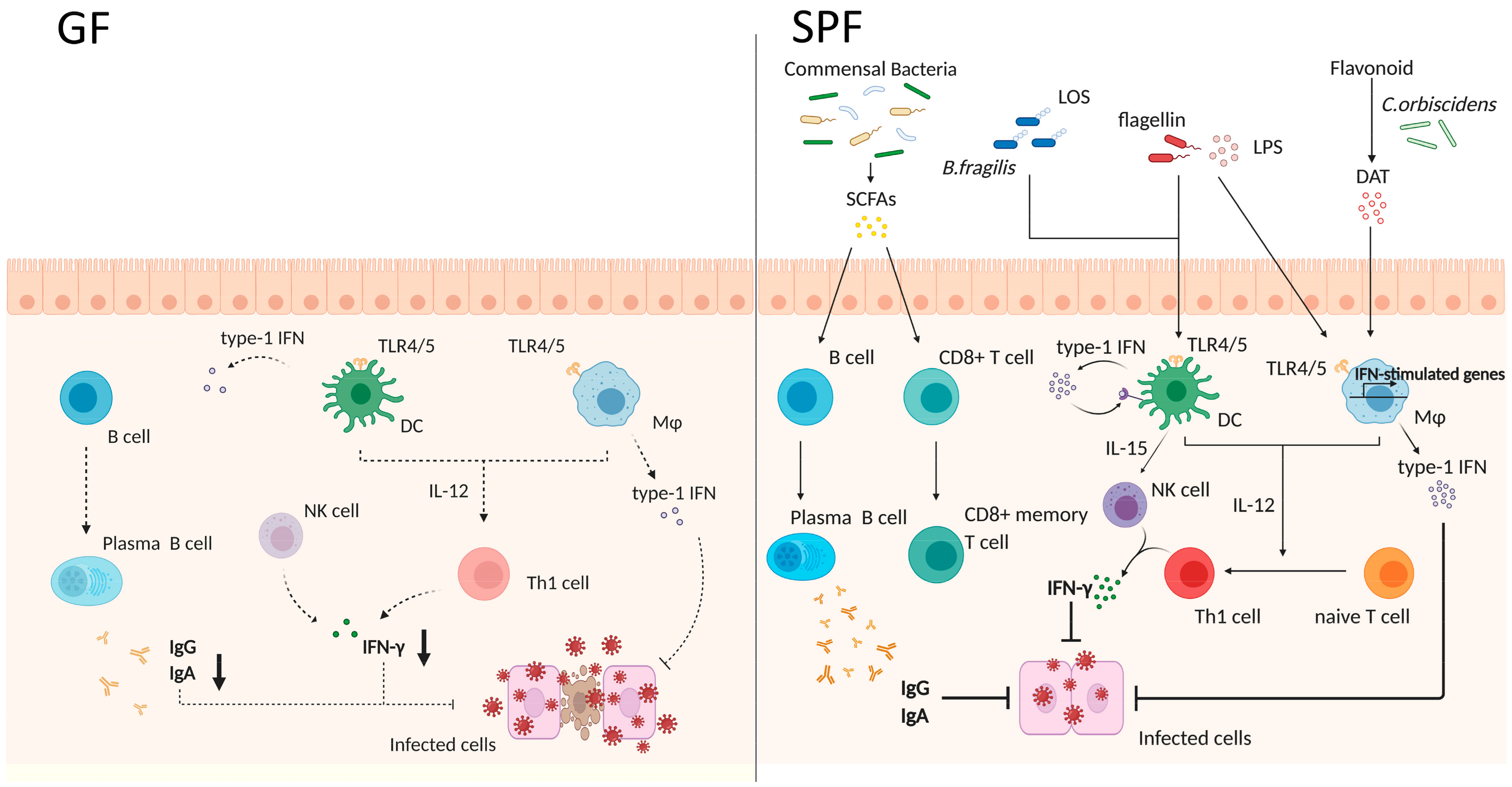
5. The Gut Microbiota and Vaccine (Adjuvant) Efficacy
5.1. The Gut Microbiota and Immunity
5.2. Vaccine Efficacy and Gut Microbiota
5.3. Gut Microbiota Acts Similar to an Adjuvant and Protects Hosts against Several Infections through the Activation of Host Immunity
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Available online: https://www.who.int/csr/resources/publications/surveillance/plague.pdf (accessed on 19 January 2021).
- Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/flu/pandemic-resources/1918-pandemic-h1n1.html (accessed on 19 January 2021).
- Our World in Data. Available online: https://ourworldindata.org/covid-deaths (accessed on 19 January 2021).
- Riedel, S. Edward Jenner and the History of Smallpox and Vaccination. Baylor Univ. Med. Cent. Proc. 2005, 18, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Coffman, R.L.; Sher, A.; Seder, R.A. Vaccine adjuvants: Putting innate immunity to work. Immunity 2010, 33, 492–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.G. Microbiota influences vaccine and mucosal adjuvant efficacy. Immune Netw. 2017, 17, 20–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Our World in Data. Available online: https://ourworldindata.org/causes-of-death (accessed on 10 December 2020).
- WHO. Available online: https://www.who.int/health-topics/tuberculosis#tab=tab_1 (accessed on 10 December 2020).
