
HPMA-Based Copolymers Carrying STAT3 Inhibitor Cucurbitacin-D as Stimulus-Sensitive Nanomedicines for Oncotherapy



 ,
, 
Abstract
:
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Synthesis of Monomers and Chain Transfer Agent
2.3. Synthesis of Polymer Precursors
2.3.1. Linear Polymer Precursor LP
2.3.2. Micellar Polymer Precursor MP
2.4. Synthesis of Polymer Conjugates
2.4.1. Polymer Conjugates Bearing CuD
2.4.2. Polymer Conjugates Bearing Dox
2.5. Size-Exclusion Chromatography
2.6. Ultraviolet–Visible Light (UV–Vis) Spectrophotometry
2.7. Nuclear Magnetic Resonance Spectrometry
2.8. Dynamic Light Scattering
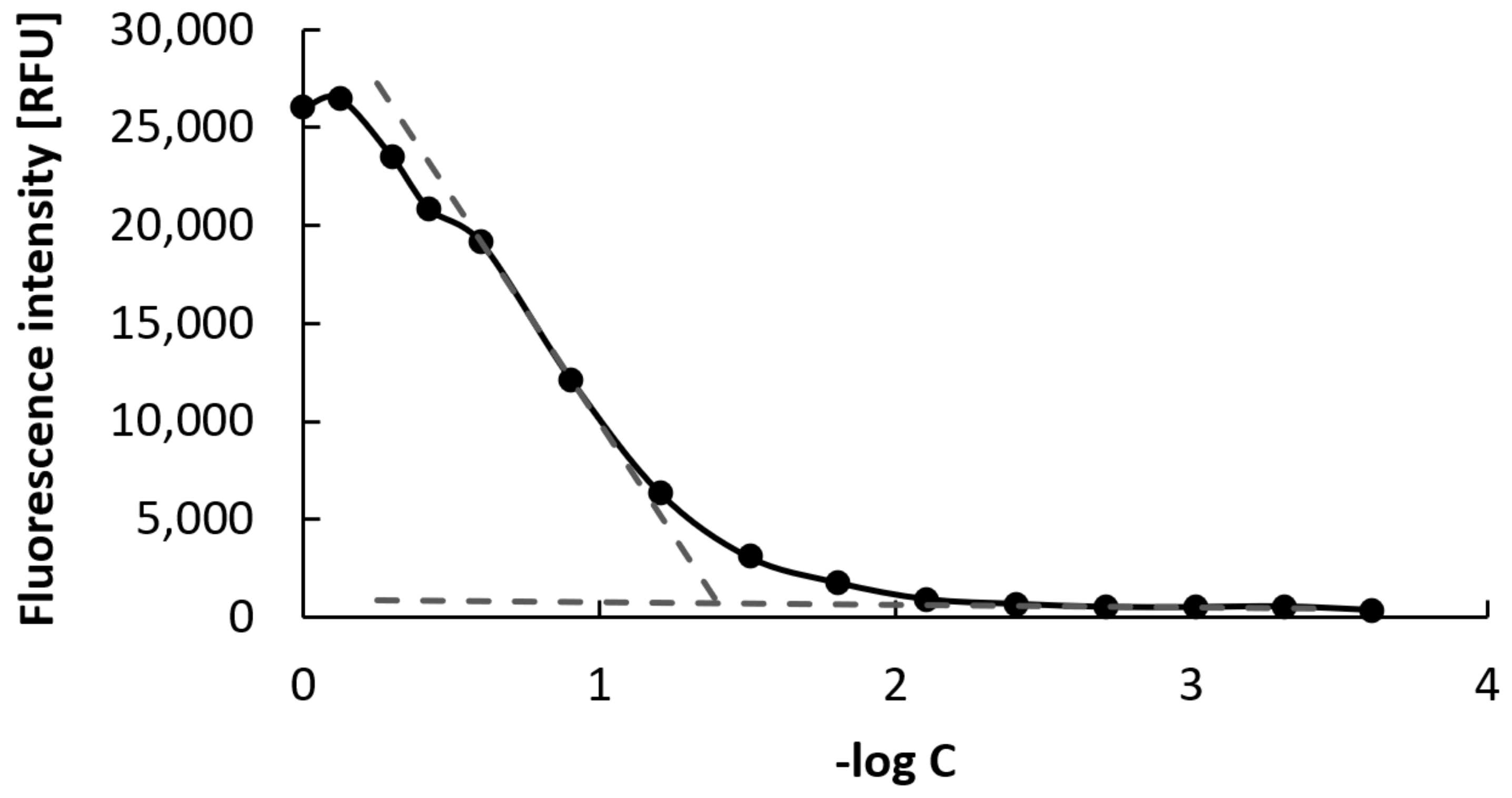
2.9. Critical Micellar Concentration Determination
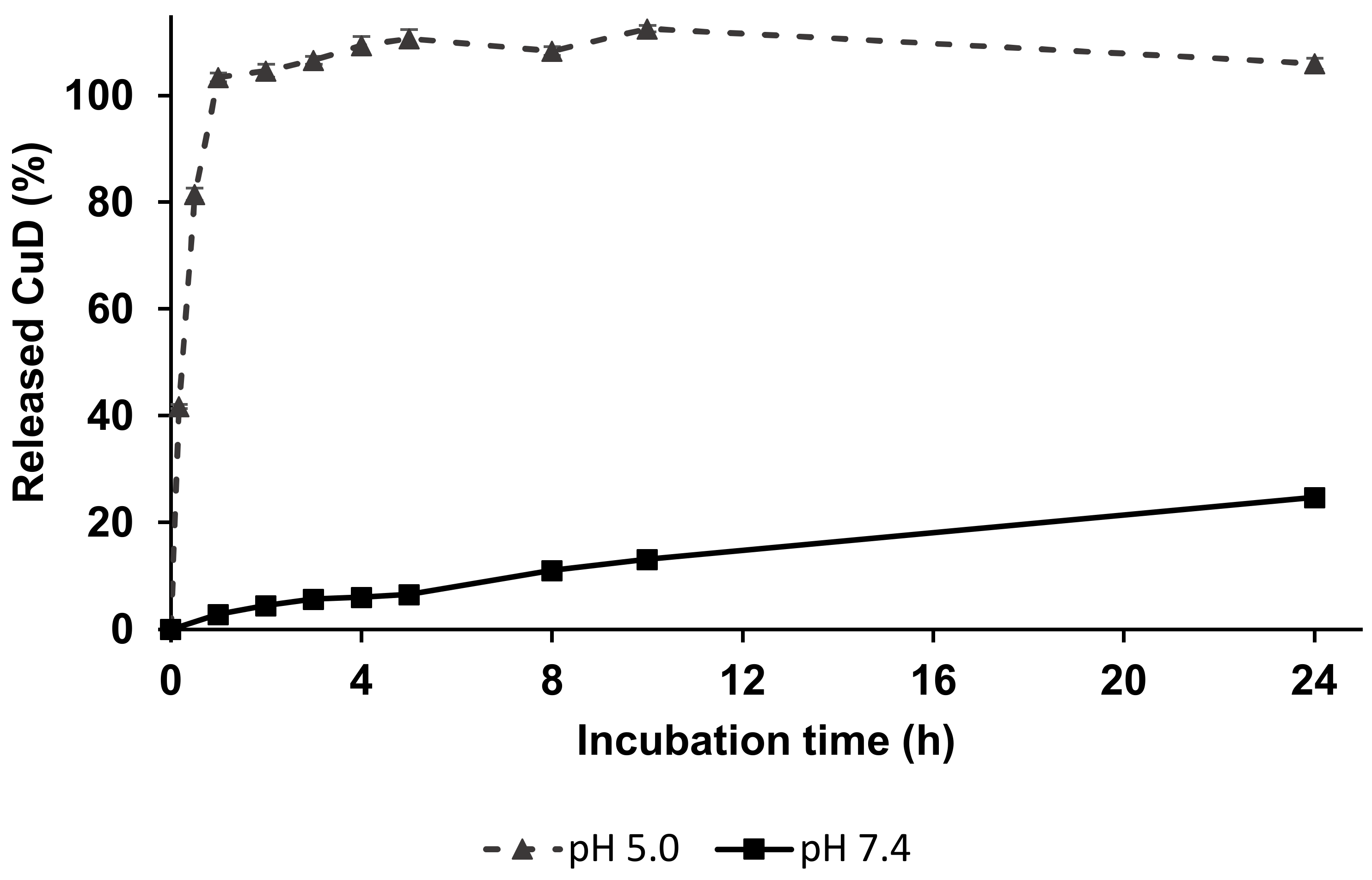
2.10. In Vitro Release of CuD from Linear Polymer–Drug Conjugate
3. Biological Evaluation
3.1. In Vitro Cytotoxicity
3.2. In Vivo Antitumor Activity
4. Results and Discussion
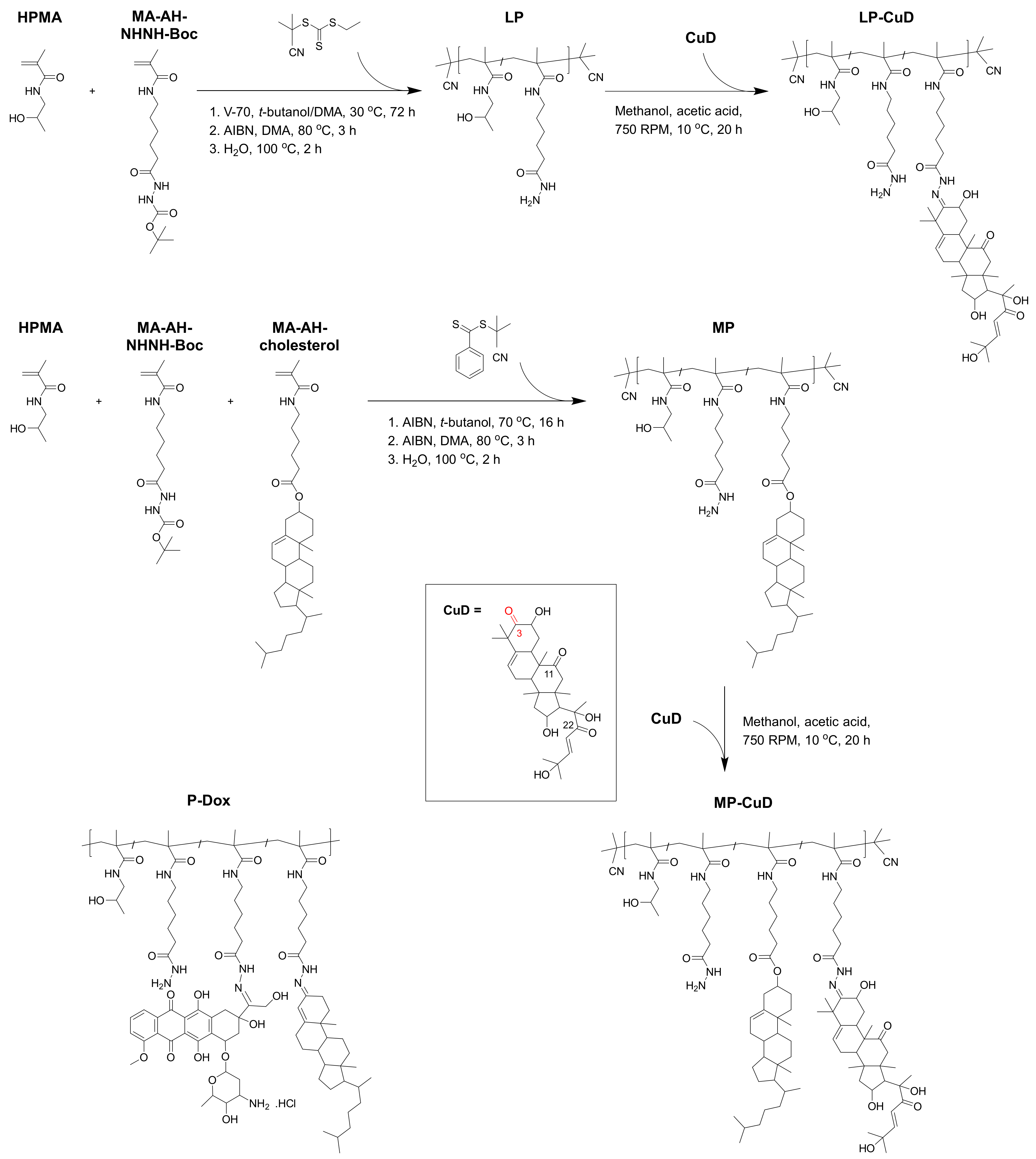
4.1. Synthesis and Physicochemical Properties of Copolymer Precursors and Polymer–Drug Conjugates
4.2. In Vitro CuD Release
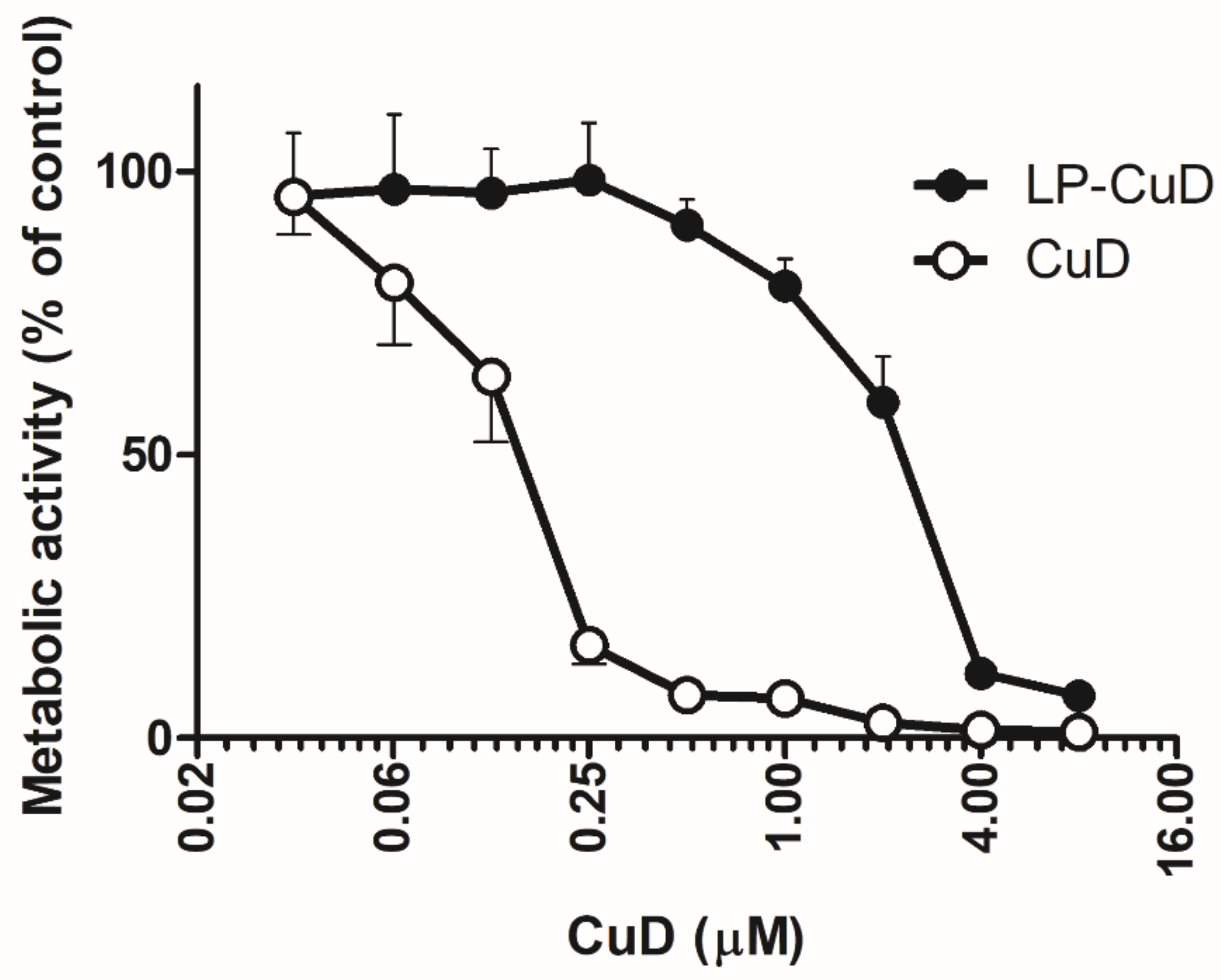
4.3. In Vitro Cytotoxicity
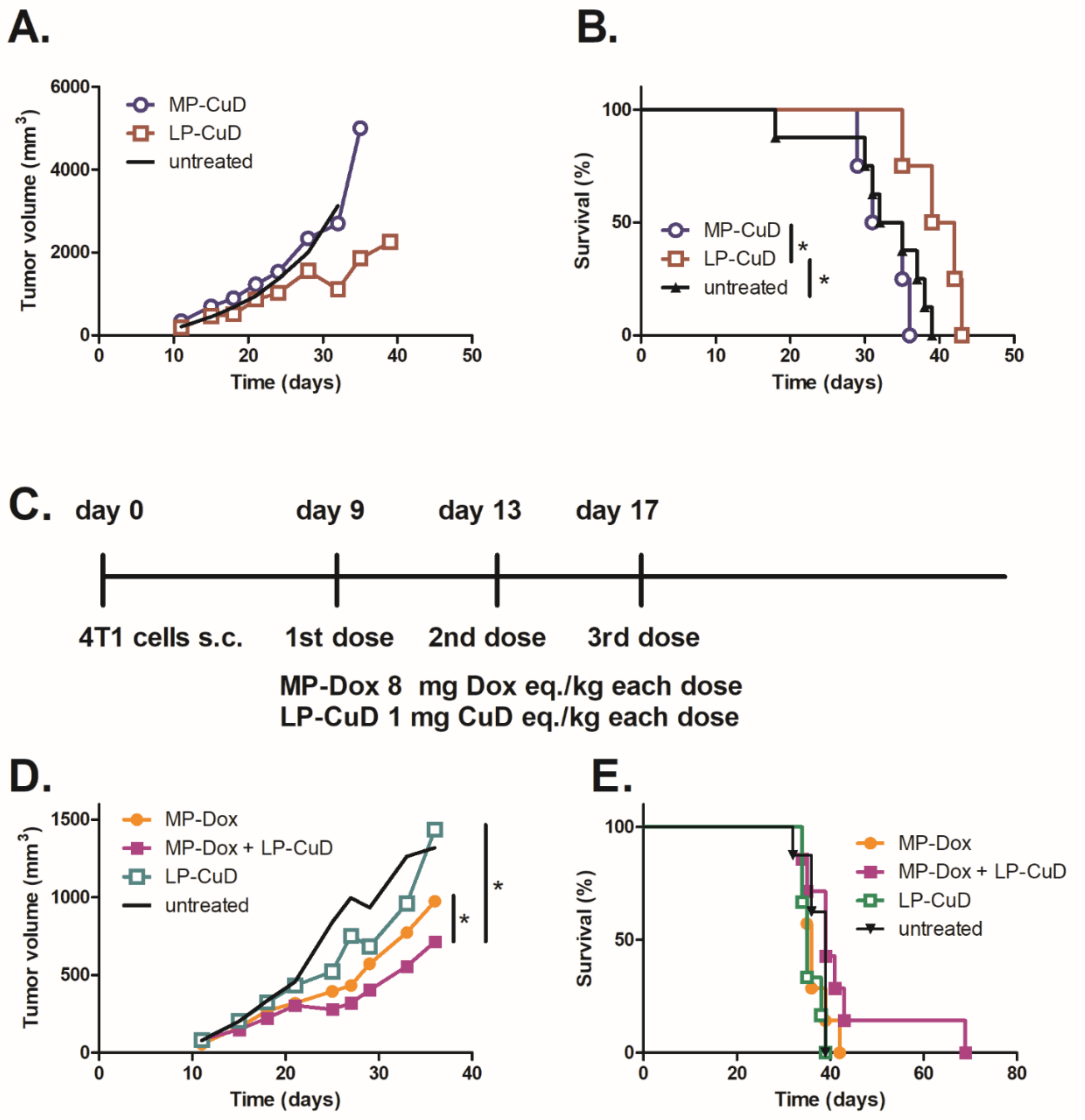
