
Development of iRGD-Modified Peptide Carriers for Suicide Gene Therapy of Uterine Leiomyoma



Abstract
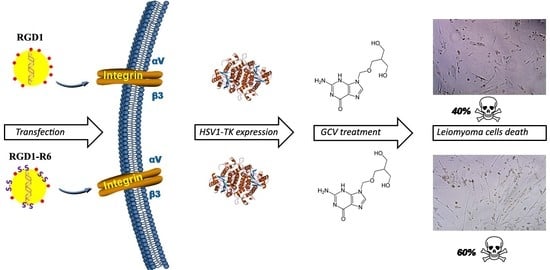
:
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Expression of αvβ3 Integrins in Leiomyoma Cells
2.2. Peptide and Reporter Plasmids
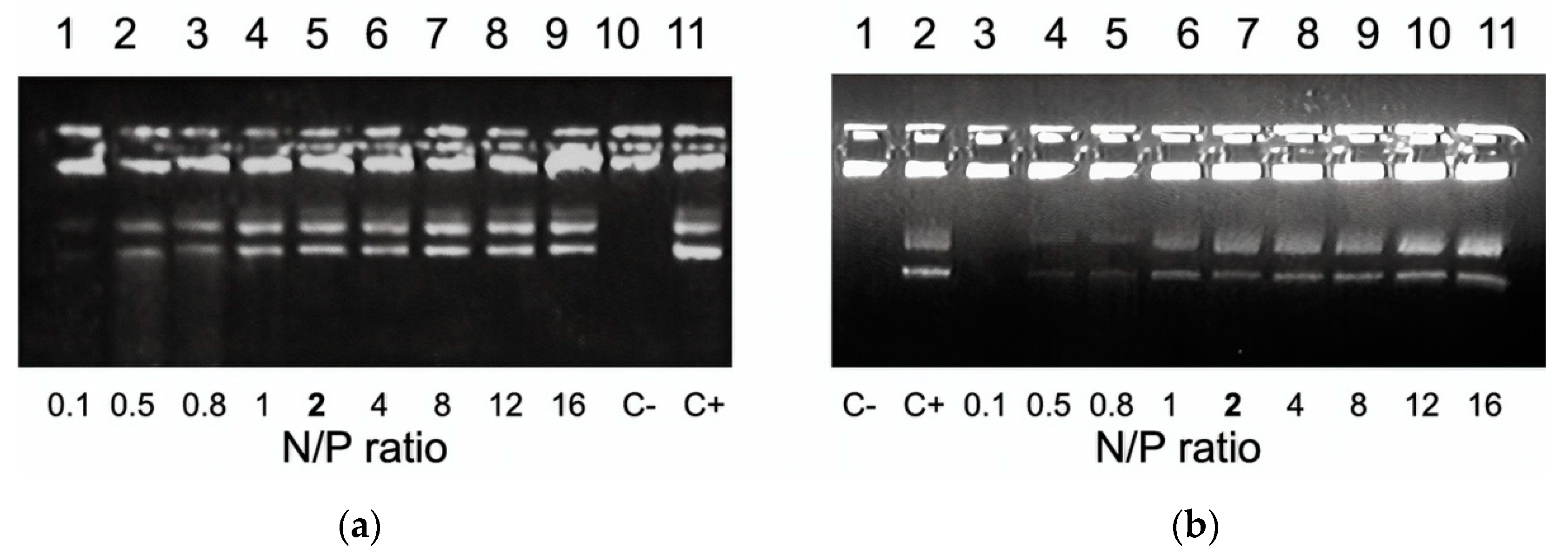
2.3. Preparation of Carrier/DNA Complexes
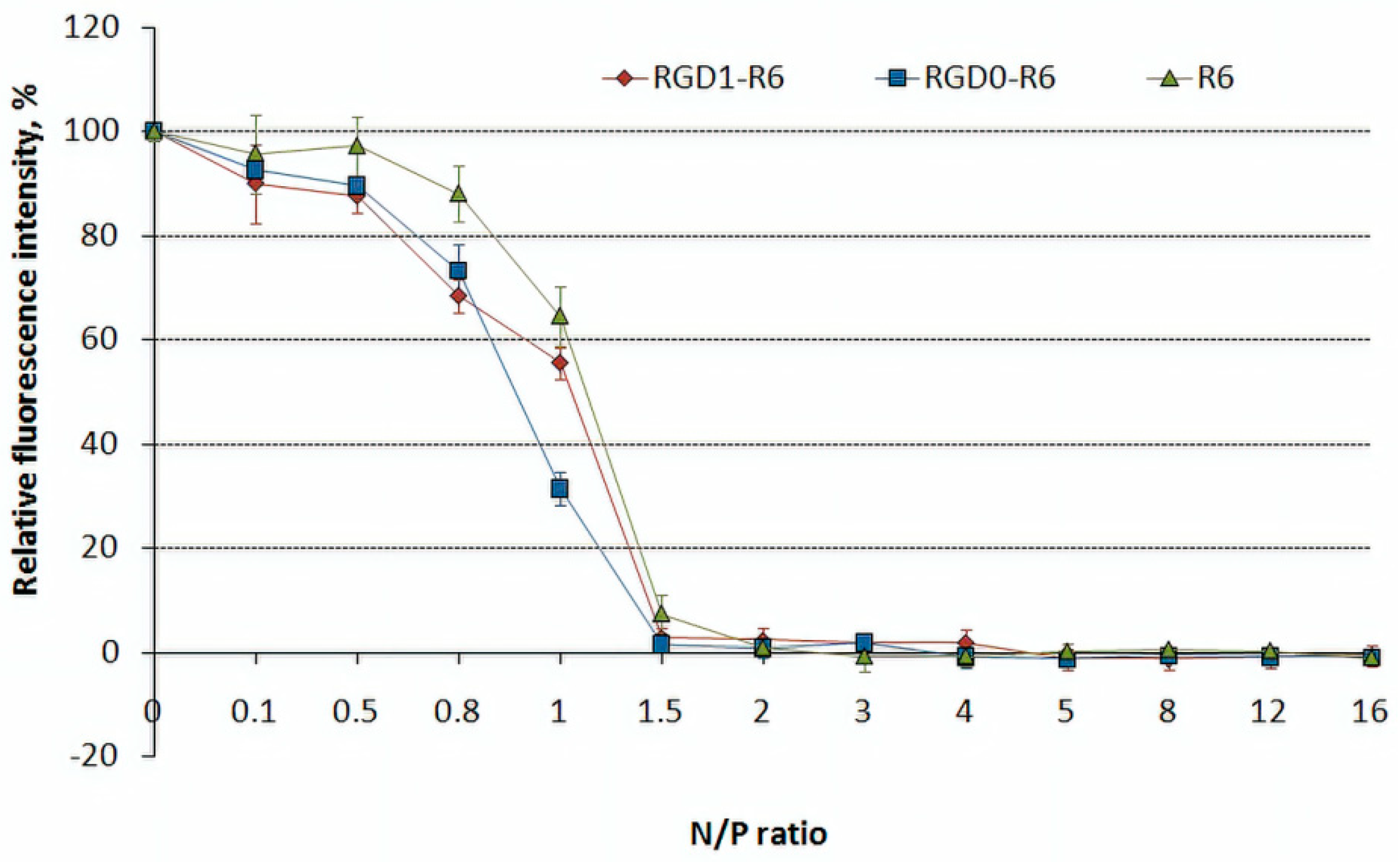
2.4. DNA Binding and DNAse I Protection Assays
2.5. Size and ʐ-Potential Measurement of Peptide/DNA Complexes
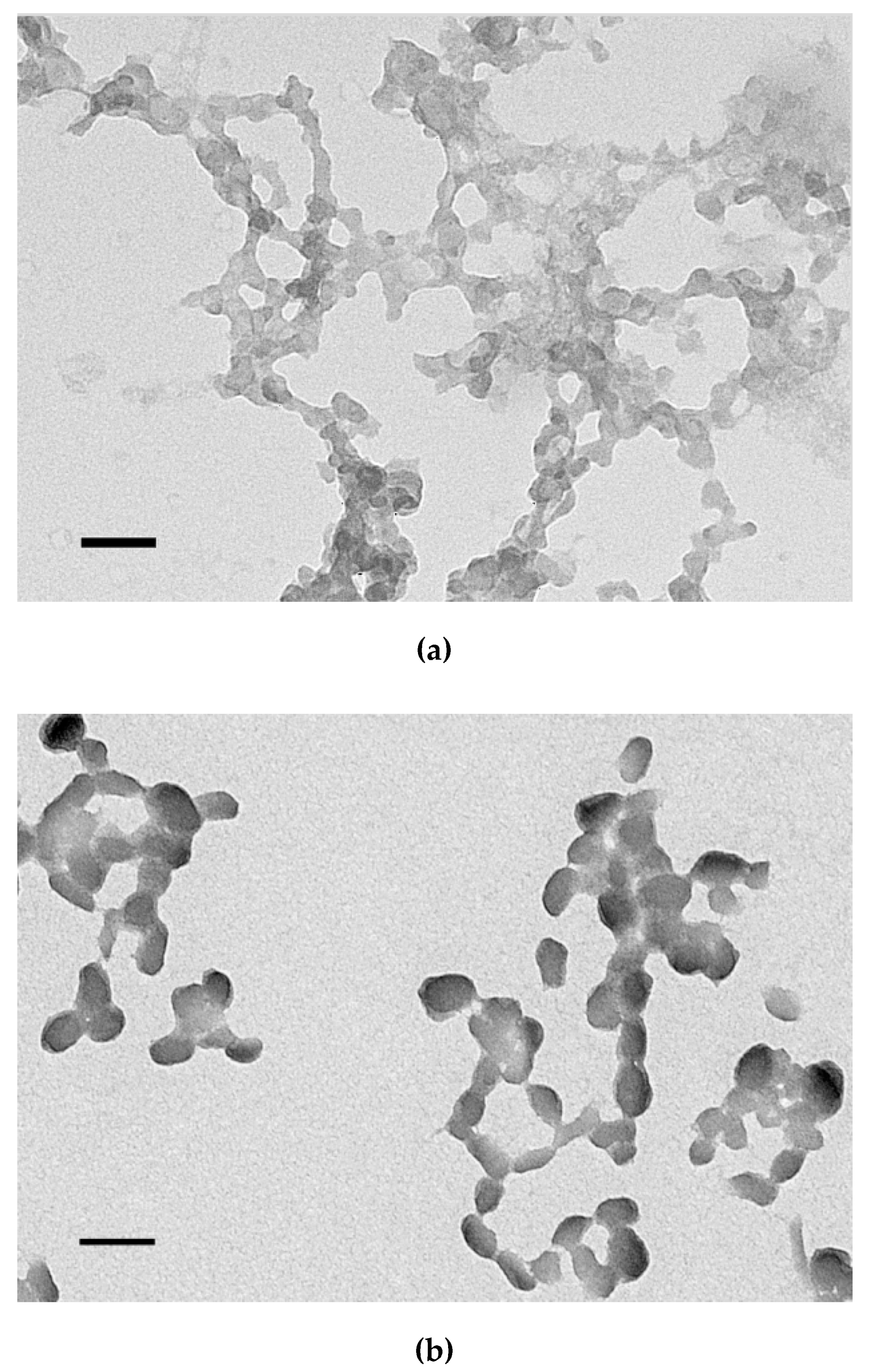
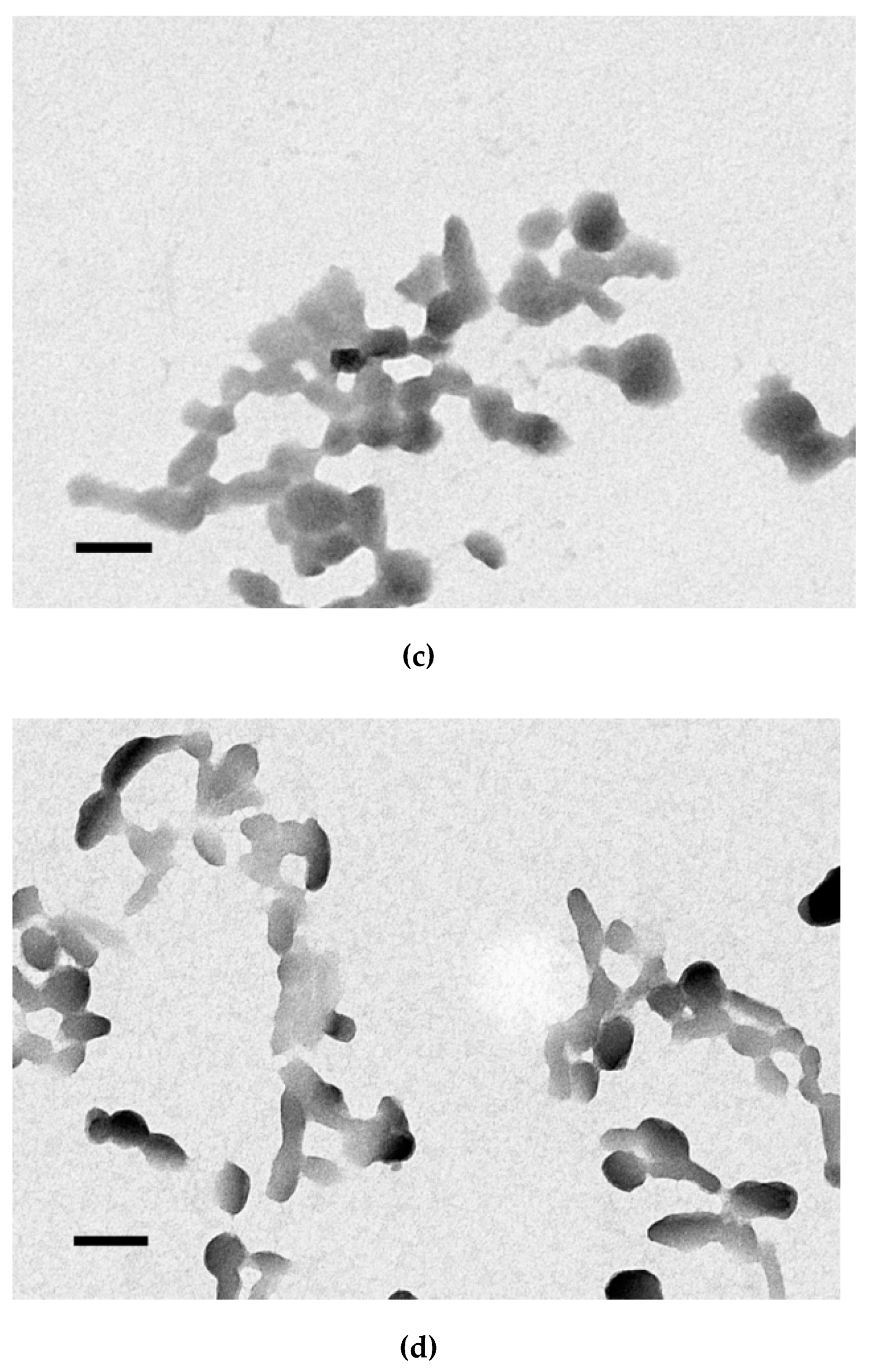
2.6. Transmission Electronic Microscopy
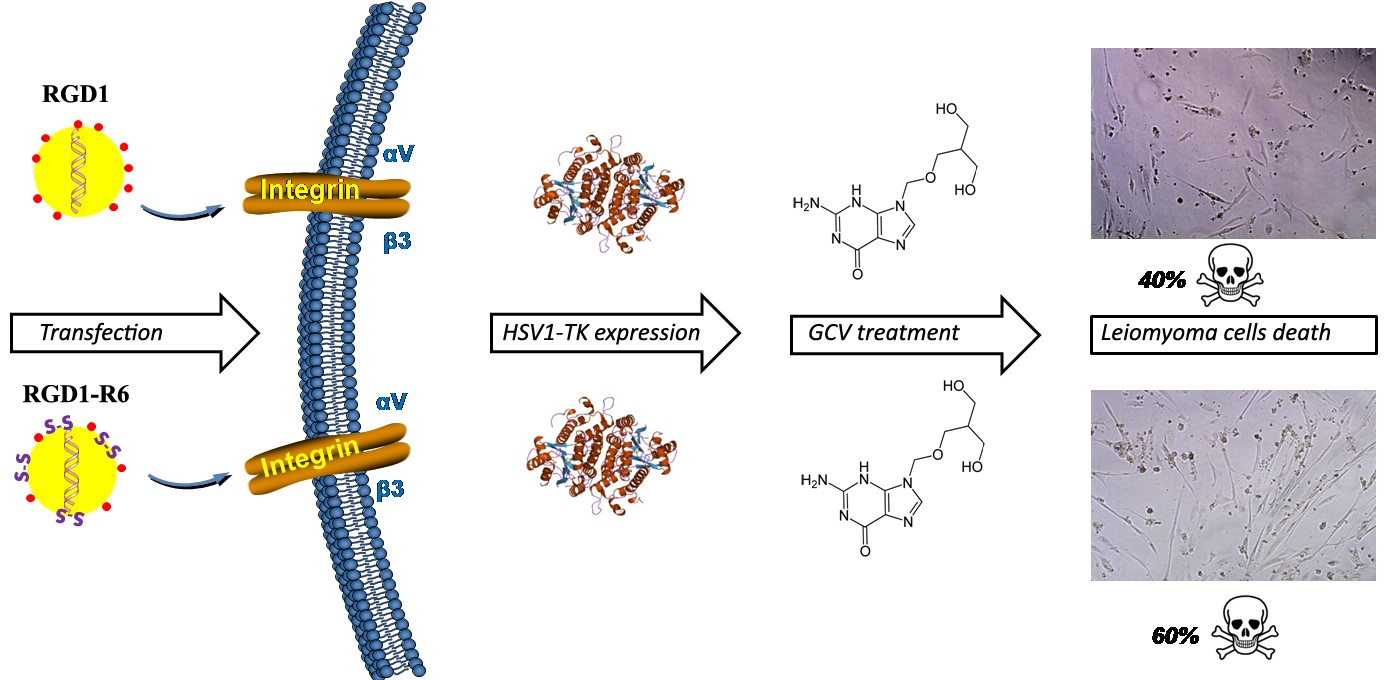
2.7. SYBR-Green Exclusion Assay
2.8. Ellman’s Assay
2.9. Relaxation of Carrier/DNA Complexes by Dextran-Sulfate and DTT Destabilization
2.10. Gene Transfer and Cytotoxicity Assays
2.11. Cellular Uptake of Peptide/DNA Complexes
2.12. Suicide Gene Therapy
2.13. Statistical Analysis
3. Results and Discussion
3.1. Carrier Design
3.2. DNA Binding and DNA Protection Properties of the Carriers
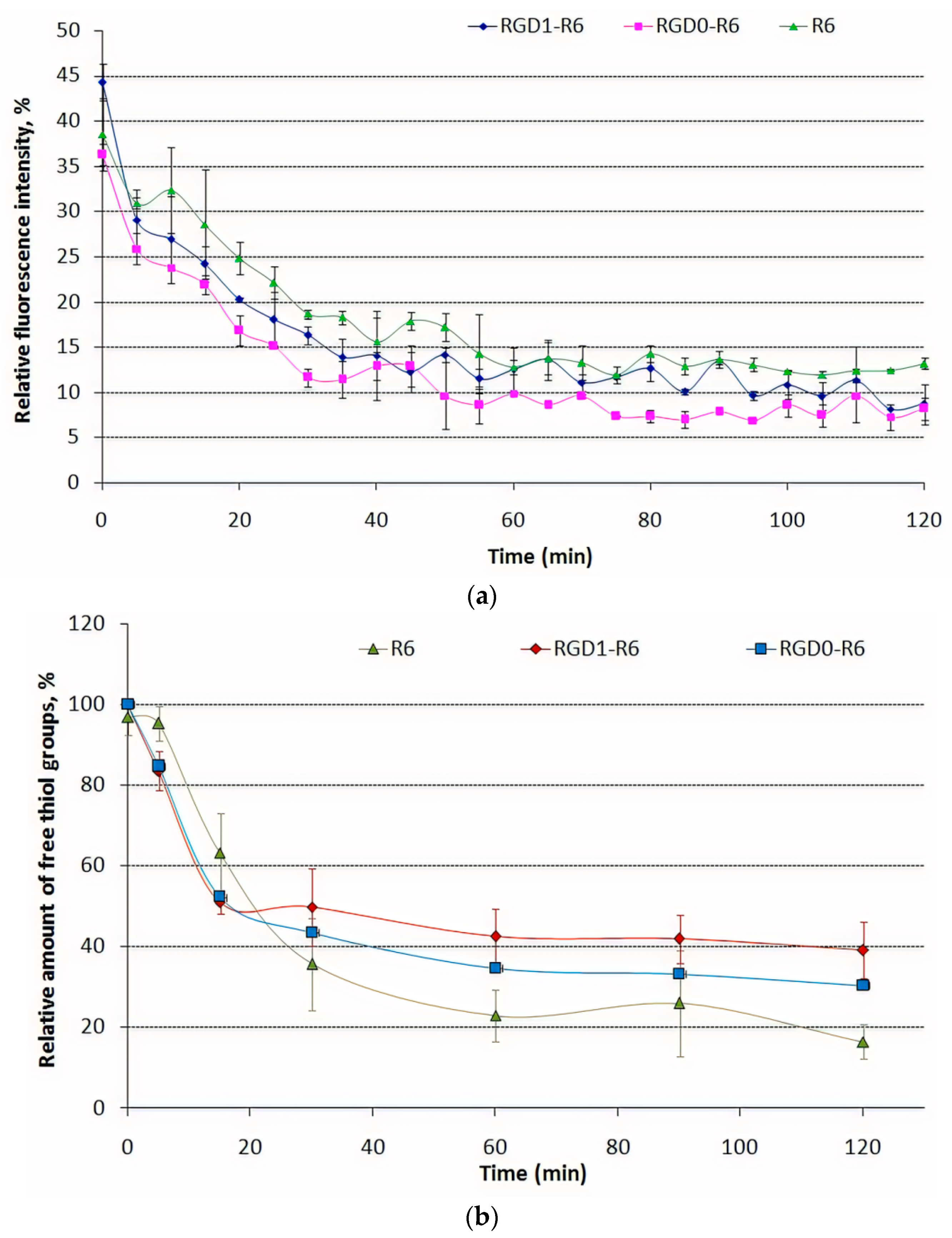
3.3. Kinetics of Carrier/DNA Complexation and Disulfide Bonds Formation during Template Polymerization
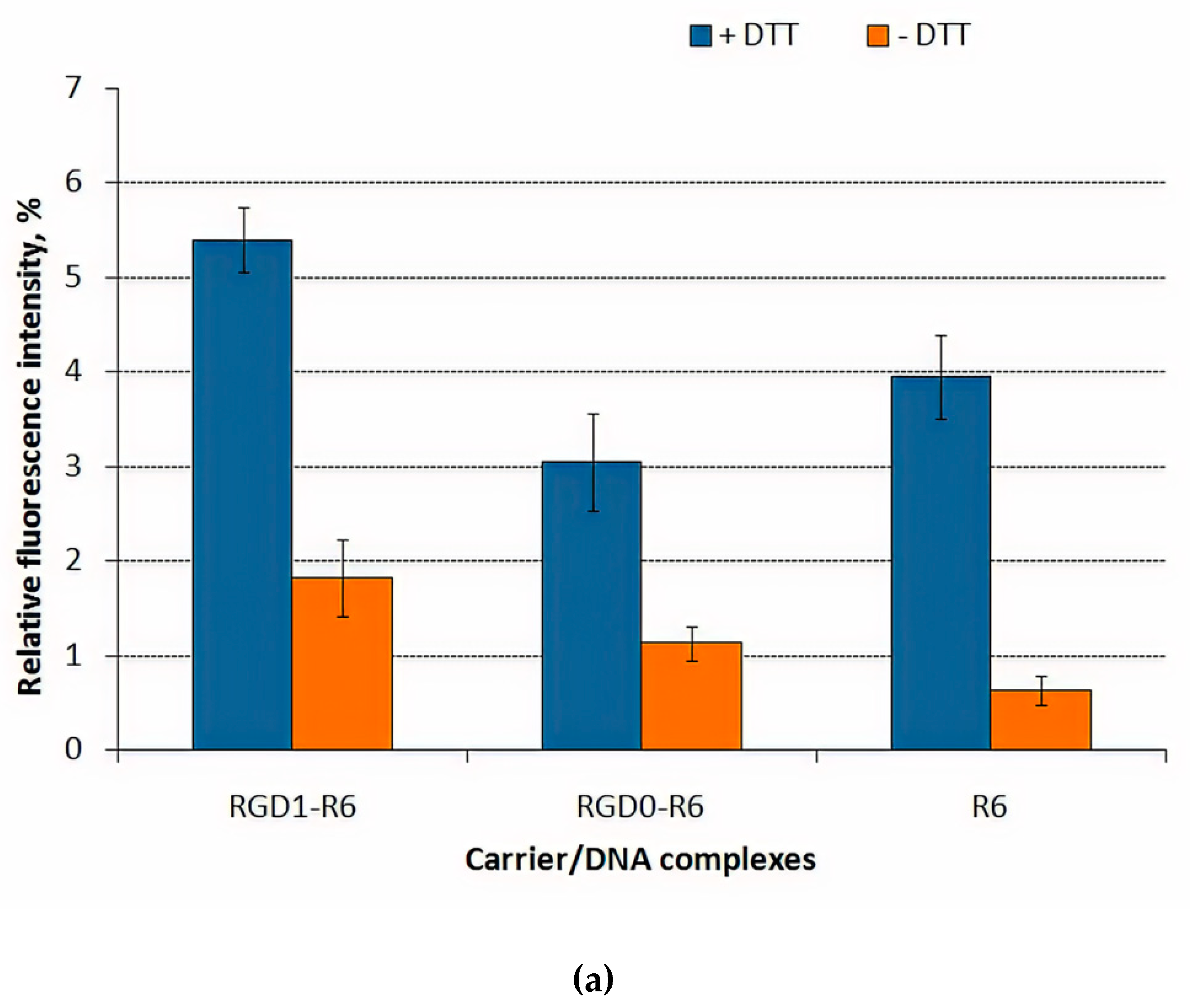
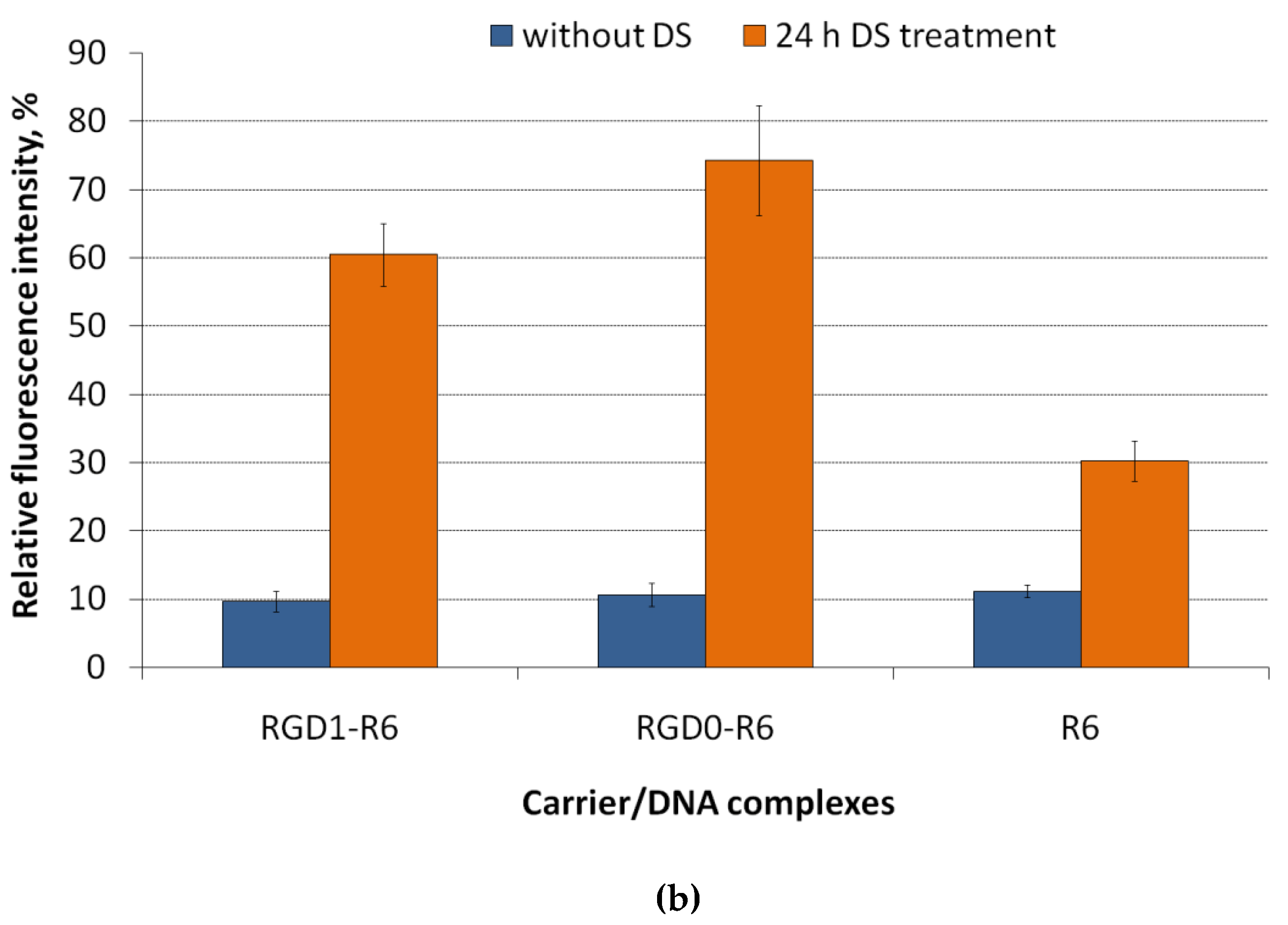
3.4. DNA Release after DTT and DS Treatment of the Polyplexes
3.5. Size and ʐ-Potential of the Carrier/DNA Complexes
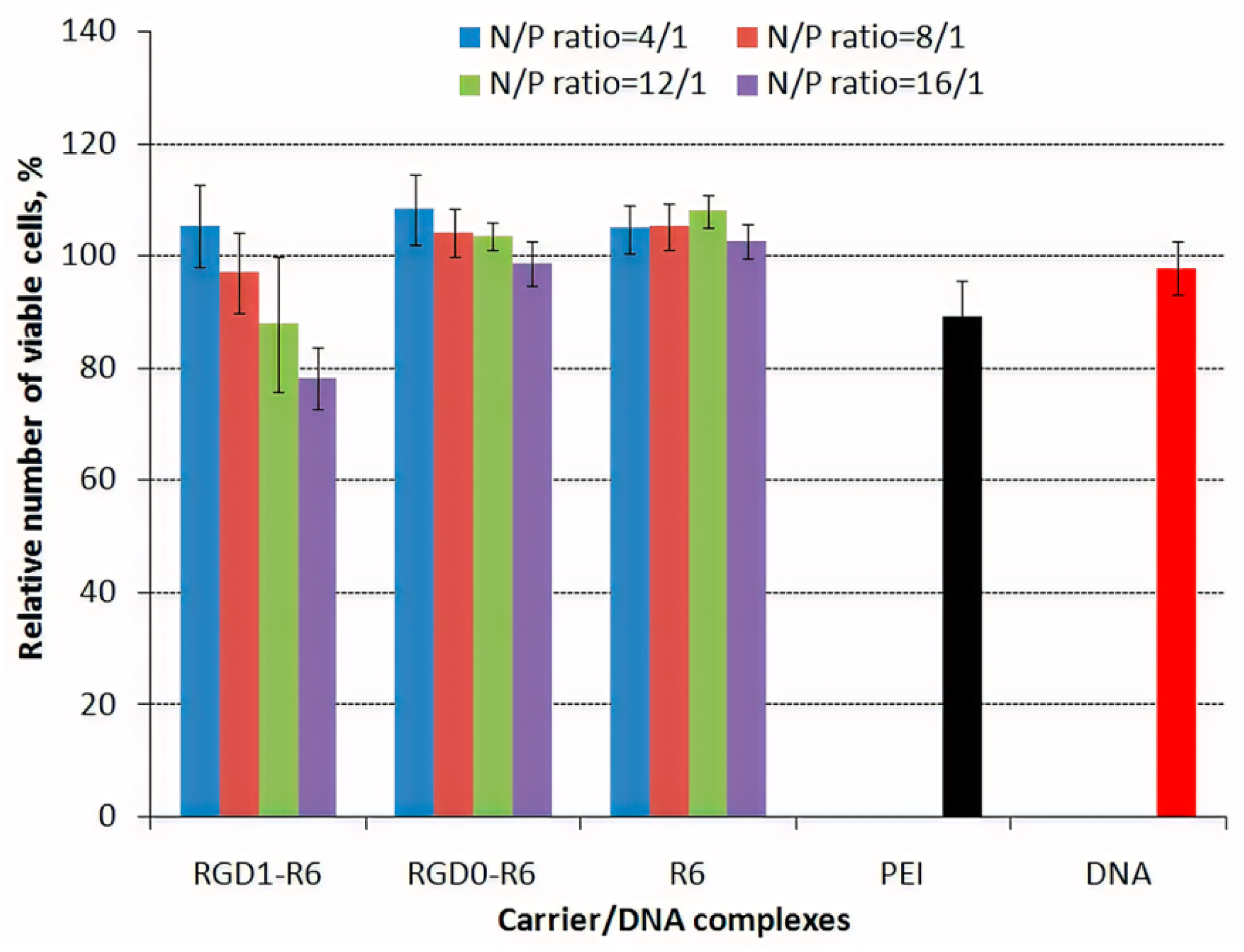
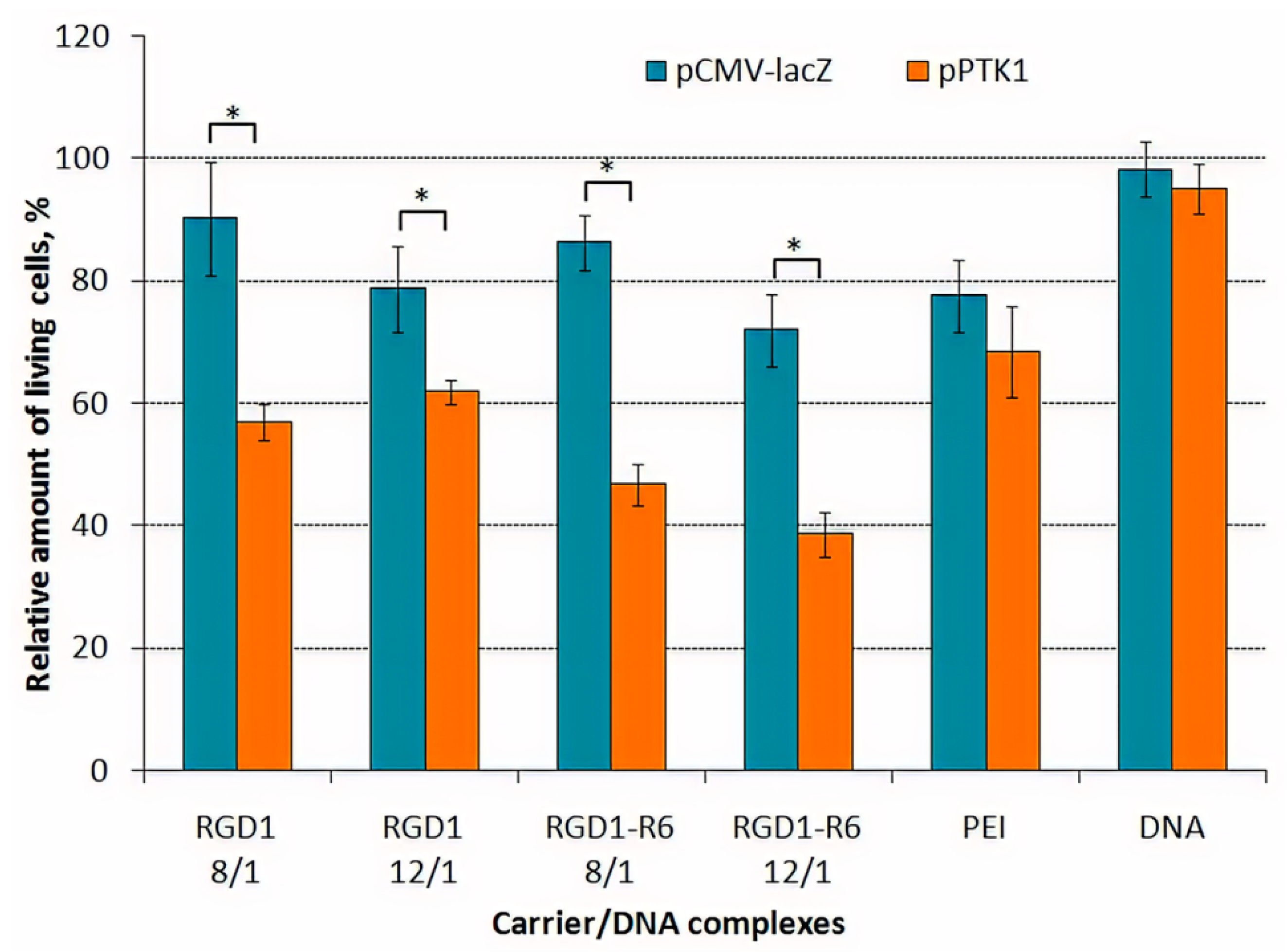
3.6. Cytotoxicity of DNA-Polyplexes
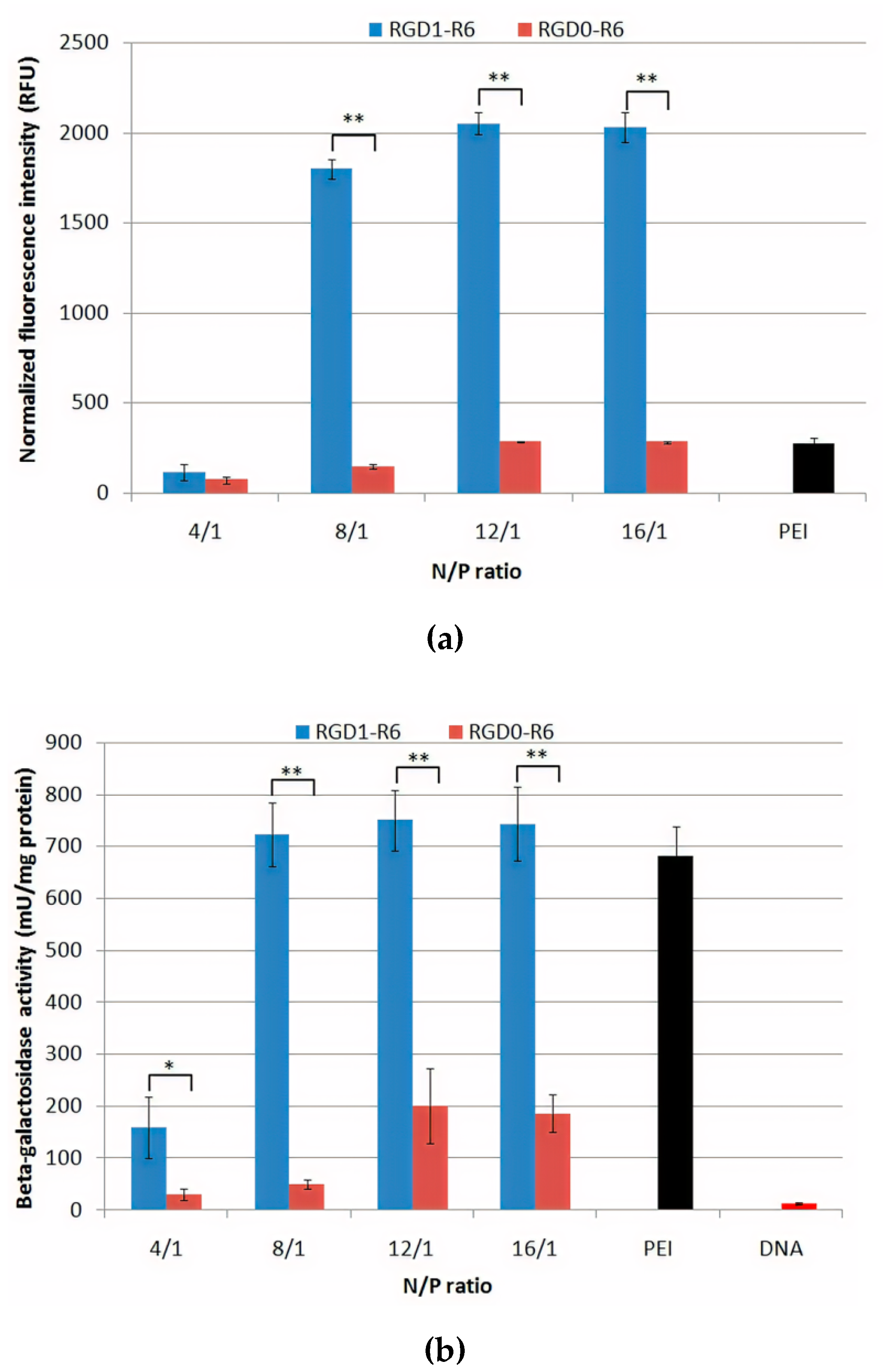
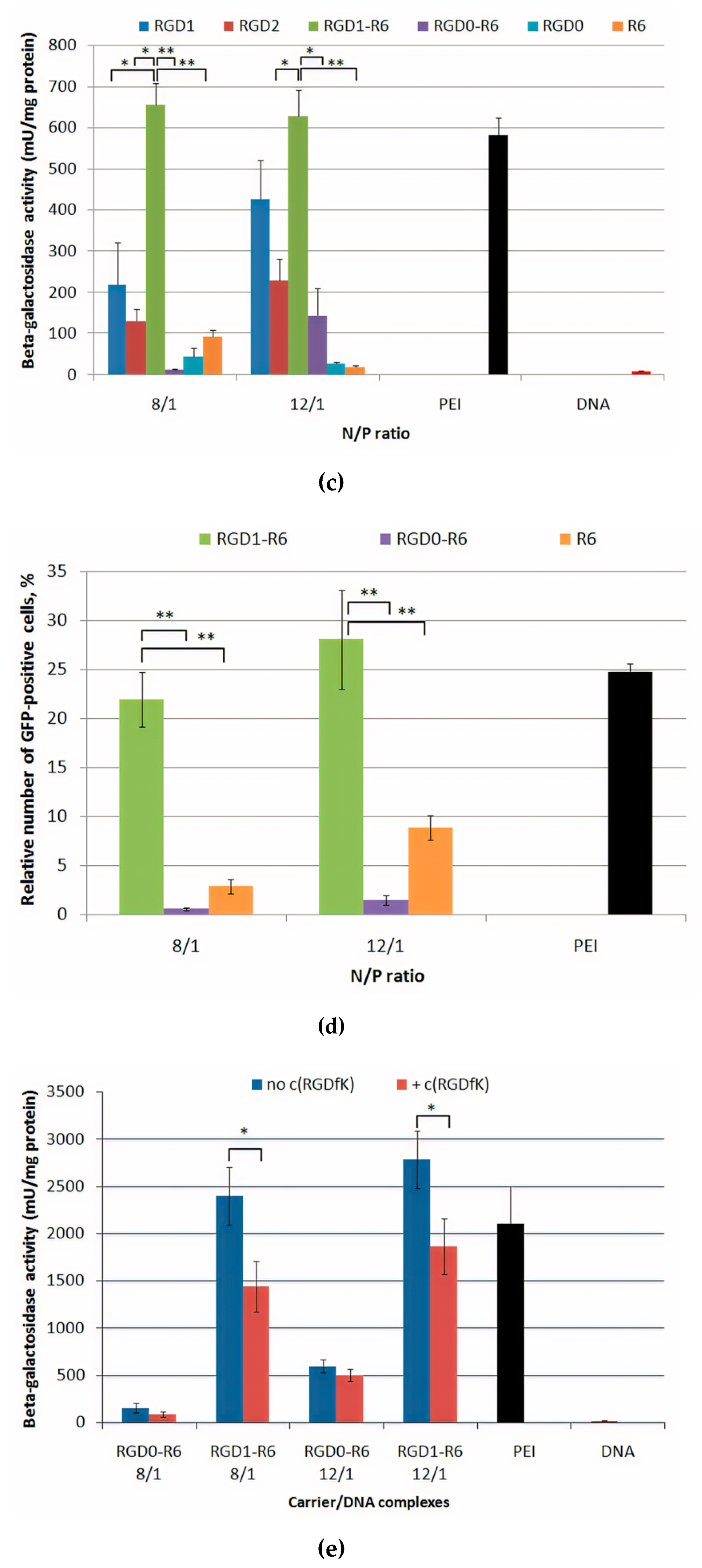
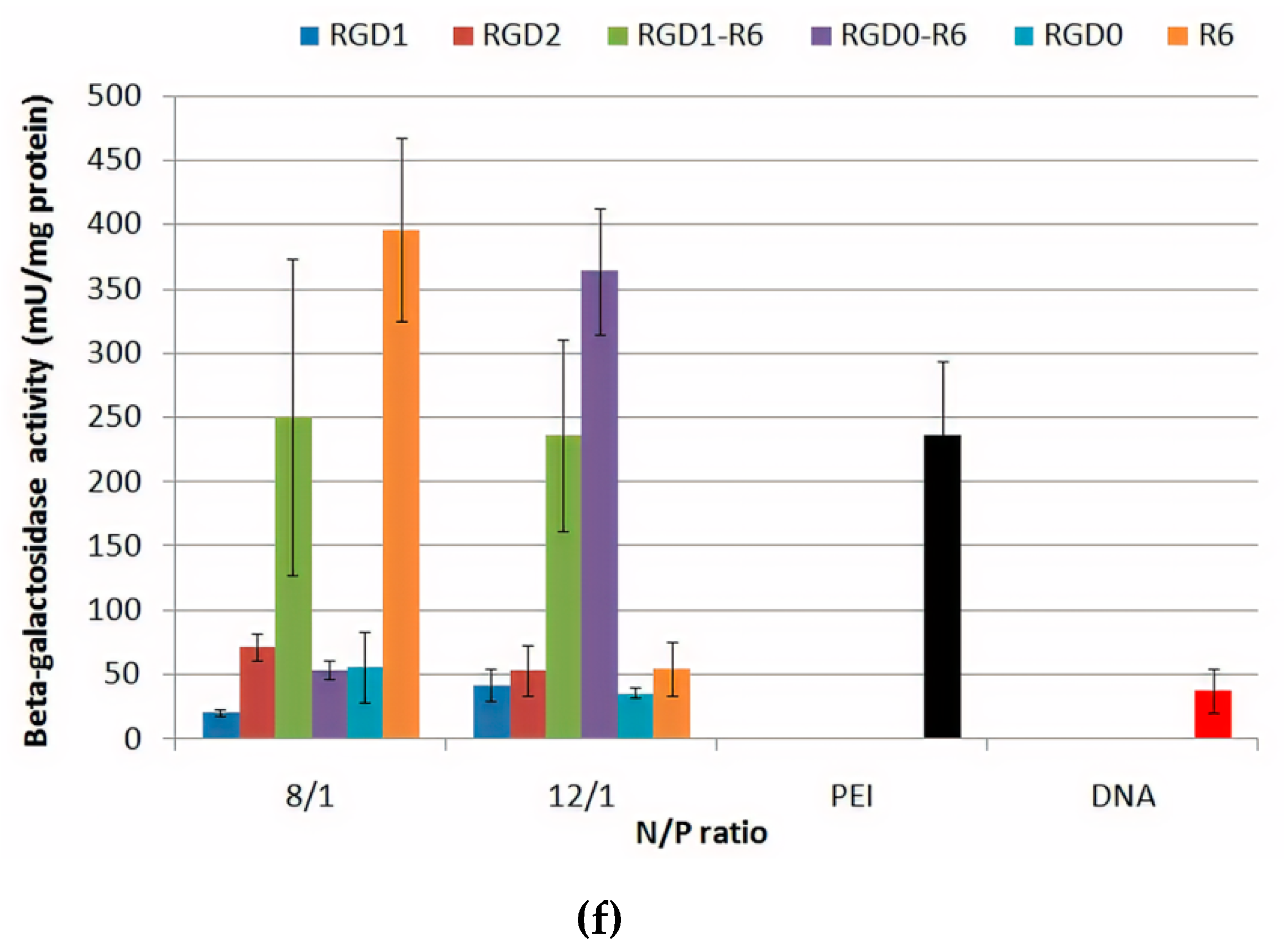
3.7. Gene Transfer
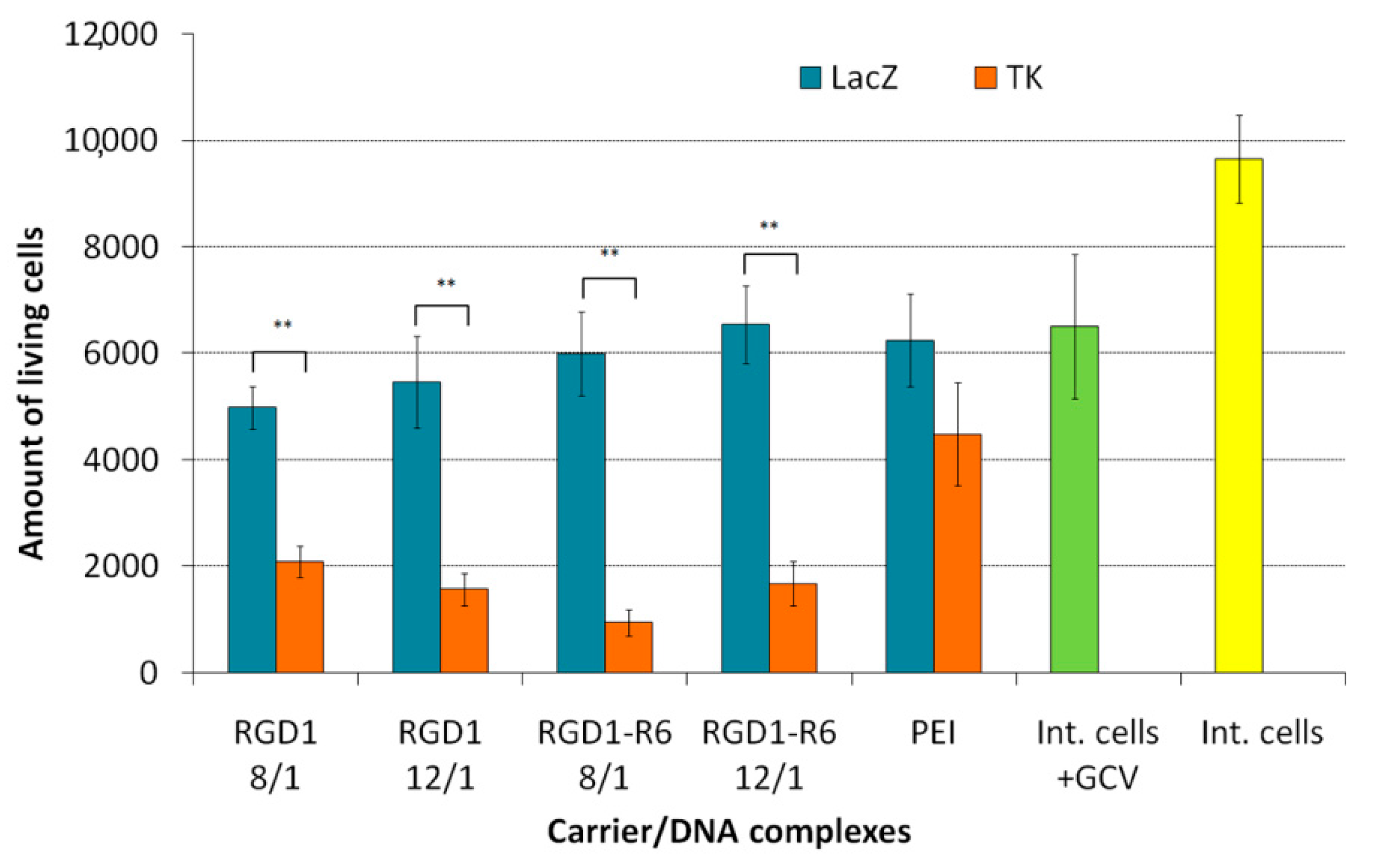
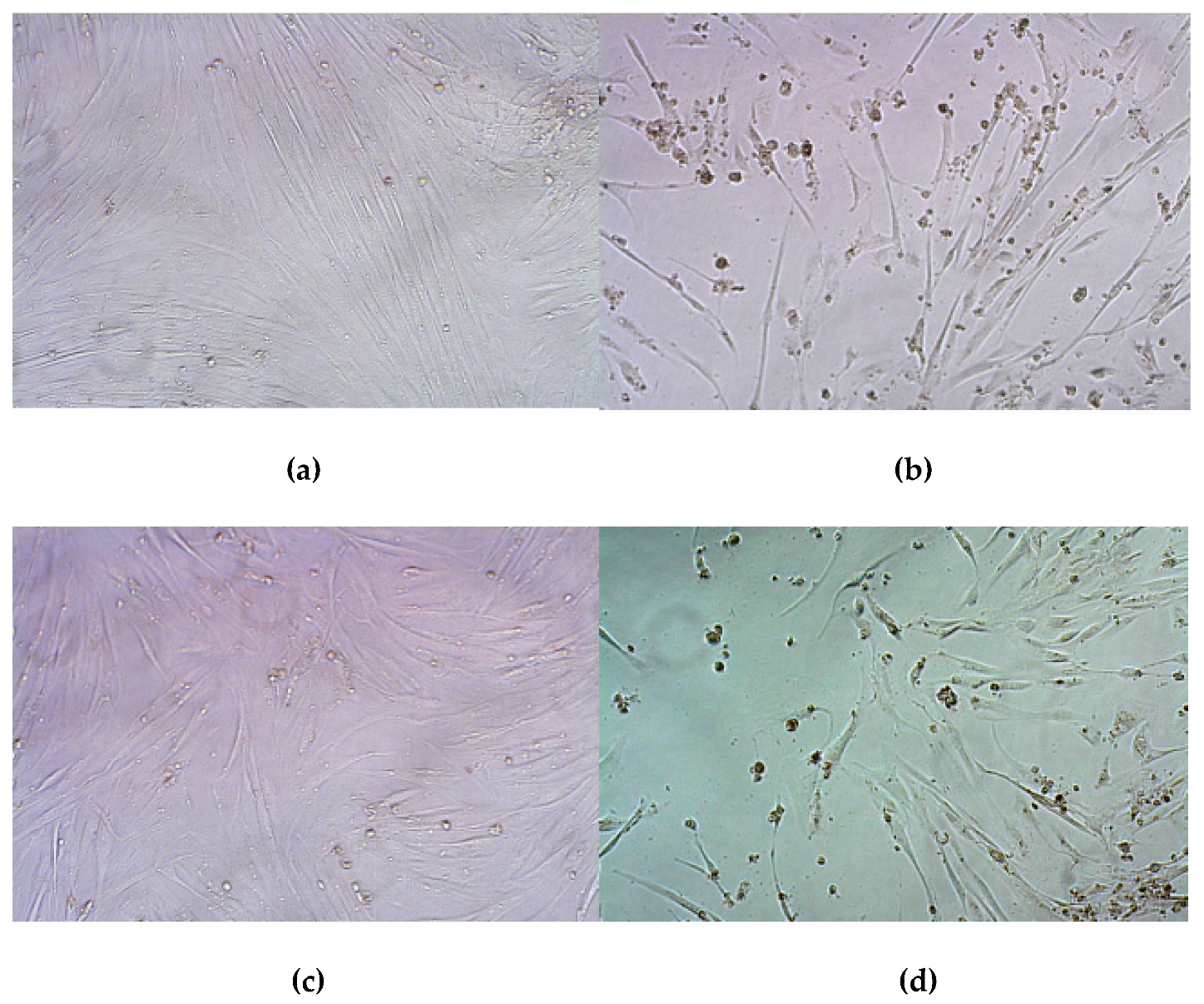
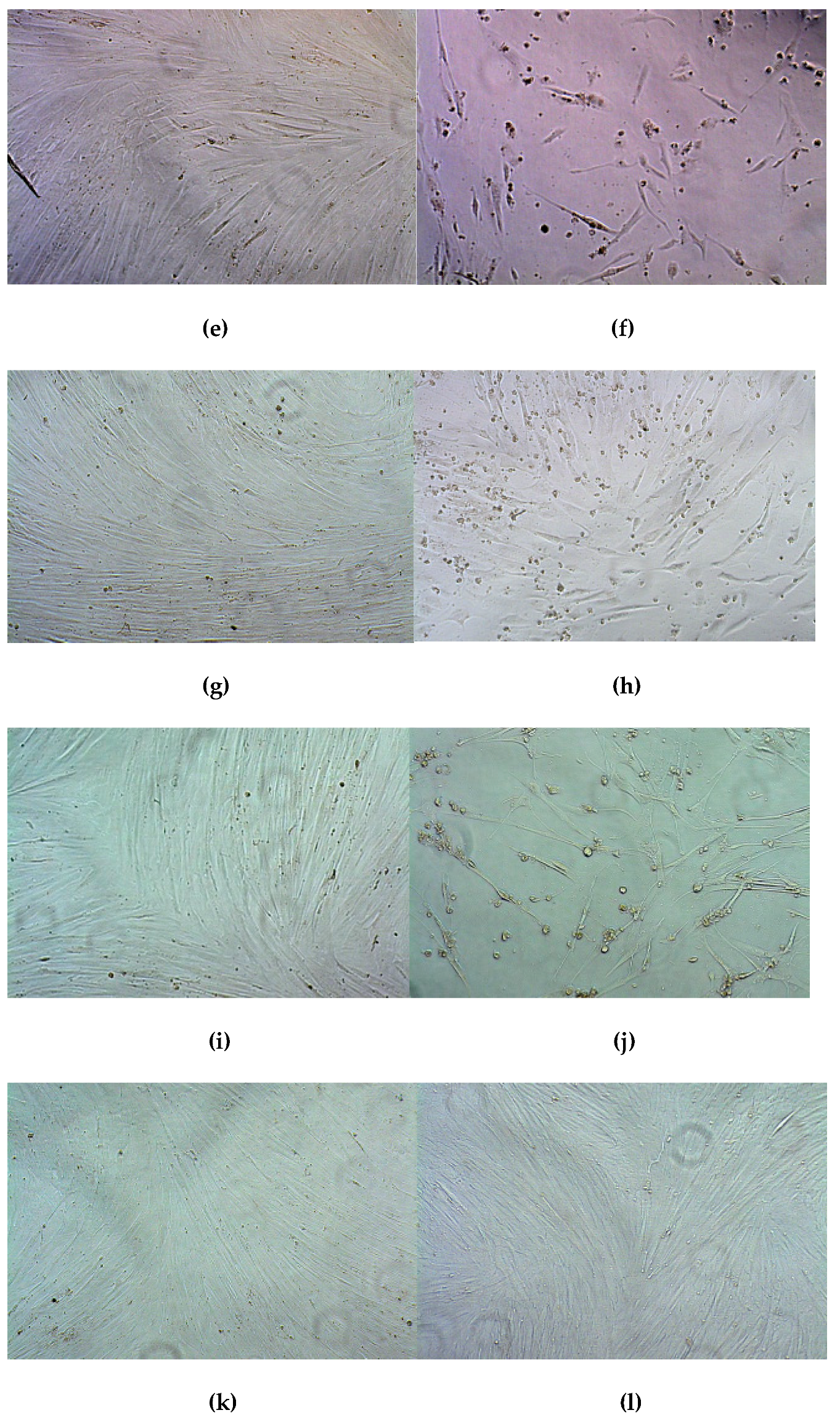
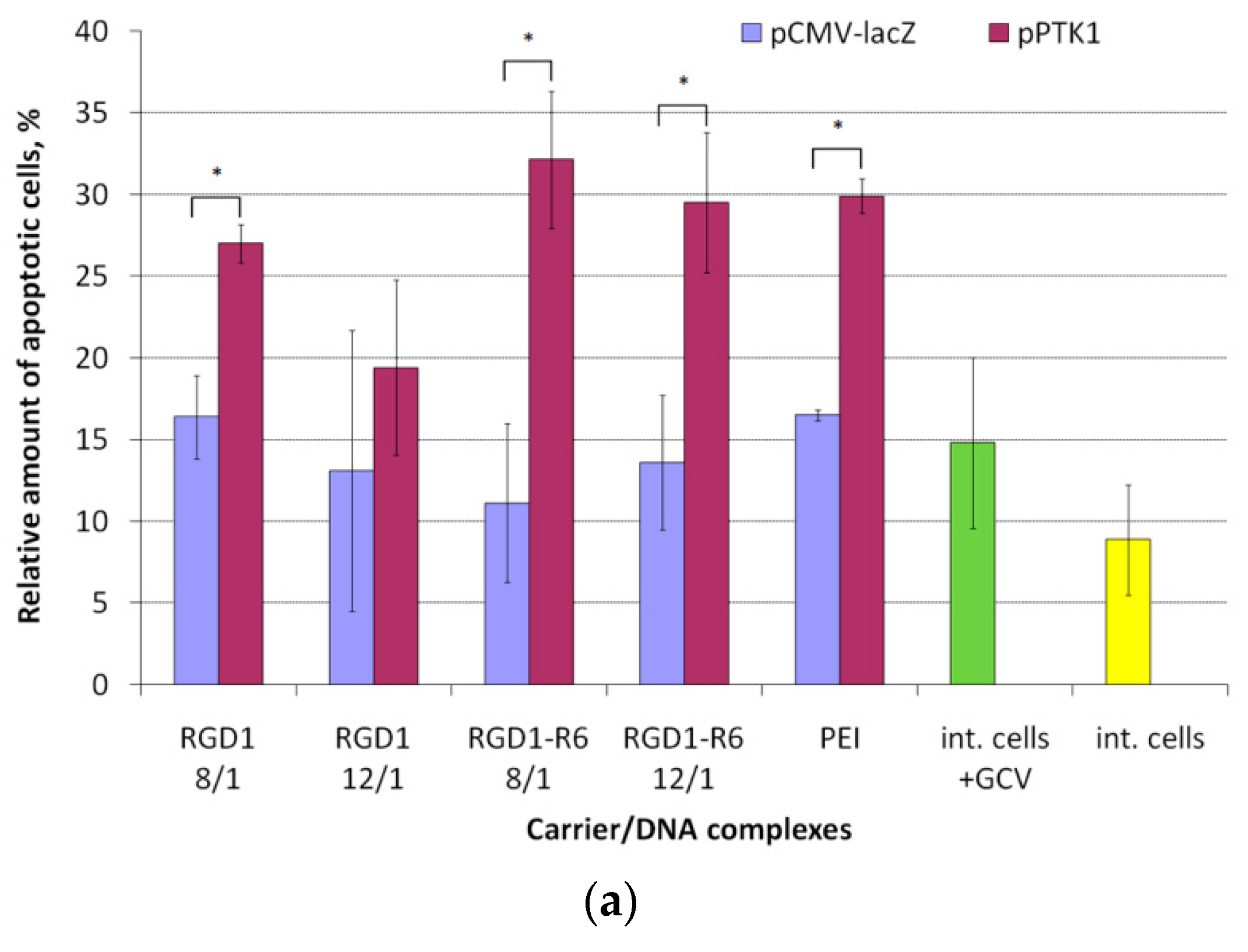
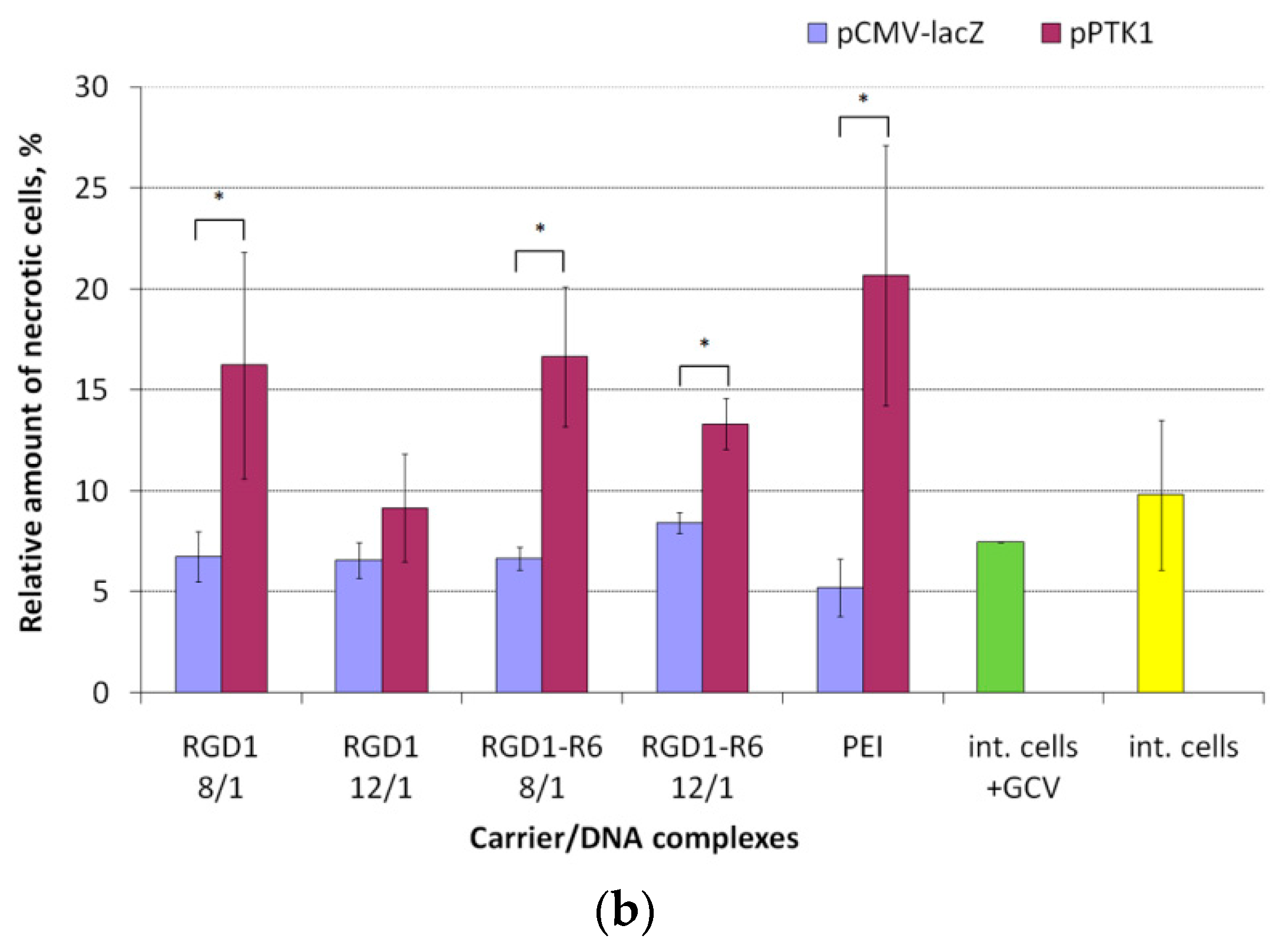
3.8. Therapeutic Effect of RGD1-R6/pPTK1 Polyplexes after Ganciclovir (GSV) Treatment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baranov, V.S.; Ivaschenko, T.E.; Yarmolinskaya, M.I. Comparative systems genetics view of endometriosis and uterine leiomyoma: Two sides of the same coin? Syst. Biol. Reprod. Med. 2016, 62, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.; Curiel, D.T.; Rajaratnam, V.; Thota, C.; Al-Hendy, A. Targeting adenoviral vectors for enhanced gene therapy of uterine leiomyomas. Hum. Reprod. 2013, 28, 2398–2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surrey, E.S.; Lietz, A.K.; Schoolcraft, W.B. Impact of intramural leiomyomata in patients with a normal endometrial cavity on in vitro fertilization–embryo transfer cycle outcome. Fertil. Steril. 2001, 75, 405–410. [Google Scholar] [CrossRef]