- Sulis, G.; Roggi, A.; Matteelli, A.; Raviglione, M.C. Tuberculosis: Epidemiology and Control. Mediterr. J. Hematol. Infect. Dis. 2014, 6, e2014070. [Google Scholar] [CrossRef]
- Randall, P.J.; Hsu, N.J.; Quesniaux, V.; Ryffel, B.; Jacobs, M. Mycobacterium tuberculosis infection of the “non-classical immune cell”. Immunol. Cell. Biol. 2015, 93, 789–795. [Google Scholar] [CrossRef]
- Zeng, G.; Zhang, G.; Chen, X. Th1 cytokines, true functional signatures for protective immunity against TB? Cell. Mol. Immunol. 2018, 15, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.M.; Dalton, D.K.; Stewart, T.A.; Griffin, J.P.; Russell, D.G.; Orme, I.M. Disseminated tuberculosis in interferon γ Gene-disrupted mice. J. Exp. Med. 1993, 178, 2243–2247. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.M.; Magram, J.; Ferrante, J.; Orme, I.M. Interleukin 12 (IL-12) is crucial to the development of protective immunity in mice intravenously infected with mycobacterium tuberculosis. J. Exp. Med. 1997, 186, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Mogues, T.; Goodrich, M.E.; Ryan, L.; LaCourse, R.; North, R.J. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J. Exp. Med. 2001, 193, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Getahun, H.; Matteelli, A.; Chaisson, R.E.; Raviglione, M. Latent Mycobacterium tuberculosis Infection. N. Engl. J. Med. 2015, 372, 2127–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. BCG vaccine: WHO position paper, February 2018; Recommendations. Vaccine 2018, 36, 3408–3410. [Google Scholar] [CrossRef] [PubMed]
- Libraty, D.H.; Zhang, L.; Woda, M.; Acosta, L.P.; Obcena, A.; Brion, J.D.; Capeding, R.Z. Neonatal BCG vaccination is associated with enhanced T-helper 1 immune responses to heterologous infant vaccines. Trials Vaccinol. 2014, 3, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boer, M.C.; Prins, C.; Van Meijgaarden, K.E.; Van Dissel, J.T.; Ottenhoff, T.H.M.; Joosten, S.A. Mycobacterium bovis BCG vaccination induces divergent proinflammatory or regulatory T cell responses in adults. Clin. Vaccine Immunol. 2015, 22, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.G.; Zelmer, A.; Blitz, R.; Fletcher, H.A.; Dockrell, H.M. Polyfunctional CD4 T-cells correlate with in vitro mycobacterial growth inhibition following Mycobacterium bovis BCG-vaccination of infants. Vaccine 2016, 34, 5298–5305. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, M.; Liao, M.; Graner, M.W.; Wu, C.; Yang, Q.; Liu, H.; Zhou, B. Reduced Th17 response in patients with tuberculosis correlates with IL-6R expression on CD4+ T cells. Am. J. Respir. Crit. Care Med. 2010, 181, 734–742. [Google Scholar] [CrossRef]
- Shen, H.; Chen, Z.W. The crucial roles of Th17-related cytokines/signal pathways in M. tuberculosis infection. Cell Mol. Immunol. 2018, 15, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Khader, S.A.; Bell, G.K.; Pearl, J.E.; Fountain, J.J.; Rangel-Moreno, J.; Cilley, G.E.; Shen, F.; Eaton, S.M.; Gaffen, S.L.; Swain, S.L.; et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol. 2007, 8, 369–377. [Google Scholar] [CrossRef]
- Lombard, R.; Doz, E.; Carreras, F.; Epardaud, M.; Le Vern, Y.; Buzoni-Gatel, D.; Winter, N. IL-17RA in non-hematopoietic cells controls CXCL-1 and 5 critical to recruit neutrophils to the lung of mycobacteria-infected mice during the adaptive immune response. PLoS ONE 2016, 11, e0149455. [Google Scholar] [CrossRef] [Green Version]
- Segueni, N.; Tritto, E.; Bourigault, M.L.; Rose, S.; Erard, F.; Le Bert, M.; Jacobs, M.; Di Padova, F.; Stiehl, D.P.; Moulin, P.; et al. Controlled Mycobacterium tuberculosis infection in mice under treatment with anti-IL-17A or IL-17F antibodies, in contrast to TNFα neutralization. Sci. Rep. 2016, 6, 36923. [Google Scholar] [CrossRef] [Green Version]
- Andersen, P.; Woodworth, J.S. Tuberculosis vaccines-rethinking the current paradigm. Trends Immunol. 2014, 35, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Brooks, L.R.K.; Mias, G.I. Streptococcus pneumoniae’s virulence and host immunity: Aging, diagnostics, and prevention. Front. Immunol. 2018, 9, 1366. [Google Scholar] [CrossRef] [PubMed]
- WHO. Available online: https://www.who.int/life-course/news/events/2017-world-pneumonia-day/en/ (accessed on 19 January 2021).
- Institute for Health Metrics and Evaluation (IHME). Available online: https://vizhub.healthdata.org/gbd-compare/ (accessed on 10 December 2020).
- Falco, V.; De Sevilla, T.F.; Alegre, J.; Barbe, J.; Ferrer, A.; Ocana, I.; Ribera, E.; Martinez-Vazquez, J.M. Bacterial pneumonia in HIV-infected patients: A prospective study of 68 episodes. Eur. Respir. J. 1994, 7, 235–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuyama, Y.; King, J.D.; Kataoka, K.; Kobayashi, R.; Gilbert, R.S.; Oishi, K.; Hollingshead, S.K.; Briles, D.E.; Fujihashi, K. Secretory-IgA Antibodies Play an Important Role in the Immunity to Streptococcus pneumoniae. J. Immunol. 2010, 185, 1755–1762. [Google Scholar] [CrossRef] [Green Version]
- Hoe, E.; Anderson, J.; Nathanielsz, J.; Toh, Z.Q.; Marimla, R.; Balloch, A.; Licciardi, P.V. The contrasting roles of Th17 immunity in human health and disease. Microbiol. Immunol. 2017, 61, 49–56. [Google Scholar] [CrossRef]
- Mook-Kanamori, B.B.; Geldhoff, M.; van der Poll, T.; van de Beek, D. Pathogenesis and pathophysiology of pneumococcal meningitis. Clin. Microbiol. Rev. 2011, 24, 557–591. [Google Scholar] [CrossRef] [Green Version]
- Aghamohammadi, A.; Cheraghi, T.; Gharagozlou, M.; Movahedi, M.; Rezaei, N.; Yeganeh, M.; Parvaneh, N.; Abolhassani, H.; Pourpak, Z.; Moin, M. IgA deficiency: Correlation between clinical and immunological phenotypes. J. Clin. Immunol. 2009, 29, 130–136. [Google Scholar] [CrossRef]
- Keller, L.E.; Robinson, D.A.; McDaniel, L.S. Nonencapsulated Streptococcus pneumoniae: Emergence and pathogenesis. MBio 2016, 7, e01792. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, Z.; Wan, Z.; Kilby, A.; Kilby, J.M.; Jiang, W. Humoral immune responses to Streptococcus pneumoniae in the setting of HIV-1 infection. Vaccine 2015, 33, 4430–4436. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention. Recommended immunization schedules for persons aged 0 through 18 years. MMWR Morb. Mortal Wkly. Rep. 2012, 61, 1–4. [Google Scholar]
- Musher Daniel, M. Available online: https://www.uptodate.com/contents/pneumococcal-vaccination-in-adults (accessed on 10 December 2020).
