Synthetic Virus-Derived Nanosystems (SVNs) for Delivery and Precision Docking of Large Multifunctional DNA Circuitry in Mammalian Cells


Abstract
:1. Introduction
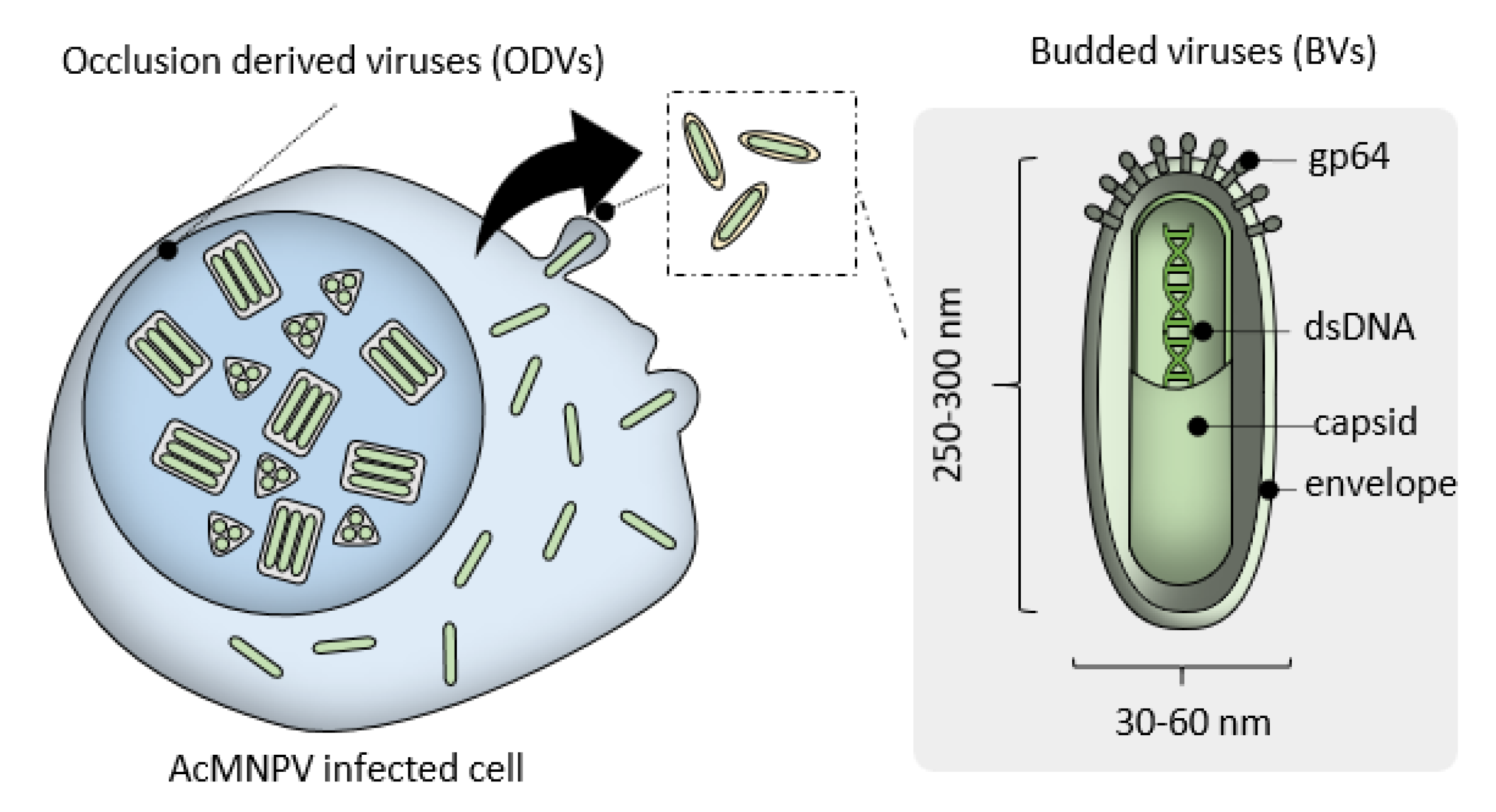
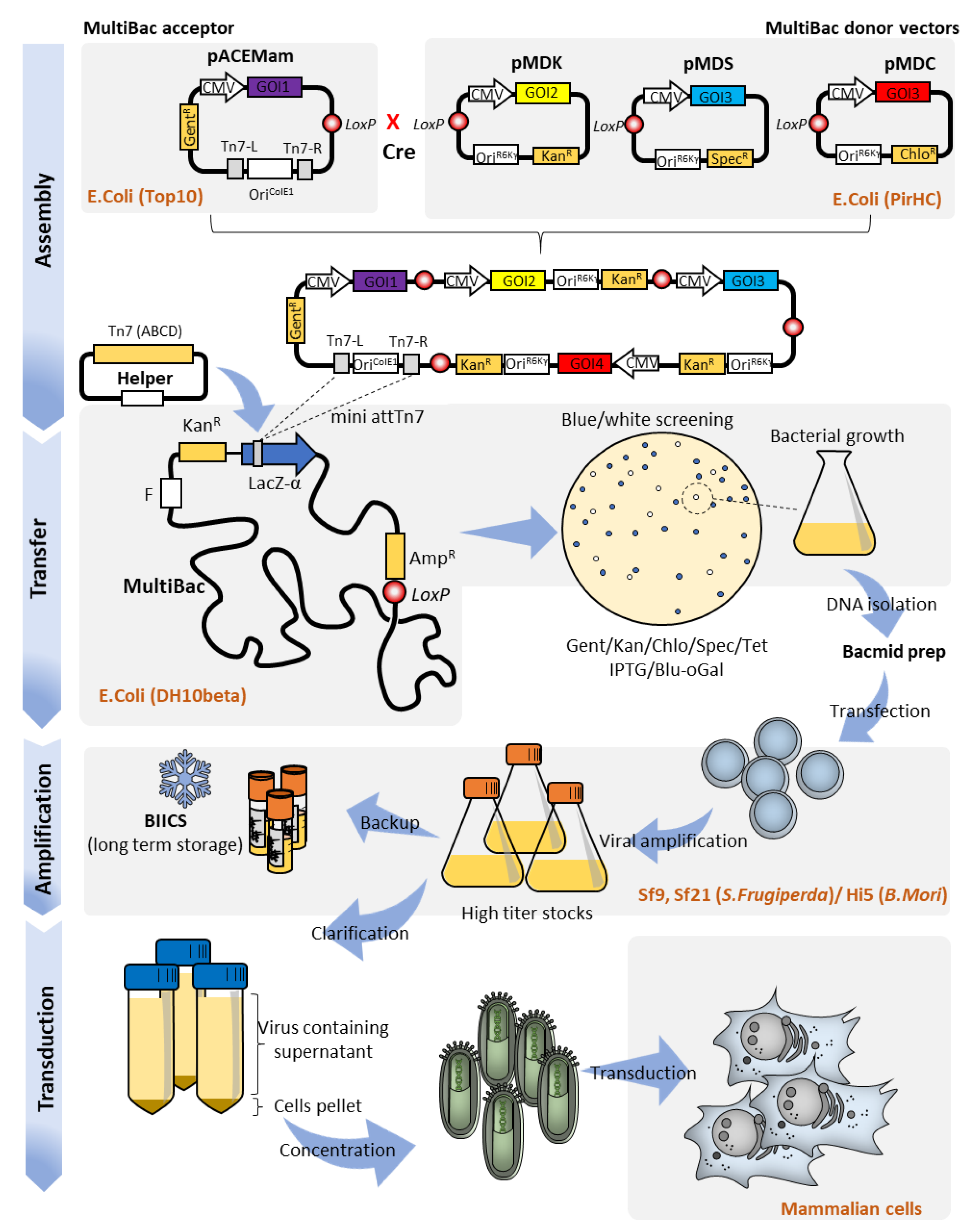
2. MultiBac Baculovirus Engineering
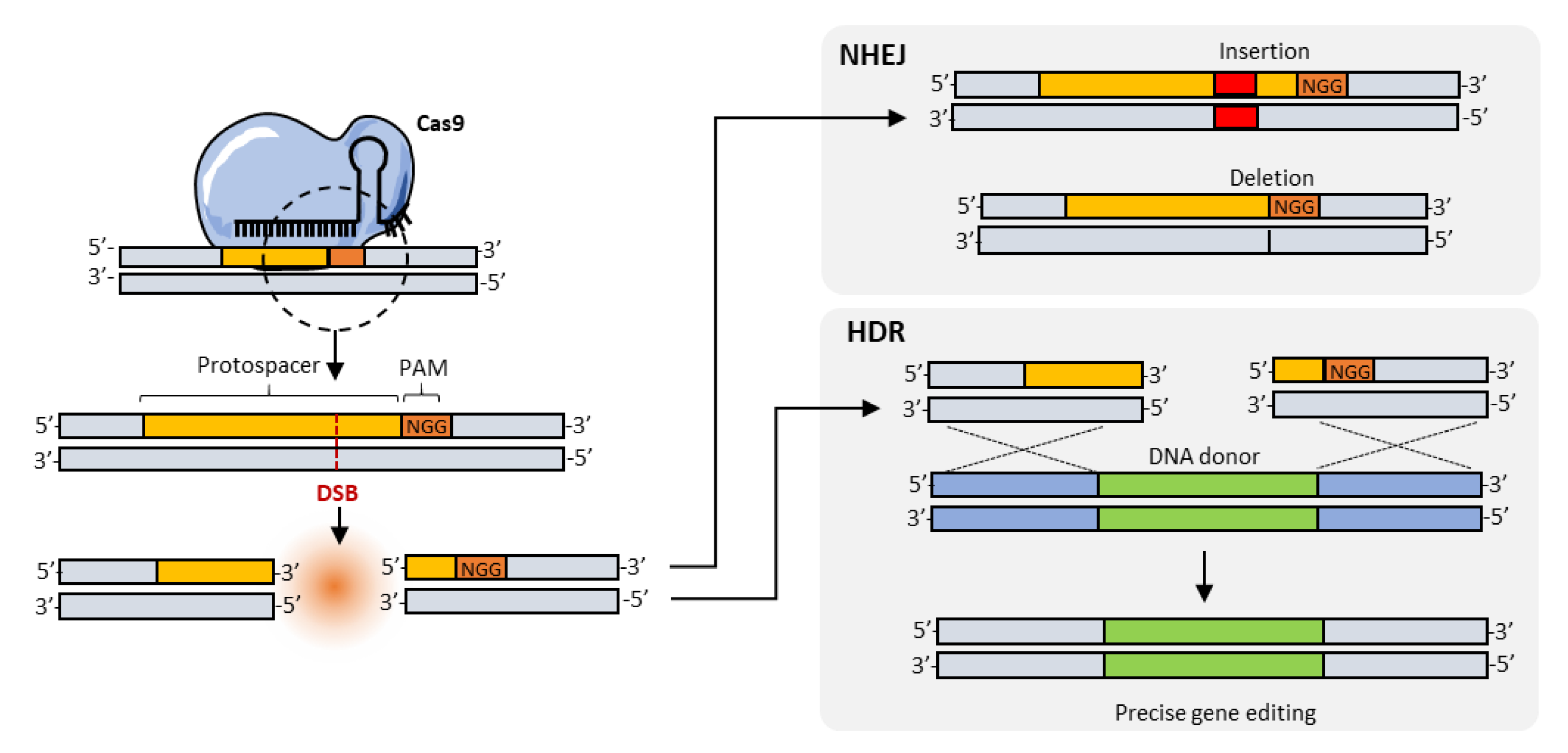
3. Homology-Directed Repair (HDR), Base Editing, and Search-and-Replace Approaches
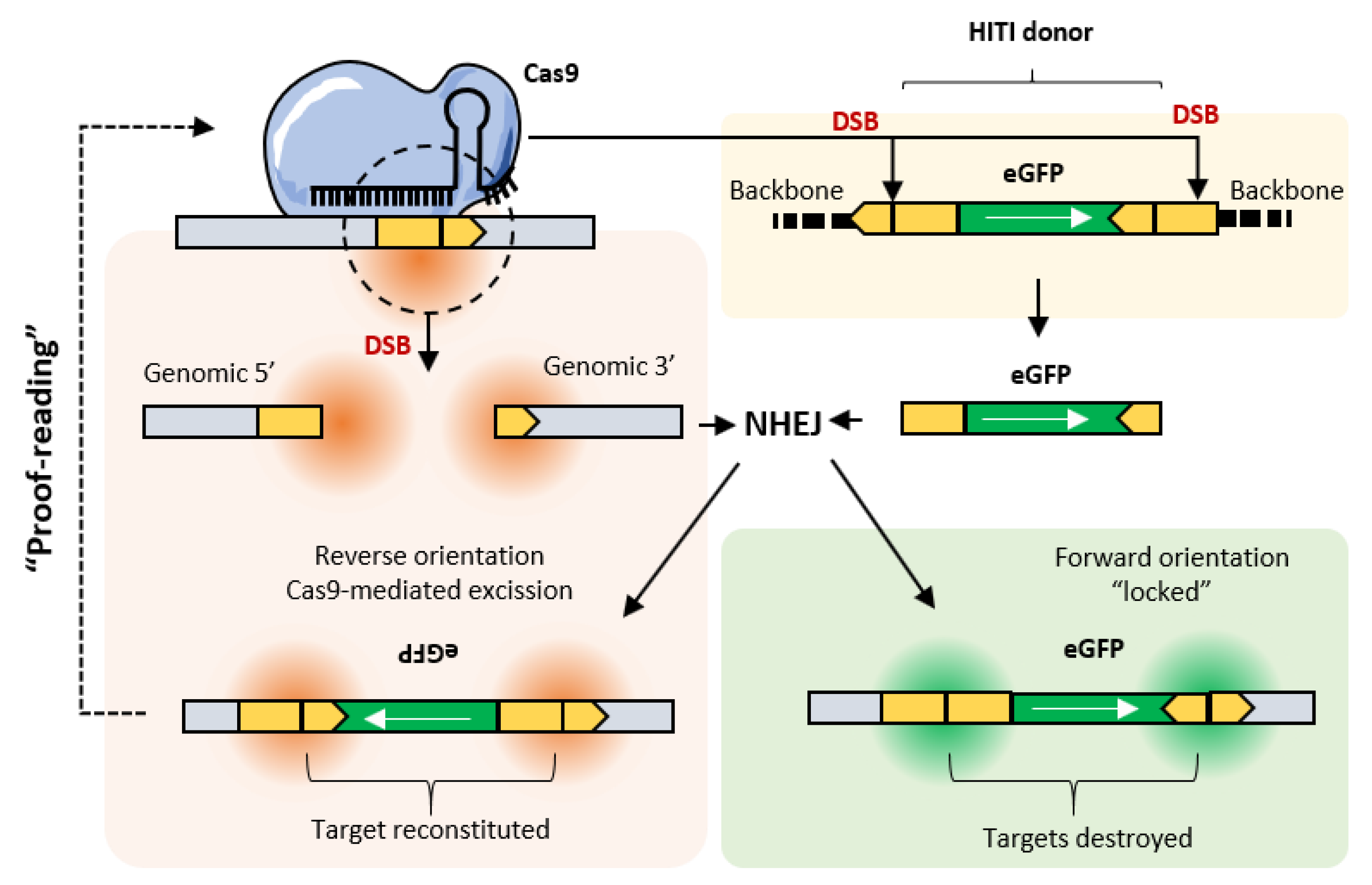
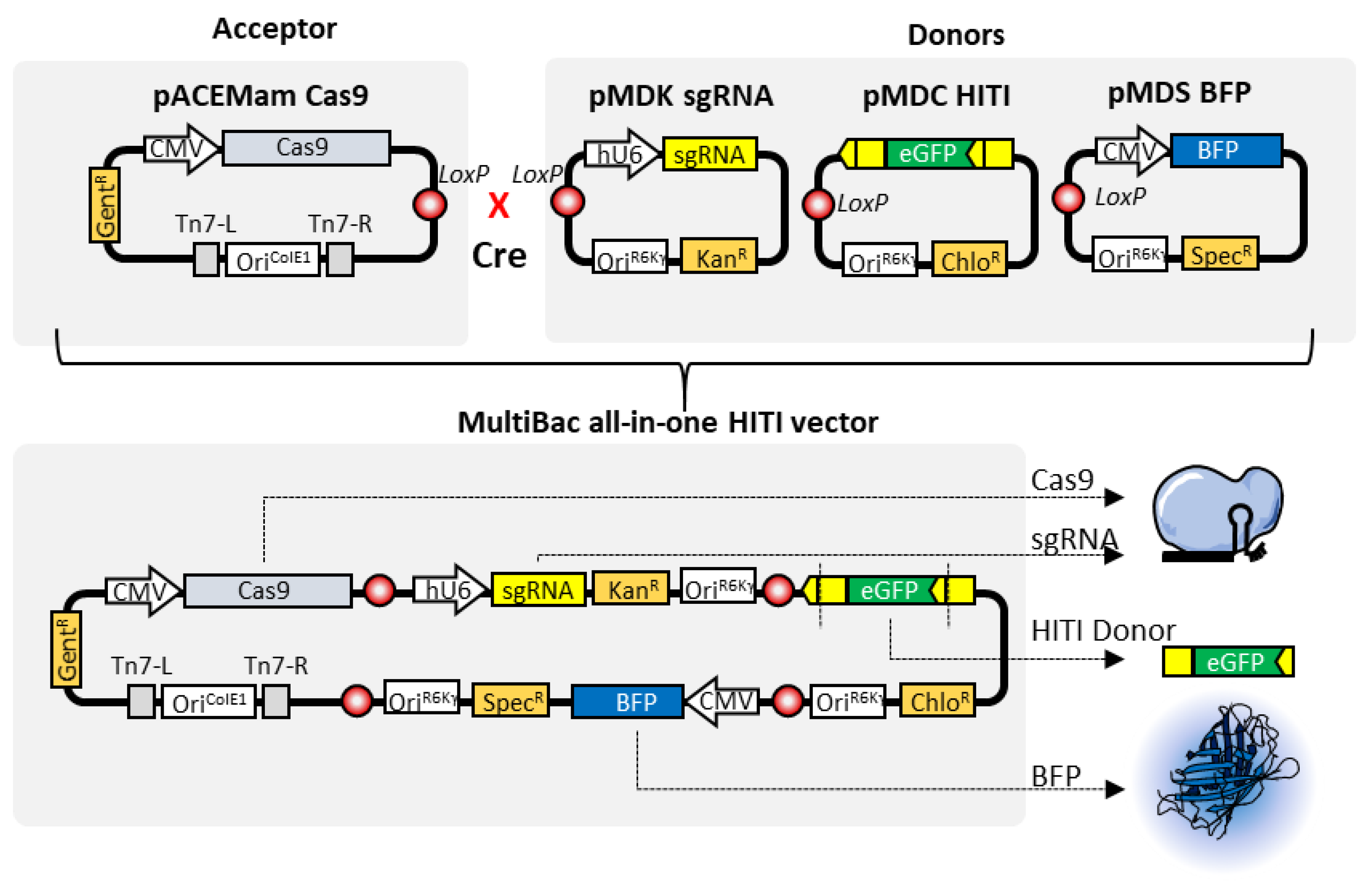
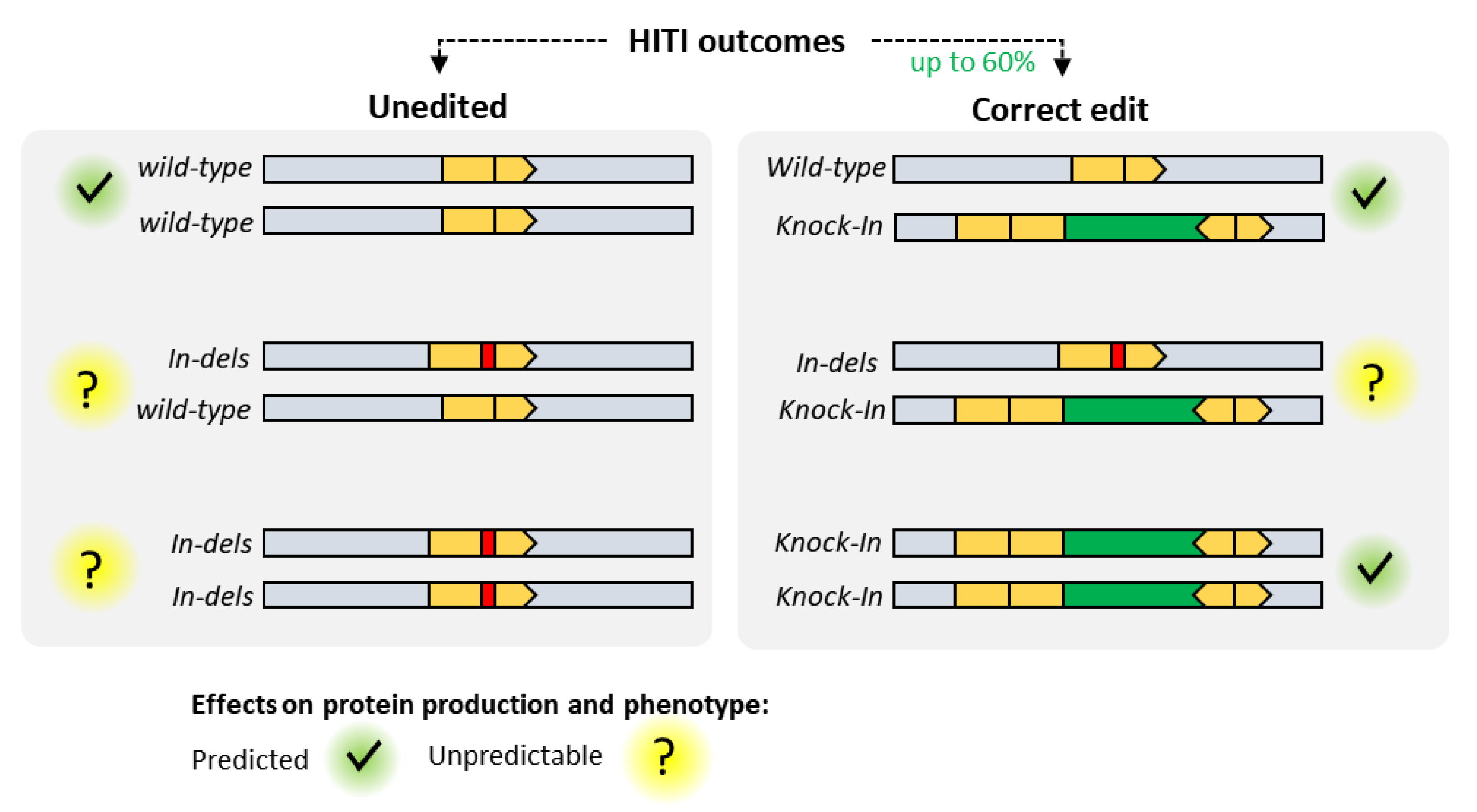
4. MultiBac and Clustered Regularly Interspaced Palindromic Repeats (CRISPR)