- Hadji, P.; Body, J.-J.; Aapro, M.S.; Brufsky, A.; Coleman, R.E.; Guise, T.; Lipton, A.; Tubiana-Hulin, M. Practical guidance for the management of aromatase inhibitor-associated bone loss. Ann. Oncol. 2008, 19, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Al-Hendy, A. Selective progesterone receptor modulators for fertility preservation in women with symptomatic uterine fibroids. Biol. Reprod. 2017, 97, 337–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hendy, A.; Salama, S. Gene therapy and uterine leiomyoma: A review. Hum. Reprod. Update 2006, 12, 385–400. [Google Scholar] [CrossRef] [Green Version]
- Hutchins, F.L. Abdominal myomectomy as a treatment for symptomatic uterine fibroids. Obstet. Gynecol. Clin. N. Am. 1995, 22, 781–789. [Google Scholar]
- Gwak, S.J.; Lee, J.S. Suicide gene therapy by amphiphilic copolymer nanocarrier for spinal cord tumor. Nanomaterials 2019, 9, 573. [Google Scholar] [CrossRef] [Green Version]
- Hattori, Y.; Maitani, Y. Folate-linked nanoparticle-mediated suicide gene therapy in human prostate cancer and nasopharyngeal cancer with herpes simplex virus thymidine kinase. Cancer Gene Ther. 2005, 12, 796–809. [Google Scholar] [CrossRef]
- Won, Y.; Kim, K.; Su, S.; Lee, M.; Ha, Y.; Kim, Y. Biomaterials Suicide gene therapy using reducible poly (oligo-d-arginine) for the treatment of spinal cord tumors. Biomaterials 2011, 32, 9766–9775. [Google Scholar] [CrossRef]
- Fillat, C.; Carrio, M.; Cascante, A.; Sangro, B. Suicide gene therapy mediated by the herpes simplex virus thymidine kinase Gene/ganciclovir system: Fifteen years of application. Curr. Gene Ther. 2003, 3, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Niu, H. Nonviral vector-mediated thymidine kinase gene transfer and ganciclovir treatment in leiomyoma cells. Obstet. Gynecol. 1998, 91, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, S.M.; Khater, M.K.; Perucho, A.M.; Mohamed, S.A.; Helwa, I.; Laknaur, A.; Lebedyeva, I.; Liu, Y.; Diamond, M.P.; Al-Hendy, A.A. Magnetic nanoparticles as a new approach to improve the efficacy of gene therapy against differentiated human uterine fibroid cells and tumor-initiating stem cells. Fertil. Steril. 2016, 105, 1638–1648.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staquicini, F.I.; Smith, T.L.; Tang, F.H.F.; Gelovani, J.G.; Giordano, R.J.; Libutti, S.K.; Sidman, R.L.; Cavenee, W.K.; Arap, W.; Pasqualini, R. Targeted AAVP-based therapy in a mouse model of human glioblastoma: A comparison of cytotoxic versus suicide gene delivery strategies. Cancer Gene Ther. 2020, 27, 301–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- De Raad, M.; Teunissen, E.A.; Mastrobattista, E. Peptide vectors for gene delivery: From single peptides to multifunctional peptide nanocarriers. Nanomedicine 2014, 9, 2217–2232. [Google Scholar] [CrossRef]