- Pollard, A.J.; Perrett, K.P.; Beverley, P.C. Maintaining protection against invasive bacteria with protein- polysaccharide conjugate vaccines. Nat. Rev. Immunol. 2009, 9, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.L.; Seon, S.H.; Rhee, D.K. Pneumonia and Streptococcus pneumoniae vaccine. Arch. Pharm. Res. 2017, 40, 885–893. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. Available online: https://www.fda.gov/media/80547/download (accessed on 10 December 2020).
- Westerink, M.A.J.; Schroeder, H.W.; Nahm, M.H. Immune responses to pneumococcal vaccines in children and adults: Rationale for age-specific vaccination. Aging Dis. 2012, 3, 51–67. [Google Scholar] [PubMed]
- WHO. Available online: https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal)#:~:text=Worldwide%2C%20these%20annual%20epidemics%20are,65%20or%20older%20(1) (accessed on 10 December 2020).
- Palese, P. Influenza: Old and new threats. Nat. Med. 2004, 10, S82–S87. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Webster, R.G.; Webby, R.J. Influenza Virus: Dealing with a Drifting and Shifting Pathogen. Viral Immunol. 2018, 31, 174–183. [Google Scholar] [CrossRef]
- Van Riet, E.; Ainai, A.; Suzuki, T.; Hasegawa, H. Mucosal IgA responses in influenza virus infections; thoughts for vaccine design. Vaccine 2012, 30, 5893–5900. [Google Scholar] [CrossRef]
- Krammer, F. The human antibody response to influenza a virus infection and vaccination. Nat. Rev. Immunol. 2019, 19, 383–397. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Palese, P.; Wilson, P.C.; Ravetch, J.V. Broadly neutralizing anti-influenza antibodies require Fc receptor engagement for in vivo protection. J. Clin. Investig. 2016, 126, 605–610. [Google Scholar] [CrossRef]
- Zens, K.D.; Chen, J.K.; Farber, D.L. Vaccine-generated lung tissue–resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight 2019, 1, e85832. [Google Scholar] [CrossRef]
- Pizzolla, A.; Nguyen, T.H.O.; Smith, J.M.; Brooks, A.G.; Kedzierska, K.; Heath, W.R.; Reading, P.C.; Wakim, L.M. Resident memory CD8+ T cells in the upper respiratory tract prevent pulmonary influenza virus infection. Sci. Immunol. 2017, 2, eaam6970. [Google Scholar] [CrossRef]
- Deng, L.; Cho, K.J.; Fiers, W.; Saelens, X. M2e-based universal influenza a vaccines. Vaccines 2015, 3, 105–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saelens, X. The Role of Matrix Protein 2 Ectodomain in the Development of Universal Influenza Vaccines. J. Infect. Dis. 2019, 219, S68–S74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.R.; Liu, Y.M.; Tseng, Y.C.; Ma, C. Better influenza vaccines: An industry perspective. J. Biomed. Sci. 2020, 27, 33. [Google Scholar] [CrossRef] [PubMed]
- HogenEsch, H. Mechanisms of stimulation of the immune response by aluminum adjuvants. Vaccine 2002, 20, S34–S39. [Google Scholar] [CrossRef]
- Glenny, A.T.; Pope, C.G. The antigenic effect of intravenous injection of diphtheria toxin. J. Pathol. Bacteriol. 1925, 28, 273–278. [Google Scholar] [CrossRef]
- Li, H.; Willingham, S.B.; Ting, J.P.-Y.; Re, F. Cutting Edge: Inflammasome Activation by Alum and Alum’s Adjuvant Effect Are Mediated by NLRP3. J. Immunol. 2008, 181, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef]
- Eisenbarth, S.C.; Colegio, O.R.; O’Connor, W.; Sutterwala, F.S.; Flavell, R.A. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 2008, 453, 1122–1126. [Google Scholar] [CrossRef]
- Kool, M.; Pétrilli, V.; De Smedt, T.; Rolaz, A.; Hammad, H.; van Nimwegen, M.; Bergen, I.M.; Castillo, R.; Lambrecht, B.N.; Tschopp, J. Cutting Edge: Alum Adjuvant Stimulates Inflammatory Dendritic Cells through Activation of the NALP3 Inflammasome. J. Immunol. 2008, 181, 3755–3759. [Google Scholar] [CrossRef] [Green Version]
- Kool, M.; Soullié, T.; Van Nimwegen, M.; Willart, M.A.M.; Muskens, F.; Jung, S.; Hoogsteden, H.C.; Hammad, H.; Lambrecht, B.N. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J. Exp. Med. 2008, 205, 869–882. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braga, T.T.; Forni, M.F.; Correa-Costa, M.; Ramos, R.N.; Barbuto, J.A.; Branco, P.; Castoldi, A.; Hiyane, M.I.; Davanso, M.R.; Latz, E.; et al. Soluble Uric Acid Activates the NLRP3 Inflammasome. Sci. Rep. 2017, 7, 39884. [Google Scholar] [CrossRef] [PubMed]
- Marichal, T.; Ohata, K.; Bedoret, D.; Mesnil, C.; Sabatel, C.; Kobiyama, K.; Lekeux, P.; Coban, C.; Akira, S.; Ishii, K.J.; et al. DNA released from dying host cells mediates aluminum adjuvant activity. Nat. Med. 2011, 17, 996–1002. [Google Scholar] [CrossRef]