5. Challenges to Baculovirus Delivery In Vivo
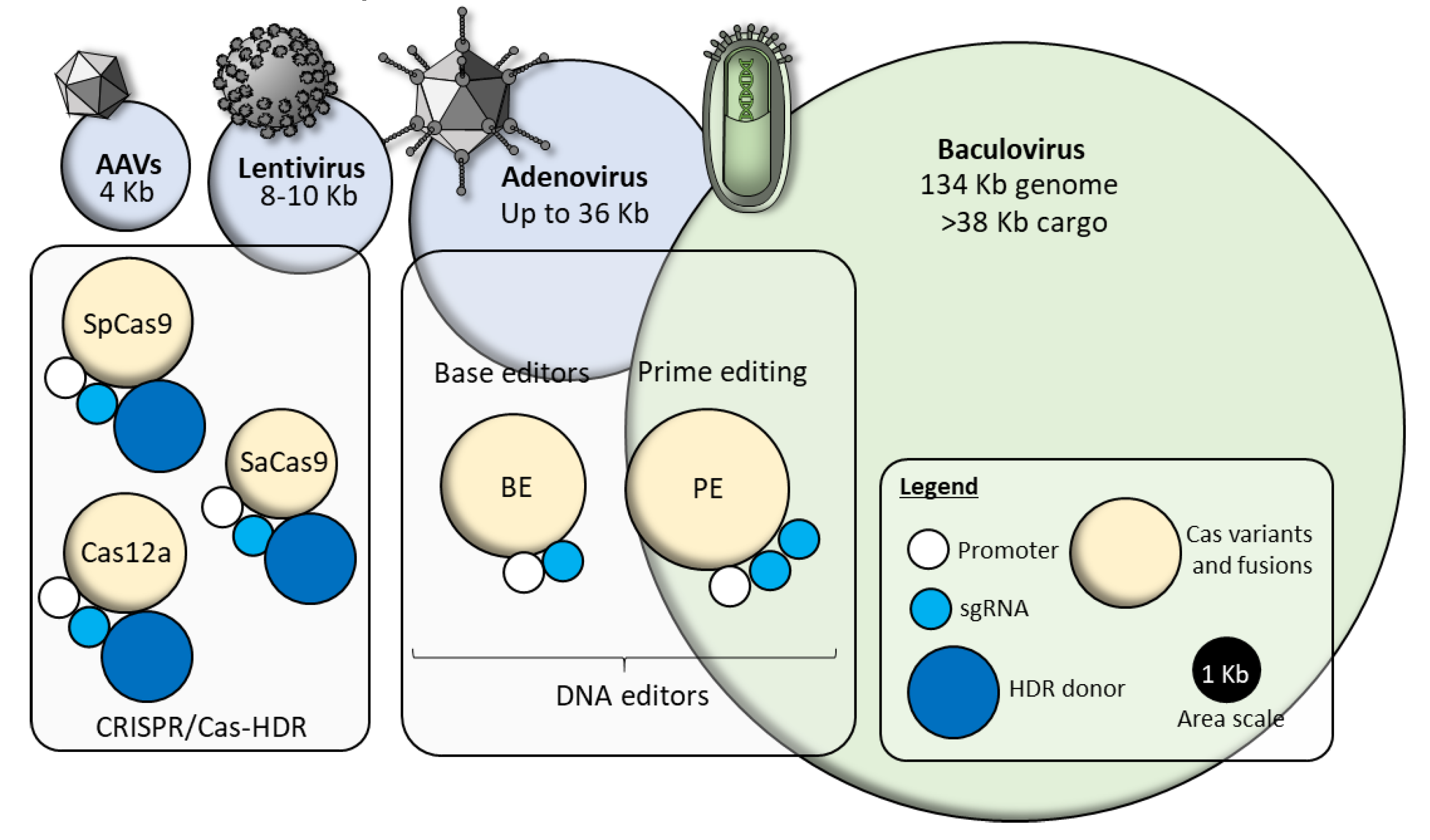
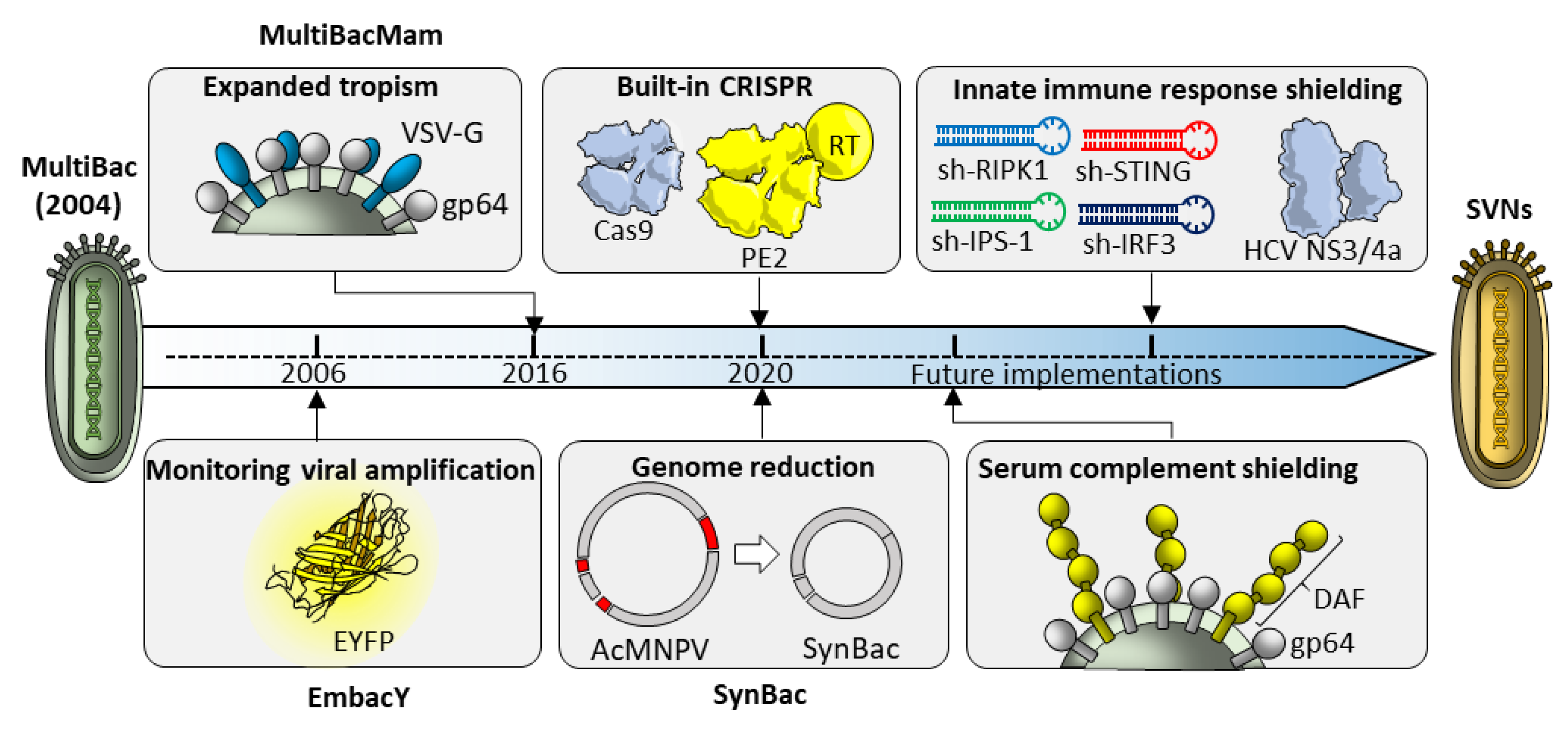
6. From MultiBac to Synthetic Virus-Derived Nanosystems (SVNs)
7. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neguembor, M.V.; Sebastian-Perez, R.; Aulicino, F.; Gomez-Garcia, P.A.; Cosma, M.P.; Lakadamyali, M. (Po)STAC (Polycistronic SunTAg modified CRISPR) enables live-cell and fixed-cell super-resolution imaging of multiple genes. Nucleic Acids Res. 2017, 46, e30. [Google Scholar] [CrossRef] [Green Version]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.