- Bennett, R.; Yakkundi, A.; McKeen, H.D.; McClements, L.; McKeogh, T.J.; McCrudden, C.M.; Arthur, K.; Robson, T.; Mccarthy, H.O. RALA-mediated delivery of FKBPL nucleic acid therapeutics. Nanomedicine 2015, 10, 2989–3001. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Liu, X.; Zhu, D.; Wang, Y.; Zhang, Z.; Zhou, X.; Qiu, N.; Chen, X.; Shen, Y. Nonviral cancer gene therapy: Delivery cascade and vector nanoproperty integration. Adv. Drug Deliv. Rev. 2017, 115, 115–154. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, J.; Zou, Y.; Wu, J.; Yao, Y.; Fan, H.; Liu, K.; Wang, J.; Gao, S. Modification of degradable nonviral delivery vehicle with a novel bifunctional peptide to enhance transfection in vivo. Nanomedicine 2018, 13, 9–24. [Google Scholar] [CrossRef]
- Abdelhamid, H.N.; Dowaidar, M.; Hällbrink, M.; Langel, Ü. Gene delivery using cell penetrating peptides-zeolitic imidazolate frameworks. Microporous Mesoporous Mater. 2020, 300, 110173. [Google Scholar] [CrossRef]
- Langlet-bertin, B.; Leborgne, C.; Scherman, D.; Bechinger, B.; Mason, A.J.; Kichler, A. Design and evaluation of histidine-rich amphipathic peptides for siRNA delivery. Pharm. Res. 2010, 1426–1436. [Google Scholar] [CrossRef] [PubMed]
- Egorova, A.; Kiselev, A. Peptide modules for overcoming barriers of nucleic acids transport to cells. Curr. Top. Med. Chem. 2016, 16, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-penetrating peptides: From basic research to clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.A.; Wan, Y.; Toth, I.; Moyle, P.M. Bombesin/oligoarginine fusion peptides for gastrin releasing peptide receptor (GRPR) targeted gene delivery. Bioorgan. Med. Chem. 2018, 26, 516–526. [Google Scholar] [CrossRef]
- Kurrikoff, K.; Freimann, K.; Veiman, K.-L.; Peets, E.M.; Piirsoo, A.; Langel, Ü. Effective lung-targeted RNAi in mice with peptide-based delivery of nucleic acid. Sci. Rep. 2019, 9, 19926. [Google Scholar] [CrossRef]
- Shirazi, A.N.; El-Sayed, N.S.; Mandal, D.; Tiwari, R.K.; Tavakoli, K.; Etesham, M.; Parang, K. Cysteine and arginine-rich peptides as molecular carriers. Bioorg. Med. Chem. Lett. 2016, 26, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M. Peptides, polypeptides and peptide-polymer hybrids as nucleic acid carriers. Biomater. Sci. 2017, 5, 2188–2211. [Google Scholar] [CrossRef]