- Brand, D.D.; Latham, K.A.; Rosloniec, E.F. Collagen-induced arthritis. Nat. Protoc. 2007, 2, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Shenderov, K.; Barber, D.L.; Mayer-Barber, K.D.; Gurcha, S.S.; Jankovic, D.; Feng, C.G.; Oland, S.; Hieny, S.; Caspar, P.; Yamasaki, S.; et al. Cord Factor and Peptidoglycan Recapitulate the Th17-Promoting Adjuvant Activity of Mycobacteria through Mincle/CARD9 Signaling and the Inflammasome. J. Immunol. 2013, 190, 5722–5730. [Google Scholar] [CrossRef]
- Su, S.B.; Silver, P.B.; Grajewski, R.S.; Agarwal, R.K.; Tang, J.; Chan, C.-C.; Caspi, R.R. Essential Role of the MyD88 Pathway, but Nonessential Roles of TLRs 2, 4, and 9, in the Adjuvant Effect Promoting Th1-Mediated Autoimmunity. J. Immunol. 2005, 175, 6303–6310. [Google Scholar] [CrossRef] [Green Version]
- Fritz, J.H.; Le Bourhis, L.; Sellge, G.; Magalhaes, J.G.; Fsihi, H.; Kufer, T.A.; Collins, C.; Viala, J.; Ferrero, R.L.; Girardin, S.E.; et al. Nod1-Mediated Innate Immune Recognition of Peptidoglycan Contributes to the Onset of Adaptive Immunity. Immunity 2007, 26, 445–459. [Google Scholar] [CrossRef] [Green Version]
- Indrigo, J.; Hunter, R.L.; Actor, J.K. Influence of trehalose 6,6′-dimycolate (TDM) during mycobacterial infection of bone marrow macrophages. Microbiology 2002, 148, 1991–1998. [Google Scholar] [CrossRef] [Green Version]
- Schoenen, H.; Bodendorfer, B.; Hitchens, K.; Manzanero, S.; Werninghaus, K.; Nimmerjahn, F.; Agger, E.M.; Stenger, S.; Andersen, P.; Ruland, J.; et al. Cutting Edge: Mincle Is Essential for Recognition and Adjuvanticity of the Mycobacterial Cord Factor and its Synthetic Analog Trehalose-Dibehenate. J. Immunol. 2010, 184, 2756–2760. [Google Scholar] [CrossRef] [Green Version]
- Thieu, V.T.; Yu, Q.; Chang, H.C.; Yeh, N.; Nguyen, E.T.; Sehra, S.; Kaplan, M.H. Signal Transducer and Activator of Transcription 4 Is Required for the Transcription Factor T-bet to Promote T Helper 1 Cell-Fate Determination. Immunity 2008, 29, 679–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, M.B.; Williams, S.J. MCL and Mincle: C-type lectin receptors that sense damaged self and pathogen-associated molecular patterns. Front. Immunol. 2014, 5, 288. [Google Scholar] [CrossRef] [PubMed]
- Magram, J.; Connaughton, S.E.; Warrier, R.R.; Carvajal, D.M.; Wu, C.Y.; Ferrante, J.; Stewart, C.; Sarmiento, U.; Faherty, D.A.; Gately, M.K. IL-12-deficient mice are defective in IFNγ production and type 1 cytokine responses. Immunity 1996, 4, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, Y.; Chang, S.H.; Martinez, G.J.; Yang, X.O.; Nurieva, R.; Kang, H.S.; Ma, L.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Critical Regulation of Early Th17 Cell Differentiation by Interleukin-1 Signaling. Immunity 2009, 30, 576–587. [Google Scholar] [CrossRef] [Green Version]
- Ciraci, C.; Janczy, J.R.; Jain, N.; Haasken, S.; ESilva, C.P.; Benjamim, C.F.; Sadler, J.J.; Olivier, A.K.; Iwakura, Y.; Shayakhmetov, D.M.; et al. Immune complexes indirectly suppress the generation of Th17 Responses In Vivo. PLoS ONE 2016, 11, e0151252. [Google Scholar] [CrossRef]
- Freytag, L.C.; Clements, J.D. Mucosal adjuvants. Vaccine 2005, 23, 1804–1813. [Google Scholar] [CrossRef]
- Porgador, A.; Staats, H.F.; Itoh, Y.; Kelsall, B.L. Intranasal immunization with cytotoxic T-lymphocyte epitope peptide and mucosal adjuvant cholera toxin: Selective augmentation of peptide-presenting dendritic cells in nasal mucosa-associated lymphoid tissue. Infect. Immun. 1998, 66, 5876–5881. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.K.; Sabet, M.; Nguyen, K.P.L.; Valdez, P.A.; Gonzalez-Navajas, J.M.; Islam, S.; Mihajlov, I.; Fierer, J.; Insel, P.A.; Webster, N.J.; et al. Mucosal adjuvant activity of cholera toxin requires Th17 cells and protects against inhalation anthrax. Proc. Natl. Acad. Sci. USA 2010, 107, 10638–10643. [Google Scholar] [CrossRef] [Green Version]
- Xu-Amano, J.; Kiyono, H.; Jackson, R.J.; Staats, H.F.; Fujihashi, K.; Burrows, P.D.; Elson, C.O.; Pillai, S.; McGhee, J.R. Helper T cell subsets for immunoglobulin a responses: Oral inmaunization with tetanus toxoid and cholera toxinn as adjuvant selectively induces th2 cells in mucosa associated tissues. J. Exp. Med. 1993, 178, 1309–1320. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Kiyono, H.; Yamamoto, M.; Imaoka, K.; Yamamoto, M.; Fujihashi, K.; Van Ginkel, F.W.; Noda, M.; Takeda, Y.; McGhee, J.R. A nontoxic mutant of cholera toxin elicits Th2-type responses for enhanced mucosal immunity. Proc. Natl. Acad. Sci. USA 1997, 94, 5267–5272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Murray, F.; Koide, N.; Goldstone, J.; Dann, S.M.; Chen, J.; Bertin, S.; Fu, G.; Weinstein, L.S.; Chen, M.; et al. Divergent requirement for Gαs and cAMP in the differentiation and inflammatory profile of distinct mouse Th subsets. J. Clin. Investig. 2012, 122, 963–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Kim, Y.G.; Seo, S.U.; Kim, D.J.; Kamada, N.; Prescott, D.; Philpott, D.J.; Rosenstiel, P.; Inohara, N.; Núñez, G. Nod2-mediated recognition of the microbiota is critical for mucosal adjuvant activity of cholera toxin. Nat. Med. 2016, 22, 524–530. [Google Scholar] [CrossRef]