-J.; Orlova, N.; Oakes, B.L.; Ma, E.; Spinner, H.B.; Baney, K.L.M.; Chuck, J.; Tan, D.; Knott, G.J.; Harrington, L.B.; et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature 2019, 566, 218–223. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Xu, Z.; Zhang, Z.; Chen, X.; Zeng, X.; Zhang, Y.; Deng, T.; Ren, M.; Sun, Z.; Jiang, R.; et al. Engineer chimeric Cas9 to expand PAM recognition based on evolutionary information. Nat. Commun. 2019, 10, 560. [Google Scholar] [CrossRef] [Green Version]
- Saha, J.; Wang, S.-Y.; Davis, A.J. Examining DNA Double-Strand Break Repair in a Cell Cycle-Dependent Manner. Methods Enzym. 2017, 591, 97–118. [Google Scholar] [CrossRef] [Green Version]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 2018, 559, 405–409. [Google Scholar] [CrossRef]
- Noureddine, A.; Maestas-Olguin, A.; Saada, E.A.; LaBauve, A.E.; Agola, J.O.; Baty, K.E.; Howard, T.; Sabo, J.K.; Espinoza, C.R.S.; Doudna, J.A.; et al. Engineering of monosized lipid-coated mesoporous silica nanoparticles for CRISPR delivery. Acta Biomater. 2020. [Google Scholar] [CrossRef] [PubMed]