- Lu, Y.; Jiang, W.; Wu, X.; Huang, S.; Huang, Z.; Shi, Y.; Dai, Q.; Chen, J.; Ren, F.; Gao, S. Peptide T7-modified polypeptide with disulfide bonds for targeted delivery of plasmid DNA for gene therapy of prostate cancer. Int. J. Nanomed. 2018, 13, 6913–6927. [Google Scholar] [CrossRef] [Green Version]
- Kiselev, A.; Egorova, A.; Laukkanen, A.; Baranov, V.; Urtti, A. Characterization of reducible peptide oligomers as carriers for gene delivery. Int. J. Pharm. 2013, 441, 736–747. [Google Scholar] [CrossRef]
- Cheng, R.; Feng, F.; Meng, F.; Deng, C.; Feijen, J.; Zhong, Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J. Control. Release 2011, 152, 2–12. [Google Scholar] [CrossRef]
- Kichler, A.; Mason, A.J.; Bechinger, B. Cationic amphipathic histidine-rich peptides for gene delivery. Biochim. Biophys. Acta Biomembr. 2006, 1758, 301–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuberg, P.; Wagner, A.; Remy, J.S.; Kichler, A. Design and evaluation of ionizable peptide amphiphiles for siRNA delivery. Int. J. Pharm. 2019, 566, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aisenbrey, C.; Douat, C.; Kichler, A.; Guichard, G.; Bechinger, B.; Bechinger, B. Characterization of the DNA and Membrane Interactions of a Bioreducible Cell-Penetrating Foldamer in its Monomeric and Dimeric Form. J. Phys. Chem. B 2020, 124, 4476–4486. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cheng, X.; Tian, X.; Guan, D.; Ao, J.; Wu, Z.; Huang, W.; Le, Z. Design and synthesis of novel dual-cyclic RGD peptides for αvβ3 integrin targeting. Bioorg. Med. Chem. Lett. 2019, 29, 896–900. [Google Scholar] [CrossRef]
- Malik, M.; Norian, J.; McCarthy-Keith, D.; Britten, J.; Catherino, W. Why leiomyomas are called fibroids: The central role of extracellular matrix in symptomatic women. Semin. Reprod. Med. 2010, 28, 169–179. [Google Scholar] [CrossRef]
- Pytela, R.; Pierschbacher, M.D.; Argraves, S.; Suzuki, S.; Ruoslahti, E. [27] Arginine-glycine-aspartic acid adhesion receptors. Methods Enzymol. 1987, 144, 475–489. [Google Scholar]
- Kim, H.A.; Nam, K.; Kim, S.W. Tumor targeting RGD conjugated bio-reducible polymer for VEGF siRNA expressing plasmid delivery. Biomaterials 2014, 35, 7543–7552. [Google Scholar] [CrossRef] [Green Version]
- Pezzoli, D.; Tarsini, P.; Melone, L.; Candiani, G. RGD-derivatized PEI-PEG copolymers: Influence of the degree of substitution on the targeting behavior. J. Drug Deliv. Sci. Technol. 2017, 37, 115–122. [Google Scholar] [CrossRef]
- Bjorge, J.D.; Pang, A.; Fujita, D.J. Delivery of gene targeting siRNAs to breast cancer cells using a multifunctional peptide complex that promotes both targeted delivery and endosomal release. PLoS ONE 2017. [Google Scholar] [CrossRef]