- Shaw, M.H.; Reimer, T.; Kim, Y.G.; Nuñez, G. NOD-like receptors (NLRs): Bona fide intracellular microbial sensors. Curr. Opin. Immunol. 2008, 20, 377–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warner, N.; Núñez, G. MyD88: A Critical Adaptor Protein in Innate Immunity Signal Transduction. J. Immunol. 2013, 190, 3–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kim, Y.M.; Kim, W.U.; Park, J.H.; Núñez, G.; Seo, S.U. Recognition of the microbiota by Nod2 contributes to the oral adjuvant activity of cholera toxin through the induction of interleukin-1β. Immunology 2019, 158, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Barbet, G.; Sander, L.E.; Geswell, M.; Leonardi, I.; Cerutti, A.; Iliev, I.; Blander, J.M. Sensing Microbial Viability through Bacterial RNA Augments T Follicular Helper Cell and Antibody Responses. Immunity 2018, 48, 584–598.e5. [Google Scholar] [CrossRef] [Green Version]
- Crotty, S. T Follicular Helper Cell Differentiation, Function, and Roles in Disease. Immunity 2014, 41, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Xiao, Y.; Huang, X.; Chen, F.; Sun, M.; Bilotta, A.J.; Xu, L.; Lu, Y.; Yao, S.; Zhao, Q.; et al. Microbiota Metabolite Short-Chain Fatty Acids Facilitate Mucosal Adjuvant Activity of Cholera Toxin through GPR43. J. Immunol. 2019, 203, 282–292. [Google Scholar] [CrossRef]
- Wang, J.; Li, P.; Yu, Y.; Fu, Y.; Jiang, H.; Lu, M.; Sun, Z.; Jiang, S.; Lu, L.; Wu, M.X. Pulmonary surfactant-biomimetic nanoparticles potentiate heterosubtypic influenza immunity. Science 2020, 367, eaau0810. [Google Scholar] [CrossRef] [PubMed]
- Han, S.H.; Mallampalli, R.K. The role of surfactant in lung disease and host defense against pulmonary infections. Ann. Am. Thorac. Soc. 2015, 12, 765–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novakovic, B.; Habibi, E.; Wang, S.Y.; Arts, R.J.W.; Davar, R.; Megchelenbrink, W.; Kim, B.; Kuznetsova, T.; Kox, M.; Zwaag, J.; et al. β-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 2016, 167, 1354–1368.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintin, J.; Saeed, S.; Martens, J.H.A.; Giamarellos-Bourboulis, E.J.; Ifrim, D.C.; Logie, C.; Jacobs, L.; Jansen, T.; Kullberg, B.J.; Wijmenga, C.; et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. 2012, 12, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.A.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef] [Green Version]
- Wahl, D.R.; Byersdorfer, C.A.; Ferrara, J.L.M.; Opipari, A.W.; Glick, G.D. Distinct metabolic programs in activated T cells: Opportunities for selective immunomodulation. Immunol. Rev. 2012, 249, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Arts, R.J.W.; Carvalho, A.; La Rocca, C.; Palma, C.; Rodrigues, F.; Silvestre, R.; Kleinnijenhuis, J.; Lachmandas, E.; Gonçalves, L.G.; Belinha, A.; et al. Immunometabolic Pathways in BCG-Induced Trained Immunity. Cell Rep. 2016, 17, 2562–2571. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, E.; Sanz, J.; Dunn, J.L.; Khan, N.; Mendonça, L.E.; Pacis, A.; Tzelepis, F.; Pernet, E.; Dumaine, A.; Grenier, J.C.; et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 2018, 172, 176–190.e19. [Google Scholar] [CrossRef] [Green Version]
- Mitroulis, I.; Ruppova, K.; Wang, B.; Chen, L.S.; Grzybek, M.; Grninko, T.; Eugster, A.; Troullinaki, M.; Palladini, A.; Kourtzelis, I.; et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 2018, 172, 147–161.e12. [Google Scholar] [CrossRef] [Green Version]
- Abate, G.; Hamzabegovic, F.; Eickhoff, C.S.; Hoft, D.F. BCG vaccination induces M. avium and M. abscessus cross-protective immunity. Front. Immunol. 2019, 10, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benn, C.S.; Netea, M.G.; Selin, L.K.; Aaby, P. A Small Jab—A Big Effect: Nonspecific Immunomodulation By Vaccines. Trends Immunol. 2013, 34, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Arts, R.J.W.; Joosten, L.A.B.; Netea, M.G. The potential role of trained immunity in autoimmune and autoinflammatory disorders. Front. Immunol. 2018, 9, 298. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Kundu, P.; Blacher, E.; Elinav, E.; Pettersson, S. Our Gut Microbiome: The Evolving Inner Self. Cell 2017, 171, 1481–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.G.; Sakamoto, K.; Seo, S.U.; Pickard, J.M.; Gillilland, M.G.; Pudlo, N.A.; Hoostal, M.; Li, X.; Wang, T.D.; Feehley, T.; et al. Neonatal acquisition of Clostridia species protects against colonization by bacterial pathogens. Science 2017, 356, 315–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef]