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2227–2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015, 33, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Alphonse, M.; Liu, Q. Strategies for nonviral nanoparticle-based delivery of CRISPR/Cas9 therapeutics. Wires Nanomed. Nanobiotechnol. 2020, 12, e1609. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, K.S.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Volak, A.; Spirig, S.E.; Muller, A.; Sousa, A.A.; Tsai, S.Q.; Bengtsson, N.E.; et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019, 10, 4439. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, N.E.; Hall, J.K.; Odom, G.L.; Phelps, M.P.; Andrus, C.R.; Hawkins, R.D.; Hauschka, S.D.; Chamberlain, J.R.; Chamberlain, J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 14454. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Suzuki, K.; Izpisua Belmonte, J.C. In vivo genome editing via the HITI method as a tool for gene therapy. J. Hum. Genet. 2018, 63, 157–164. [Google Scholar] [CrossRef]
- Mansouri, M.; Bellon-Echeverria, I.; Rizk, A.; Ehsaei, Z.; Cianciolo Cosentino, C.; Silva, C.S.; Xie, Y.; Boyce, F.M.; Davis, M.W.; Neuhauss, S.C.; et al. Highly efficient baculovirus-mediated multigene delivery in primary cells. Nat. Commun. 2016, 7, 11529. [Google Scholar] [CrossRef] [Green Version]
- Mansouri, M.; Ehsaei, Z.; Taylor, V.; Berger, P. Baculovirus-based genome editing in primary cells. Plasmid 2017, 90, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Sung, L.-Y.; Chen, C.-L.; Lin, S.-Y.; Li, K.-C.; Yeh, C.-L.; Chen, G.-Y.; Lin, C.-Y.; Hu, Y.-C. Efficient gene delivery into cell lines and stem cells using baculovirus. Nat. Protoc. 2014, 9, 1882–1899. [Google Scholar] [CrossRef] [PubMed]
- Rohrmann, G.F. Baculovirus Molecular Biology, 4th ed.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2019.