4.4. In Vivo Activity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, H.; Chen, J. Current status and future directions of cancer immunotherapy. J. Cancer 2018, 9, 1773–1781. [Google Scholar] [CrossRef] [Green Version]
- Marshall, H.T.; Djamgoz, M.B.A. Immuno-oncology: Emerging targets and combination therapies. Front. Oncol. 2018, 8, 318. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef]
- Nam, J.; Son, S.; Park, K.S.; Zou, W.; Shea, L.D.; Moon, J.J. Cancer nanomedicine for combination cancer immunotherapy. Nat. Rev. Mater. 2019, 4, 398–414. [Google Scholar] [CrossRef]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. [Google Scholar] [CrossRef] [Green Version]
- Říhová, B.; Kovář, L.; Kovář, M.; Hovorka, O. Cytotoxicity and immunostimulation: Double attack on cancer cells with polymeric therapeutics. Trends Biotechnol. 2009, 27, 11–17. [Google Scholar] [CrossRef]
- Yu, H.; Jove, R. The stats of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mulé, J.; Kerr, W.G.; et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef]
- Niu, G.; Bowman, T.; Huang, M.; Shivers, S.; Reintgen, D.; Daud, A.; Chang, A.; Kraker, A.; Jove, R.; Yu, H. Roles of activated Src and Stat3 signaling in melanoma tumor cell growth. Oncogene 2002, 21, 7001–7010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaskovich, M.A.; Sun, J.; Cantor, A.; Turkson, J.; Jove, R.; Sebti, S.M. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003, 63, 1270–1279. [Google Scholar] [PubMed]
- Turkson, J.; Zhang, S.; Mora, L.B.; Burns, A.; Sebti, S.; Jove, R. A novel platinum compound inhibits constitutive Stat3 signaling and induces cell cycle arrest and apoptosis of malignant cells. J. Biol. Chem. 2005, 280, 32979–32988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagard, R.; Metelev, V.; Souissi, I.; Baran-Marszak, F. STAT3 inhibitors for cancer therapy: Have all roads been explored? Jak-Stat 2013, 2, e22882/1–e22882/9. [Google Scholar] [CrossRef] [Green Version]
- Ge, W.; Chen, X.; Han, F.; Liu, Z.; Wang, T.; Wang, M.; Chen, Y.; Ding, Y.; Zhang, Q. Synthesis of cucurbitacin B derivatives as potential anti-hepatocellular carcinoma agents. Molecules 2018, 23, 3345. [Google Scholar] [CrossRef] [Green Version]
- Jayaprakasam, B.; Seeram, N.P.; Nair, M.G. Anticancer and antiinflammatory activities of cucurbitacins from Cucurbita andreana. Cancer Lett. 2003, 189, 11–16. [Google Scholar] [CrossRef]
- Kaushik, U.; Aeri, V.; Mir, S.R. Cucurbitacins—An insight into medicinal leads from nature. Pharmacogn. Rev. 2015, 9, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Sikander, M.; Malik, S.; Chauhan, N.; Khan, P.; Kumari, S.; Kashyap, V.K.; Khan, S.; Ganju, A.; Halaweish, F.T.; Yallapu, M.M.; et al. Cucurbitacin D reprograms glucose metabolic network in prostate cancer. Cancers 2019, 11, 364. [Google Scholar] [CrossRef] [Green Version]
- Sikander, M.; Hafeez, B.B.; Malik, S.; Alsayari, A.; Halaweish, F.T.; Yallapu, M.M.; Chauhan, S.C.; Jaggi, M. Cucurbitacin D exhibits potent anti-cancer activity in cervical cancer. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Ku, J.M.; Hong, S.H.; Kim, H.I.; Lim, Y.S.; Lee, S.J.; Kim, M.; Seo, H.S.; Shin, Y.C.; Ko, S.-G. Cucurbitacin D exhibits its anti-cancer effect in human breast cancer cells by inhibiting Stat3 and Akt signaling. Eur. J. Inflamm. 2018, 16, 1721727X17751809. [Google Scholar] [CrossRef]
- Molavi, O.; Ma, Z.; Mahmud, A.; Alshamsan, A.; Samuel, J.; Lai, R.; Kwon, G.S.; Lavasanifar, A. Polymeric micelles for the solubilization and delivery of STAT3 inhibitor cucurbitacins in solid tumors. Int. J. Pharm. 2008, 347, 118–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [CrossRef] [PubMed]