- Abdel-gadir, A.; Stephen-victor, E.; Gerber, G.K.; Rivas, M.N.; Wang, S.; Harb, H.; Wang, L.; Li, N.; Crestani, E.; Spielman, S.; et al. Microbiota therapy acts via a regulatory T cell MyD88 / ROR γ t pathway to suppress food allergy. Nat. Med. 2019, 25, 1458. [Google Scholar] [CrossRef]
- Kim, Y.-G.; Udayanga, K.G.S.; Totsuka, N.; Weinberg, J.B.; Núñez, G.; Shibuya, A. Gut Dysbiosis Promotes M2 Macrophage Polarization and Allergic Airway Inflammation via Fungi-Induced PGE2. Cell Host Microbe 2014, 15, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariño, E.; Richards, J.L.; Mcleod, K.H.; Stanley, D.; Yap, Y.A.; Knight, J.; McKenzie, C.; Kranich, J.; Oliveira, A.C.; Rossello, F.J.; et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat. Immunol. 2017, 18, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Wittung-stafshede, P.; Knight, R.; Mazmanian, S.K.; Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isobe, J.; Maeda, S.; Obata, Y.; Iizuka, K.; Nakamura, Y.; Fujimura, Y.; Kimizuka, T.; Hattori, K.; Kim, Y.-G.; Morita, T.; et al. Commensal-bacteria-derived butyrate promotes the T-cell-independent IgA response in the colon. Int. Immunol. 2020, 32, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; Alvarado, L.J.; Kim, T.; Lehmann, M.L.; Cho, H.; He, J.; Li, P.; Kim, B.H.; Larochelle, A.; Kelsall, B.L. Commensal microbiota drive the functional diversification of colon macrophages. Mucosal Immunol. 2020, 13, 216–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catanzaro, J.R.; Strauss, J.D.; Bielecka, A.; Porto, A.F.; Lobo, F.M.; Urban, A.; Schofield, W.B.; Palm, N.W. IgA-deficient humans exhibit gut microbiota dysbiosis despite secretion of compensatory IgM. Sci. Rep. 2019, 9, 13574. [Google Scholar] [CrossRef]
- Kawamoto, S.; Maruya, M.; Kato, L.M.; Suda, W.; Atarashi, K.; Doi, Y.; Tsutsui, Y.; Qin, H.; Honda, K.; Okada, T.; et al. Foxp3+T Cells Regulate Immunoglobulin A Selection and Facilitate Diversification of Bacterial Species Responsible for Immune Homeostasis. Immunity 2014, 41, 152–165. [Google Scholar] [CrossRef] [Green Version]
- Earley, A.M.; Graves, C.L.; Shiau, C.E. Critical Role for a Subset of Intestinal Macrophages in Shaping Gut Microbiota in Adult Zebrafish. Cell Rep. 2018, 25, 424–436. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell. Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Di, Y.u.; Schilter, H.C.; Rolph, M.S.; MacKay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, C.E.; Kim, C.; Weyand, C.M.; Goronzy, J.J. Influence of immune aging on vaccine responses. J. Allergy Clin. Immunol. 2020, 145, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Ellwanger, J.H.; Chies, J.A.B. Host genetic factors can impact vaccine immunogenicity and effectiveness. Lancet Infect. Dis. 2019, 19, 359–360. [Google Scholar] [CrossRef] [Green Version]
- Lamousé-Smith, E.S.; Tzeng, A.; Starnbach, M.N. The intestinal flora is required to support antibody responses to systemic immunization in infant and germ free mice. PLoS ONE 2011, 6, e27662. [Google Scholar] [CrossRef] [Green Version]
- Nadeem, S.; Maurya, S.K.; Das, D.K.; Khan, N.; Agrewala, J.N. Gut Dysbiosis Thwarts the Efficacy of Vaccine Against Mycobacterium tuberculosis. Front. Immunol. 2020, 11, 726. [Google Scholar] [CrossRef]
- Huda, M.N.; Lewis, Z.; Kalanetra, K.M.; Rashid, M.; Ahmad, S.M.; Raqib, R.; Qadri, F.; Underwood, M.A.; Mills, D.A.; Stephensen, C.B. Stool microbiota and vaccine responses of infants. Pediatrics 2014, 134, e362–e372. [Google Scholar] [CrossRef] [Green Version]
- Harris, V.; Ali, A.; Fuentes, S.; Korpela, K.; Kazi, M.; Tate, J.; Parashar, U.; Wiersinga, W.J.; Giaquinto, C.; de Weerth, C.; et al. Rotavirus vaccine response correlates with the infant gut microbiota composition in Pakistan. Gut Microbes 2018, 9, 93–101. [Google Scholar] [CrossRef]
- Hagan, T.; Cortese, M.; Rouphael, N.; Boudreau, C.; Linde, C.; Maddur, M.S.; Das, J.; Wang, H.; Guthmiller, J.; Zheng, N.Y.; et al. Antibiotics-Driven Gut Microbiome Perturbation Alters Immunity to Vaccines in Humans. Cell 2019, 178, 1313–1328.e13. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Chen, H.; Zhang, S.; Zhuang, J.; Li, Q.; Feng, Z.-C. Intestinal Microbiota in Early Life and Its Implications on Childhood Health. Genom. Proteom. Bioinform. 2019, 17, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferretti, P.; Pasolli, E.; Tett, A.; Asnicar, F.; Gorfer, V.; Fedi, S.; Armanini, F.; Truong, D.T.; Manara, S.; Zolfo, M.; et al. Mother-to-Infant Microbial Transmission from Different Body Sites Shapes the Developing Infant Gut Microbiome. Cell Host Microbe 2018, 24, 133–145. [Google Scholar] [CrossRef]