- Zuo, H. iRGD: A Promising Peptide for Cancer Imaging and a Potential Therapeutic Agent for Various Cancers. J. Oncol. 2019, 2019, 1–15. [Google Scholar] [CrossRef]
- Gregory, J.V.; Kadiyala, P.; Doherty, R.; Cadena, M.; Habeel, S.; Ruoslahti, E.; Lowenstein, P.R.; Castro, M.G.; Lahann, J. Systemic brain tumor delivery of synthetic protein nanoparticles for glioblastoma therapy. Nat. Commun. 2020, 11, 5687. [Google Scholar] [CrossRef] [PubMed]
- Egorova, A.; Selutin, A.; Maretina, M.; Selkov, S.; Baranov, V.; Kiselev, A. Characterization of iRGD-ligand modified arginine-histidine-rich peptides for nucleic acid therapeutics delivery to αvβ3 integrin-expressing cancer cells. Pharmaceuticals 2020, 13, 300. [Google Scholar] [CrossRef] [PubMed]
- Egorova, A.A.; Shtykalova, S.V.; Maretina, M.A.; Selyutin, A.V.; Shved, N.Y.; Krylova, N.V.; Ilina, A.V.; Pyankov, I.A.; Freund, S.A.; Selkov, S.A.; et al. Cys-flanked cationic peptides for cell delivery of the herpes simplex virus thymidine kinase gene for suicide gene therapy of uterine leiomyoma. Mol. Biol. 2020, 54, 436–448. [Google Scholar] [CrossRef]
- Egorova, A.; Bogacheva, M.; Shubina, A.; Baranov, V.; Kiselev, A. Development of a receptor-targeted gene delivery system using CXCR4 ligand-conjugated cross-linking peptides. J. Gene Med. 2014, 16, 336–351. [Google Scholar] [CrossRef] [PubMed]
- Aitken, A.; Learmonth, M. Estimation of disulfide bonds using Ellman’s reagent. In The Protein Protocols Handbook; Humana Press: Totowa, NJ, USA, 1996; pp. 487–488. [Google Scholar]
- Chen, H.; Wang, L.; Yeh, J.; Wu, X.; Cao, Z.; Wang, Y.A.; Zhang, M.; Yang, L.; Mao, H. Reducing non-specific binding and uptake of nanoparticles and improving cell targeting with an antifouling PEO-b-PγMPS copolymer coating. Biomaterials 2010, 31, 5397–5407. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Kanazawa, T.; Horiuchi, S.; Ando, T.; Sugawara, K.; Takashima, Y.; Seta, Y.; Okada, H. Cytoplasm-responsive nanocarriers conjugated with a functional cell-penetrating peptide for systemic siRNA delivery. Int. J. Pharm. 2013, 455, 40–47. [Google Scholar] [CrossRef]
- Kanazawa, T.; Hamasaki, T.; Endo, T.; Tamano, K.; Sogabe, K.; Seta, Y.; Ohgi, T.; Okada, H. Functional peptide nanocarriers for delivery of novel anti-RelA RNA interference agents as a topical treatment of atopic dermatitis. Int. J. Pharm. 2015, 489, 261–267. [Google Scholar] [CrossRef]
- Kanazawa, T.; Endo, T.; Arima, N.; Ibaraki, H.; Takashima, Y.; Seta, Y. Systemic delivery of small interfering RNA targeting nuclear factor κB in mice with collagen-induced arthritis using arginine-histidine-cysteine based oligopeptide-modified polymer nanomicelles. Int. J. Pharm. 2016, 515, 315–323. [Google Scholar] [CrossRef]