- Etrych, T.; Šubr, V.; Strohalm, J.; Šírová, M.; Říhová, B.; Ulbrich, K. HPMA copolymer-doxorubicin conjugates: The effects of molecular weight and architecture on biodistribution and in vivo activity. J. Control. Release 2012, 164, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Chytil, P.; Šírová, M.; Kudláčová, J.; Říhová, B.; Ulbrich, K.; Etrych, T. Bloodstream Stability Predetermines the Antitumor Efficacy of Micellar Polymer-Doxorubicin Drug Conjugates with pH-Triggered Drug Release. Mol. Pharm. 2018, 15, 3654–3663. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics:a review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Maeda, H.; Bharate, G.Y.; Daruwalla, J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur. J. Pharm. Biopharm. 2009, 71, 409–419. [Google Scholar] [CrossRef]
- Chytil, P.; Koziolová, E.; Etrych, T.; Ulbrich, K. HPMA Copolymer-Drug Conjugates with Controlled Tumor-Specific Drug Release. Macromol. Biosci. 2018, 18, 1700209. [Google Scholar] [CrossRef]
- Larson, N.; Ghandehari, H. Polymeric conjugates for drug delivery. Chem. Mater. 2012, 24, 840–853. [Google Scholar] [CrossRef] [Green Version]
- Ulbrich, K.; Holá, K.; Šubr, V.; Bakandritsos, A.; Tuček, J.; Zbořil, R. Targeted Drug Delivery with Polymers and Magnetic Nanoparticles: Covalent and Noncovalent Approaches, Release Control, and Clinical Studies. Chem. Rev. 2016, 116, 5338–5431. [Google Scholar] [CrossRef]