- Kristensen, K.; Henriksen, L. Cesarean section and disease associated with immune function. J. Allergy Clin. Immunol. 2016, 137, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Fehr, K.; Moossavi, S.; Sbihi, H.; Boutin, R.C.; Bode, L.; Robertson, B.; Yonemitsu, C.; Field, C.J.; Becker, A.B.; Mandhane, P.J.; et al. Breastmilk Feeding Practices Are Associated with the Co-Occurrence of Bacteria in Mothers’ Milk and the Infant Gut: The CHILD Cohort Study. Cell Host Microbe 2020, 28, 285–297. [Google Scholar] [CrossRef]
- Lynn, M.A.; Tumes, D.J.; Choo, J.M.; Sribnaia, A.; Blake, S.J.; Leong, L.E.X.; Young, G.P.; Marshall, H.S.; Wesselingh, S.L.; Rogers, G.B.; et al. Early-Life Antibiotic-Driven Dysbiosis Leads to Dysregulated Vaccine Immune Responses in Mice. Cell Host Microbe 2018, 23, 653–660. [Google Scholar] [CrossRef] [Green Version]
- Vos, A.P.; Haarman, M.; Buco, A.; Govers, M.; Knol, J.; Garssen, J.; Stahl, B.; Boehm, G.; M’Rabet, L. A specific prebiotic oligosaccharide mixture stimulates delayed-type hypersensitivity in a murine influenza vaccination model. Int. Immunopharmacol. 2006, 6, 1277–1286. [Google Scholar] [CrossRef]
- Xiao, L.; Engen, P.A.; Leusink-Muis, T.; Van Ark, I.; Stahl, B.; Overbeek, S.A.; Garssen, J.; Naqib, A.; Green, S.J.; Keshavarzian, A.; et al. The combination of 2-fucosyllactose with short-chain galacto-oligosaccharides and long-chain fruc-to-oligosaccharides that enhance influenza vaccine responses is associated with mucosal immune regulation in mice. J. Nutr. 2019, 149, 856–869. [Google Scholar] [CrossRef]
- Di Luccia, B.; Ahern, P.P.; Griffin, N.W.; Cheng, J.; Guruge, J.L.; Byrne, A.E.; Rodionov, D.A.; Leyn, S.A.; Osterman, A.L.; Ahmed, T.; et al. Combined Prebiotic and Microbial Intervention Improves Oral Cholera Vaccination Responses in a Mouse Model of Childhood Undernutrition. Cell Host Microbe 2020, 27, 899–908.e5. [Google Scholar] [CrossRef]
- Akatsu, H.; Iwabuchi, N.; Xiao, J.-Z.; Matsuyama, Z.; Kurihara, R.; Okuda, K.; Yamamoto, T.; Maruyama, M. Clinical Effects of Probiotic Bifidobacterium longum BB536 on Immune Function and Intestinal Microbiota in Elderly Patients Receiving Enteral Tube Feeding. J. Parenter. Enter. Nutr. 2013, 37, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.-B.; Yang, Y.; Xu, X.; Wang, W.-P. Effects of Bifidobacterium supplementation on intestinal microbiota composition and the immune response in healthy infants. World J. Pediatr. 2015, 12, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Boge, T.; Rémigy, M.; Vaudaine, S.; Tanguy, J.; Bourdet-Sicard, R.; Van Der Werf, S. A probiotic fermented dairy drink improves antibody response to influenza vaccination in the elderly in two randomised controlled trials. Vaccine 2009, 27, 5677–5684. [Google Scholar] [CrossRef] [PubMed]
- Bosch, M.; Méndez, M.; Pérez, M.; Farran, A.; Fuentes, M.C.; Cuñé, J. Lactobacillus plantarum CECT7315 and CECT7316 stimulate immunoglobulin production after influenza vaccination in elderly. Nutrición Hospitalaria 2012, 27, 504–509. [Google Scholar] [PubMed]
- Van Puyenbroeck, K.; Hens, N.; Coenen, S.; Michiels, B.; Beunckens, C.; Molenberghs, G.; Van Royen, P.; Verhoeven, V. Efficacy of daily intake of Lactobacillus casei Shirota on respiratory symptoms and influenza vaccination immune response: A randomized, double-blind, placebo-controlled trial in healthy elderly nursing home residents. Am. J. Clin. Nutr. 2012, 95, 1165–1171. [Google Scholar] [CrossRef]
- Jespersen, L.; Tarnow, I.; Eskesen, D.; Morberg, C.M.; Michelsen, B.; Bügel, S.; Dragsted, L.O.; Rijkers, G.T.; Calder, P.C. Effect of Lactobacillus paracasei subsp. paracasei, L. casei 431 on immune response to influenza vaccination and upper respiratory tract infections in healthy adult volunteers: A randomized, double-blind, placebo-controlled, parallel-group study. Am. J. Clin. Nutr. 2015, 101, 1188–1196. [Google Scholar] [CrossRef] [Green Version]
- De Jong, S.E.; Olin, A.; Pulendran, B. The Impact of the Microbiome on Immunity to Vaccination in Humans. Cell Host Microbe. 2020, 28, 169–179. [Google Scholar] [CrossRef]
- Dabbagh, K.; Lewis, D.B. Toll-like receptors and T-helper-1/T-helper-2 responses. Curr. Opin. Infect. Dis. 2003, 16, 199–204. [Google Scholar] [CrossRef]