- Ibaraki, H.; Kanazawa, T.; Takashima, Y.; Okada, H.; Seta, Y. Transdermal anti-nuclear kappaB siRNA therapy for atopic dermatitis using a combination of two kinds of functional oligopeptide. Int. J. Pharm. 2018, 542, 213–220. [Google Scholar] [CrossRef]
- Egorova, A.; Petrosyan, M.; Maretina, M.; Balashova, N.; Polyanskih, L.; Baranov, V.; Kiselev, A. Anti-angiogenic treatment of endometriosis via anti-VEGFA siRNA delivery by means of peptide-based carrier in a rat subcutaneous model. Gene Ther. 2018, 25, 548–555. [Google Scholar] [CrossRef]
- Niidome, T.; Ohmori, N.; Ichinose, A.; Wada, A.; Mihara, H.; Hirayama, T.; Aoyagi, H. Binding of cationic α-helical peptides to plasmid DNA and their gene transfer abilities into cells. J. Biol. Chem. 1997, 272, 15307–15312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloomfield, V.A. DNA condensation by multivalent cations. Biopolymers 1997, 44, 269–282. [Google Scholar] [CrossRef]
- Männistö, M.; Vanderkerken, S.; Toncheva, V.; Elomaa, M.; Ruponen, M.; Schacht, E.; Urtti, A. Structure-activity relationships of poly(l-lysines): Effects of pegylation and molecular shape on physicochemical and biological properties in gene delivery. J. Control. Release 2002, 83, 169–182. [Google Scholar] [CrossRef]
- Ruponen, M.; Honkakoski, P.; Tammi, M.; Urtti, A. Cell-surface glycosaminoglycans inhibit cation-mediated gene transfer. J. Gene Med. 2004, 6, 405–414. [Google Scholar] [CrossRef]
- Lehtinen, J.; Hyvönen, Z.; Subrizi, A.; Bunjes, H.; Urtti, A. Glycosaminoglycan-resistant and pH-sensitive lipid-coated DNA complexes produced by detergent removal method. J. Control. Release 2008, 131, 145–149. [Google Scholar] [CrossRef]
- Huth, S.; Hoffmann, F.; von Gersdorff, K.; Laner, A.; Reinhardt, D.; Rosenecker, J.; Rudolph, C. Interaction of polyamine gene vectors with RNA leads to the dissociation of plasmid DNA-carrier complexes. J. Gene Med. 2006, 8, 1416–1424. [Google Scholar] [CrossRef]
- Mounkes, L.C.; Zhong, W.; Cipres-Palacin, G.; Heath, T.D.; Debs, R.J. Proteoglycans mediate cationic liposome-DNA complex-based gene delivery in vitro and in vivo. J. Biol. Chem. 1998. [Google Scholar] [CrossRef] [Green Version]
- Lai, W.F.; Wong, W.T. Design of polymeric gene carriers for effective intracellular delivery. Trends Biotechnol. 2018, 36, 713–728. [Google Scholar] [CrossRef]
- Patel, S.; Kim, J.; Herrera, M.; Mukherjee, A.; Kabanov, A.V.; Sahay, G. Brief update on endocytosis of nanomedicines. Adv. Drug Deliv. Rev. 2019, 144, 90–111. [Google Scholar] [CrossRef]