- Randárová, E.; Nakamura, H.; Islam, R.; Studenovský, M.; Mamoru, H.; Fang, J.; Chytil, P.; Etrych, T. Highly effective anti-tumor nanomedicines based on HPMA copolymer conjugates with pirarubicin prepared by controlled RAFT polymerization. Acta Biomater. 2020, 106, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Kostka, L.; Kotrchová, L.; Šubr, V.; Libánská, A.; Ferreira, C.A.; Malátová, I.; Lee, H.J.; Barnhart, T.E.; Engle, J.W.; Cai, W.; et al. HPMA-based star polymer biomaterials with tuneable structure and biodegradability tailored for advanced drug delivery to solid tumours. Biomaterials 2020, 235, 119728. [Google Scholar] [CrossRef] [PubMed]
- Dozono, H.; Yanazume, S.; Nakamura, H.; Etrych, T.; Chytil, P.; Ulbrich, K.; Fang, J.; Arimura, T.; Douchi, T.; Kobayashi, H.; et al. HPMA Copolymer-Conjugated Pirarubicin in Multimodal Treatment of a Patient with Stage IV Prostate Cancer and Extensive Lung and Bone Metastases. Target. Oncol. 2016, 11, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Lead Compound: SDX-7320. Available online: https://syndevrx.com/lead-compound-sdx-7320/ (accessed on 22 January 2021).