- Oh, J.Z.; Ravindran, R.; Chassaing, B.; Carvalho, F.A.; Maddur, M.S.; Bower, M.; Hakimpour, P.; Gill, K.P.; Nakaya, H.I.; Yarovinsky, F.; et al. TLR5-Mediated Sensing of Gut Microbiota Is Necessary for Antibody Responses to Seasonal Influenza Vaccination. Immunity 2014, 41, 478–492. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.-M.; Yoo, D.-G.; Kim, M.-C.; Song, J.-M.; Park, M.-K.; O, E.; Quan, F.-S.; Akira, S.; Compans, R.W. MyD88 Plays an Essential Role in Inducing B Cells Capable of Differentiating into Antibody-Secreting Cells after Vaccination. J. Virol. 2011, 85, 11391–11400. [Google Scholar] [CrossRef] [Green Version]
- Abt, M.C.; Osborne, L.C.; Monticelli, L.A.; Doering, T.A.; Alenghat, T.; Sonnenberg, G.F.; Paley, M.A.; Antenus, M.; Williams, K.L.; Erikson, J.; et al. Commensal Bacteria Calibrate the Activation Threshold of Innate Antiviral Immunity. Immunity 2012, 37, 158–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganal, S.C.; Sanos, S.L.; Kallfass, C.; Oberle, K.; Johner, C.; Kirschning, C.; Lienenklaus, S.; Weiss, S.; Staeheli, P.; Aichele, P.; et al. Priming of Natural Killer Cells by Nonmucosal Mononuclear Phagocytes Requires Instructive Signals from Commensal Microbiota. Immunity 2012, 37, 171–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, M.; Schachterle, W.; Oberle, K.; Aichele, P.; Diefenbach, A. Dendritic Cells Prime Natural Killer Cells by trans-Presenting Interleukin 15. Immunity 2007, 26, 503–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefan, K.L.; Kim, M.V.; Iwasaki, A.; Kasper, D.L. Commensal Microbiota Modulation of Natural Resistance to Virus Infection. Cell 2020, 183, 1312–1324. [Google Scholar] [CrossRef]
- Steed, A.L.; Christophi, G.P.; Kaiko, G.E.; Sun, L.; Goodwin, V.M.; Jain, U.; Esaulova, E.; Artyomov, M.N.; Morales, D.J.; Holtzman, M.J.; et al. The microbial metabolite desaminotyrosine protects from influenza through type I in-terferon. Science. 2017, 357, 498–502. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Qie, Y.; Park, J.; Kim, C.H. Gut Microbial Metabolites Fuel Host Antibody Responses. Cell Host Microbe 2016, 20, 202–214. [Google Scholar] [CrossRef] [Green Version]
- Schulthess, J.; Pandey, S.; Capitani, M.; Rue-Albrecht, K.C.; Arnold, I.; Franchini, F.; Chomka, A.; Ilott, N.E.; Johnston, D.G.; Pires, E.; et al. The Short Chain Fatty Acid Butyrate Imprints an Antimicrobial Program in Macrophages. Immunity 2019, 50, 432–445.e7. [Google Scholar] [CrossRef] [Green Version]
- Bachem, A.; Makhlouf, C.; Binger, K.J.; Souza, D.P.; Tull, D.; Hochheiser, K.; Whitney, P.G.; Fernandez-Ruiz, D.; Dähling, S.; Kastenmüller, W.; et al. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8+ T Cells. Immunity 2019, 51, 285–297.e5. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [Green Version]


| Diseases | Pathogenic Bacteria | Current Vaccine (Components) | Key Immune Responses for Protection |
|---|---|---|---|
| Tuberculosis | Mycobacterium tuberculosis | BCG vaccine (live-attenuated M. bovis) | ・IFN-γ+ Th1 and IL-17+ Th17 cells ・Activating phagocytes and cytotoxic T cells |
| Pneumococcal pneumonia | Streptococcus pneumoniae | PPSV23 (23-valent polysaccharides only) PCV13 (carrier protein-conjugated 13-valent polysaccharides) | ・Systemic IgG and mucosal sIgA production ・IL-17+ Th17 cells ・Elimination by monocytes and macrophages recruited by IL-17-induced chemokines expression |
| Influenza virus infection | Influenza virus | Live-attenuated or inactivated (split or subunit) influenza vaccine (influenza virus A: H1N1, H3N2) (influenza virus B: 1 or 2 strains) | ・Systemic IgG and mucosal sIgA secretion against HA and NA of influenza virus ・Inducing CD8+ TRM cells in nasal and lung |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yakabe, K.; Uchiyama, J.; Akiyama, M.; Kim, Y.-G. Understanding Host Immunity and the Gut Microbiota Inspires the New Development of Vaccines and Adjuvants. Pharmaceutics 2021, 13, 163. https://doi.org/10.3390/pharmaceutics13020163
Yakabe K, Uchiyama J, Akiyama M, Kim Y-G. Understanding Host Immunity and the Gut Microbiota Inspires the New Development of Vaccines and Adjuvants. Pharmaceutics. 2021; 13(2):163. https://doi.org/10.3390/pharmaceutics13020163
Chicago/Turabian StyleYakabe, Kyosuke, Jun Uchiyama, Masahiro Akiyama, and Yun-Gi Kim. 2021. "Understanding Host Immunity and the Gut Microbiota Inspires the New Development of Vaccines and Adjuvants" Pharmaceutics 13, no. 2: 163. https://doi.org/10.3390/pharmaceutics13020163
APA StyleYakabe, K., Uchiyama, J., Akiyama, M., & Kim, Y. -G. (2021). Understanding Host Immunity and the Gut Microbiota Inspires the New Development of Vaccines and Adjuvants. Pharmaceutics, 13(2), 163. https://doi.org/10.3390/pharmaceutics13020163