- Midoux, P.; Breuzard, G.; Gomez, J.; Pichon, C. Polymer-based gene delivery: A current review on the uptake and intracellular trafficking of polyplexes. Curr. Gene Ther. 2008, 8, 335–352. [Google Scholar] [CrossRef]
- Jones, N.A.; Hill, I.R.C.; Stolnik, S.; Bignotti, F.; Davis, S.S.; Garnett, M.C. Polymer chemical structure is a key determinant of physicochemical and colloidal properties of polymer–DNA complexes for gene delivery. Biochim. Biophys. Acta Gene Struct. Expr. 2000, 1517, 1–18. [Google Scholar] [CrossRef]
- Haladjova, E.; Halacheva, S.; Posheva, V.; Peycheva, E.; Moskova-Doumanova, V.; Topouzova-Hristova, T.; Doumanov, J.; Rangelov, S. Comblike polyethylenimine-based polyplexes: Balancing toxicity, cell internalization, and transfection efficiency via polymer chain topology. Langmuir 2015, 31, 10017–10025. [Google Scholar] [CrossRef] [PubMed]
- Kargaard, A.; Sluijter, J.P.G.; Klumperman, B. Polymeric siRNA gene delivery—Transfection efficiency versus cytotoxicity. J. Control. Release 2019, 316, 263–291. [Google Scholar] [CrossRef] [PubMed]
- Swenson, S.; Ramu, S.; Markland, F. Anti-Angiogenesis and RGD-Containing Snake Venom Disintegrins. Curr. Pharm. Des. 2007, 13, 2860–2871. [Google Scholar] [CrossRef] [PubMed]
- Vachutinsky, Y.; Oba, M.; Miyata, K.; Hiki, S.; Kano, M.R.; Nishiyama, N.; Koyama, H.; Miyazono, K.; Kataoka, K. Antiangiogenic gene therapy of experimental pancreatic tumor by sFlt-1 plasmid DNA carried by RGD-modified crosslinked polyplex micelles. J. Control. Release 2011, 149, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Onoue, S.; Yamada, S.; Chan, H.K. Nanodrugs: Pharmacokinetics and safety. Int. J. Nanomed. 2014, 9, 1025–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritton, K.; Borahay, M.A. New and Emerging Therapies for Uterine Fibroids. Semin. Reprod. Med. 2017, 35, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Rodríguez, R.A.; Arango-Rodríguez, M.L.; Escobedo, L.; Hernandez-Baltazar, D.; Gompel, A.; Forgez, P.; Martínez-Fong, D. Suicide HSVtk gene delivery by neurotensin-polyplex nanoparticles via the bloodstream and GCV treatment specifically inhibit the growth of human MDA-MB-231 triple negative breast cancer tumors xenografted in athymic mice. PLoS ONE 2014. [Google Scholar] [CrossRef]
- Andrea, P.; Jana, J.; Ursula, A.; Cestmir, A. Suicide Gene Therapy Mediated with Exosomes. Cancers 2020, 12, 1096. [Google Scholar]
- Andersen, J.; Grine, E.; Eng, C.L.Y.; Zhao, K.; Barbieri, R.L.; Chumas, J.C.; Brink, P.R. Expression of connexin-43 in human myometrium and leiomyoma. Am. J. Obstet. Gynecol. 1993, 169, 1266–1276. [Google Scholar] [CrossRef]