- Chytil, P.; Etrych, T.; Konák, Č.; Šírová, M.; Mrkvan, T.; Bouček, J.; Ríhová, B.; Ulbrich, K. New HPMA copolymer-based drug carriers with covalently bound hydrophobic substituents for solid tumour targeting. J. Control. Release 2008, 127, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Chytil, P.; Etrych, T.; Kostka, L.; Ulbrich, K. Hydrolytically degradable polymer micelles for anticancer drug delivery to solid tumors. Macromol. Chem. Phys. 2012, 213, 858–867. [Google Scholar] [CrossRef]
- Filippov, S.K.; Chytil, P.; Konarev, P.V.; Dyakonova, M.; Papadakis, C.; Zhigunov, A.; Plestil, J.; Stepanek, P.; Etrych, T.; Ulbrich, K.; et al. Macromolecular HPMA-based nanoparticles with cholesterol for solid-tumor targeting: Detailed study of the inner structure of a highly efficient drug delivery system. Biomacromolecules 2012, 13, 2594–2604. [Google Scholar] [CrossRef]
- Filippov, S.K.; Vishnevetskaya, N.S.; Niebuur, B.-J.; Koziolová, E.; Lomkova, E.A.; Chytil, P.; Etrych, T.; Papadakis, C.M. Influence ofmolar mass, dispersity, and type and location of hydrophobic side chain moieties on the critical micellar concentration and stability ofamphiphilic HPMA-based polymer drug carriers. Colloid Polym. Sci. 2017, 295, 1313–1325. [Google Scholar] [CrossRef]
- Lidický, O.; Šírová, M.; Etrych, T. HPMA copolymer-based polymer conjugates for the delivery and controlled release of retinoids. Physiol. Res. 2016, 65, S233–S241. [Google Scholar] [CrossRef]
- Chytil, P.; Etrych, T.; Kříz, J.; Šubr, V.; Ulbrich, K. N-(2-Hydroxypropyl)methacrylamide-based polymer conjugates with pH-controlled activation of doxorubicin for cell-specific or passive tumour targeting. Synthesis by RAFT polymerisation and physicochemical characterisation. Eur. J. Pharm. Sci. 2010, 41, 473–482. [Google Scholar] [CrossRef]
- Ulbrich, K.; Etrych, T.; Chytil, P.; Jelínková, M.; Ríhová, B. Antibody-targeted Polymer—Doxorubicin Conjugates with pH-controlled Activation. J. Drug Target. 2004, 12, 477–489. [Google Scholar] [CrossRef]
- Ishitake, K.; Satoh, K.; Kamigaito, M.; Okamoto, Y. Stereogradient polymers formed by controlled/living radical polymerization of bulky methacrylate monomers. Angew. Chem. Int. Ed. 2009, 48, 1991–1994. [Google Scholar] [CrossRef] [PubMed]
- Perrier, S.; Takolpuckdee, P.; Mars, C.A. Reversible Addition−Fragmentation Chain Transfer Polymerization: End Group Modification for Functionalized Polymers and Chain Transfer Agent Recovery. Macromolecules 2005, 38, 2033–2036. [Google Scholar] [CrossRef]
- Koziolová, E.; Kostka, L.; Kotrchová, L.; Šubr, V.; Konefal, R.; Nottelet, B.; Etrych, T. N-(2-Hydroxypropyl)methacrylamide-Based Linear, Diblock, and Starlike Polymer Drug Carriers: Advanced Process for Their Simple Production. Biomacromolecules 2018, 19, 4003–4013. [Google Scholar] [CrossRef] [PubMed]
- Etrych, T.; Mrkvan, T.; Chytil, P.; Konak, Č.; Říhová, B.; Ulbrich, K. N-(2-Hydroxypropyl)methacrylamide-Based Polymer Conjugates with pH-Controlled Activation of Doxorubicin. I. New Synthesis, Physicochemical Characterization and Preliminary Biological Evaluation. J. Appl. Polym. Sci. 2008, 109, 3050–3061. [Google Scholar] [CrossRef]
- Braunová, A.; Kaňa, M.; Kudláčová, J.; Kostka, L.; Bouček, J.; Betka, J.; Šírová, M.; Etrych, T. Micelle-Forming Block Copolymers Tailored for Inhibition of P-gp-Mediated Multidrug Resistance: Structure to Activity Relationship. Pharmaceutics 2019, 11, 579–600. [Google Scholar] [CrossRef] [Green Version]
- Trousil, J.; Syrová, Z.; Dal, N.J.K.; Rak, D.; Konefał, R.; Pavlova, E.; Matějková, J.; Cmarko, D.; Kubíčková, P.; Pavliš, O.; et al. Rifampicin Nanoformulation Enhances Treatment of Tuberculosis in Zebrafish. Biomacromolecules 2019, 20, 1798–1815. [Google Scholar] [CrossRef]
- Braunová, A.; Chytil, P.; Laga, R.; Šírová, M.; Machová, D.; Parnica, J.; Říhová, B.; Janoušková, O.; Etrych, T. Polymer nanomedicines based on micelle-forming amphiphilic or water-soluble polymer-doxorubicin conjugates: Comparative study of in vitro and in vivo properties related to the polymer carrier structure, composition, and hydrodynamic properties. J. Control. Release 2020, 321, 718–733. [Google Scholar] [CrossRef]