- Andrade-Rozental, A.; Rozental, R.; Hopperstad, M.; Wu, J.; Vrionis, F.; Spray, D. Gap junctions: The “kiss of death” and the “kiss of life”. Brain Res. Rev. 2000, 32, 308–315. [Google Scholar] [CrossRef]
- Huang, Q.; Liu, X.-Z.; Kang, C.-S.; Wang, G.-X.; Zhong, Y.; Pu, P.-Y. The anti-glioma effect of suicide gene therapy using BMSC expressing HSV/TK combined with overexpression of Cx43 in glioma cells. Cancer Gene Ther. 2010, 17, 192–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koopman, G.; Reutelingsperger, C.; Kuijten, G.; Keehnen, R.; Pals, S.; van Oers, M. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 1994, 84, 1415–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinnapireddy, S.R.; Duse, L.; Strehlow, B.; Schäfer, J.; Bakowsky, U. Composite liposome-PEI/nucleic acid lipopolyplexes for safe and efficient gene delivery and gene knockdown. Colloids Surf. B Biointerfaces 2017, 158, 93–101. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carrier | Composition (mol%) |
|---|---|
| RGD0-R6 | RRRRRRRRRHHHH (50 mol%) + CHRRRRRRHC (50 mol%) |
| RGD1-R6 | RRRRRRRRRHHHH-CRGDRGPDC (50 mol%) + CHRRRRRRHC (50 mol%) |__________| |
| Carrier | Charge Ratio | Size (nm) ± S.D. | ʐ-Potential (mV) ± S.D. |
|---|---|---|---|
| RGD1-R6 | 4/1 8/1 | 304.8 ± 0.69 98.8 ± 0.49 | 4.7 ± 0.7 16.2 ± 0.1 |
| 12/1 16/1 | 102.0 ± 0.38 121.2 ± 0.25 | 24.0 ± 0.3 25.2 ± 0.1 | |
| RGD0-R6 | 4/1 8/1 | 308.0 ± 0.35 97.5 ± 0.45 | 5.1 ± 0.6 20.2 ± 0.5 |
| 12/1 16/1 | 101.1 ± 0.51 129.6 ± 0.42 | 25.1 ± 0.5 26.9 ± 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egorova, A.; Shtykalova, S.; Selutin, A.; Shved, N.; Maretina, M.; Selkov, S.; Baranov, V.; Kiselev, A. Development of iRGD-Modified Peptide Carriers for Suicide Gene Therapy of Uterine Leiomyoma. Pharmaceutics 2021, 13, 202. https://doi.org/10.3390/pharmaceutics13020202
Egorova A, Shtykalova S, Selutin A, Shved N, Maretina M, Selkov S, Baranov V, Kiselev A. Development of iRGD-Modified Peptide Carriers for Suicide Gene Therapy of Uterine Leiomyoma. Pharmaceutics. 2021; 13(2):202. https://doi.org/10.3390/pharmaceutics13020202
Chicago/Turabian StyleEgorova, Anna, Sofia Shtykalova, Alexander Selutin, Natalia Shved, Marianna Maretina, Sergei Selkov, Vladislav Baranov, and Anton Kiselev. 2021. "Development of iRGD-Modified Peptide Carriers for Suicide Gene Therapy of Uterine Leiomyoma" Pharmaceutics 13, no. 2: 202. https://doi.org/10.3390/pharmaceutics13020202
APA StyleEgorova, A., Shtykalova, S., Selutin, A., Shved, N., Maretina, M., Selkov, S., Baranov, V., & Kiselev, A. (2021). Development of iRGD-Modified Peptide Carriers for Suicide Gene Therapy of Uterine Leiomyoma. Pharmaceutics, 13(2), 202. https://doi.org/10.3390/pharmaceutics13020202