- Ku, J.M.; Kim, S.R.; Hong, S.H.; Choi, H.-S.; Seo, H.S.; Shin, Y.C.; Ko, S.-G. Cucurbitacin D induces cell cycle arrest and apoptosis by inhibiting STAT3 and NF-κB signaling in doxorubicin-resistant human breast carcinoma (MCF7/ADR) cells. Mol. Cell. Biochem. 2015, 409, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Sikander, M.; Malik, S.; Khan, S.; Kumari, S.; Chauhan, N.; Khan, P.; Halaweish, F.T.; Chauhan, B.; Yallapu, M.M.; Jaggi, M.; et al. Novel Mechanistic Insight into the Anticancer Activity of Cucurbitacin D against Pancreatic Cancer (Cuc D Attenuates Pancreatic Cancer). Cells 2020, 9, 103. [Google Scholar] [CrossRef] [Green Version]
- Ding, N.; Yamashita, U.; Matsuoka, H.; Sugiura, T.; Tsukada, J.; Noguchi, J.; Yoshida, Y. Apoptosis induction through proteasome inhibitory activity of cucurbitacin D in human T-cell leukemia. Cancer 2011, 117, 2735–2746. [Google Scholar] [CrossRef]
- Nakanishi, T.; Song, Y.; He, C.; Wang, D.; Morita, K.; Tsukada, J.; Kanazawa, T.; Yoshida, Y. Autophagy is associated with cucurbitacin D-induced apoptosis in human T cell leukemia cells. Med. Oncol. 2016, 33, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Etrych, T.; Šírova, M.; Starovoytova, L.; Řihova, B.; Ulbrich, K. HPMA copolymer conjugates of paclitaxel and docetaxel with pH-controlled drug release. Mol. Pharm. 2010, 7, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Koziolová, E.; Etrych, T.; Chytil, P.; Fang, J.; Ulbrich, K.; Maeda, H. Comparison between linear and star-like HPMA conjugated pirarubicin (THP) in pharmacokinetics and antitumor activity in tumor bearing mice. Eur. J. Pharm. Biopharm. 2015, 90, 90–96. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Content of Hydrazide (mol.%) a | Content of Hydrophobic Moiety (mol.%) b | Content of Drug (wt.%) c | Mn (g·mol−1) d | Mw (g·mol−1) d | Ðd | Dh ± SD e (nm) |
|---|---|---|---|---|---|---|---|
| LP | 4.0 | - | - | 25,100 | 26,500 | 1.05 | 8.8 ± 0.2 |
| LP-CuD | - | - | 6.2 | 27,000 | 36,200 | 1.34 | 12.4 ± 1.0 |
| MP | 5.0 | 2.1 | - | 21,500 | 26,400 | 1.23 | 19.8 ± 0.4 |
| MP-CuD | - | 2.1 | 6.7 | 25,700 | 39,000 | 1.51 | 32.2 ± 1.6 |
| MP-Dox f | - | 2.0 | 8.1 | 14,200 | 25,500 | 1.80 | 29.6 ± 1.0 |
| CT26 a | 4T1 a | EL4 b | |
|---|---|---|---|
| MP-CuD | 0.386 ± 0.037 c | 0.293 ± 0.033 | 0.380 ± 0.189 |
| LP-CuD | 0.521 ± 0.226 | 0.512 ± 0.258 | 0.175 ± 0.002 |
| CuD | 0.125 ± 0.020 | 0.175 ± 0.025 | 0.048 ± 0.013 |
| SK-OV-3 | OVCAR-3 | DU145 | |
|---|---|---|---|
| MP-CuD | 0.185 ± 0.037 a | 0.277 ± 0.099 | 0.517 ± 0.155 |
| LP-CuD | 0.183 ± 0.009 | 0.190 ± 0.057 | 0.272 ± 0.057 |
| CuD | 0.025 ± 0.007 | 0.053 ± 0.014 | 0.062 ± 0.022 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tavares, M.R.; Hrabánková, K.; Konefał, R.; Kaňa, M.; Říhová, B.; Etrych, T.; Šírová, M.; Chytil, P. HPMA-Based Copolymers Carrying STAT3 Inhibitor Cucurbitacin-D as Stimulus-Sensitive Nanomedicines for Oncotherapy. Pharmaceutics 2021, 13, 179. https://doi.org/10.3390/pharmaceutics13020179
Tavares MR, Hrabánková K, Konefał R, Kaňa M, Říhová B, Etrych T, Šírová M, Chytil P. HPMA-Based Copolymers Carrying STAT3 Inhibitor Cucurbitacin-D as Stimulus-Sensitive Nanomedicines for Oncotherapy. Pharmaceutics. 2021; 13(2):179. https://doi.org/10.3390/pharmaceutics13020179
Chicago/Turabian StyleTavares, Marina R., Klára Hrabánková, Rafał Konefał, Martin Kaňa, Blanka Říhová, Tomáš Etrych, Milada Šírová, and Petr Chytil. 2021. "HPMA-Based Copolymers Carrying STAT3 Inhibitor Cucurbitacin-D as Stimulus-Sensitive Nanomedicines for Oncotherapy" Pharmaceutics 13, no. 2: 179. https://doi.org/10.3390/pharmaceutics13020179
APA StyleTavares, M. R., Hrabánková, K., Konefał, R., Kaňa, M., Říhová, B., Etrych, T., Šírová, M., & Chytil, P. (2021). HPMA-Based Copolymers Carrying STAT3 Inhibitor Cucurbitacin-D as Stimulus-Sensitive Nanomedicines for Oncotherapy. Pharmaceutics, 13(2), 179. https://doi.org/10.3390/pharmaceutics13020179