- Luckow, V.A.; Lee, S.C.; Barry, G.F.; Olins, P.O. Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli. J. Virol. 1993, 67, 4566–4579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kost, T.A.; Condreay, J.P.; Jarvis, D.L. Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nat. Biotechnol. 2005, 23, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, D.J.; Berger, P.; Schaffitzel, C.; Yamada, K.; Richmond, T.J.; Berger, I. Protein complex expression by using multigene baculoviral vectors. Nat. Methods 2006, 3, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Bieniossek, C.; Imasaki, T.; Takagi, Y.; Berger, I. MultiBac: Expanding the research toolbox for multiprotein complexes. Trends Biochem. Sci. 2012, 37, 49–57. [Google Scholar] [CrossRef]
- Gupta, K.; Tölzer, C.; Sari-Ak, D.; Fitzgerald, D.J.; Schaffitzel, C.; Berger, I. MultiBac: Baculovirus-Mediated Multigene DNA Cargo Delivery in Insect and Mammalian Cells. Viruses 2019, 11, 198. [Google Scholar] [CrossRef] [Green Version]
- Pelosse, M.; Crocker, H.; Gorda, B.; Lemaire, P.; Rauch, J.; Berger, I. MultiBac: From protein complex structures to synthetic viral nanosystems. BMC Biol. 2017, 15, 99. [Google Scholar] [CrossRef] [Green Version]
- Cory, J.S.; Bishop, D.H. Use of baculoviruses as biological insecticides. Mol. Biotechnol. 1997, 7, 303–313. [Google Scholar] [CrossRef]
- Heigwer, F.; Kerr, G.; Boutros, M. E-CRISP: Fast CRISPR target site identification. Nat. Methods 2014, 11, 122–123. [Google Scholar] [CrossRef]
- Paquet, D.; Kwart, D.; Chen, A.; Sproul, A.; Jacob, S.; Teo, S.; Olsen, K.M.; Gregg, A.; Noggle, S.; Tessier-Lavigne, M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016, 533, 125–129. [Google Scholar] [CrossRef]
- Heyer, W.-D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Shi, Z.; Guo, X.; Jiang, B.; Wang, G.; Luo, D.; Chen, Y.; Zhu, Y.-S. Ligase IV inhibitor SCR7 enhances gene editing directed by CRISPR–Cas9 and ssODN in human cancer cells. Cell Biosci. 2018, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canny, M.D.; Moatti, N.; Wan, L.C.K.; Fradet-Turcotte, A.; Krasner, D.; Mateos-Gomez, P.A.; Zimmermann, M.; Orthwein, A.; Juang, Y.-C.; Zhang, W.; et al. Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR–Cas9 genome-editing efficiency. Nat. Biotechnol. 2018, 36, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Bajar, B.T.; Lam, A.J.; Badiee, R.K.; Oh, Y.-H.; Chu, J.; Zhou, X.X.; Kim, N.; Kim, B.B.; Chung, M.; Yablonovitch, A.L.; et al. Fluorescent indicators for simultaneous reporting of all four cell cycle phases. Nat. Methods 2016, 13, 993–996. [Google Scholar] [CrossRef] [Green Version]
- Gutschner, T.; Haemmerle, M.; Genovese, G.; Draetta, G.F.; Chin, L. Post-translational Regulation of Cas9 during G1 Enhances Homology-Directed Repair. Cell Rep. 2016, 14, 1555–1566. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Scavuzzo, M.A.; Chmielowiec, J.; Sharp, R.; Bajic, A.; Borowiak, M. Enrichment of G2/M cell cycle phase in human pluripotent stem cells enhances HDR-mediated gene repair with customizable endonucleases. Sci. Rep. 2016, 6, 21264. [Google Scholar] [CrossRef] [Green Version]
- Casini, A.; Olivieri, M.; Petris, G.; Montagna, C.; Reginato, G.; Maule, G.; Lorenzin, F.; Prandi, D.; Romanel, A.; Demichelis, F.; et al. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat. Biotechnol. 2018, 36, 265–271. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [Green Version]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hu, S.; Chen, X. Non-viral delivery systems for CRISPR/Cas9-based genome editing: Challenges and opportunities. Biomaterials 2018, 171, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Hindriksen, S.; Bramer, A.J.; Truong, M.A.; Vromans, M.J.M.; Post, J.B.; Verlaan-Klink, I.; Snippert, H.J.; Lens, S.M.A.; Hadders, M.A. Baculoviral delivery of CRISPR/Cas9 facilitates efficient genome editing in human cells. PLoS ONE 2017, 12, e0179514. [Google Scholar] [CrossRef] [PubMed]
- Maresca, M.; Lin, V.G.; Guo, N.; Yang, Y. Obligate ligation-gated recombination (ObLiGaRe): Custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Res. 2013, 23, 539–546. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef]
- Artegiani, B.; Hendriks, D.; Beumer, J.; Kok, R.; Zheng, X.; Joore, I.; Chuva de Sousa Lopes, S.; van Zon, J.; Tans, S.; Clevers, H. Fast and efficient generation of knock-in human organoids using homology-independent CRISPR–Cas9 precision genome editing. Nat. Cell Biol. 2020, 22, 321–331. [Google Scholar] [CrossRef]
- Kelly, J.J.; Saee-Marand, M.; Nyström, N.N.; Chen, Y.; Evans, M.M.; Hamilton, A.M.; Ronald, J.A. A Safe Harbor-Targeted CRISPR/Cas9 Homology Independent Targeted Integration (HITI) System for Multi-Modality Reporter Gene-Based Cell Tracking. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Monsma, S.A.; Oomens, A.G.; Blissard, G.W. The GP64 envelope fusion protein is an essential baculovirus protein required for cell-to-cell transmission of infection. J. Virol. 1996, 70, 4607–4616. [Google Scholar] [CrossRef] [Green Version]
- Oomens, A.G.; Blissard, G.W. Requirement for GP64 to drive efficient budding of Autographa californica multicapsid nucleopolyhedrovirus. Virology 1999, 254, 297–314. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, C.; Kaname, Y.; Taguwa, S.; Abe, T.; Fukuhara, T.; Tani, H.; Moriishi, K.; Matsuura, Y. Baculovirus GP64-mediated entry into mammalian cells. J. Virol. 2012, 86, 2610–2620. [Google Scholar] [CrossRef] [Green Version]
- Blissard, G.W.; Wenz, J.R. Baculovirus gp64 envelope glycoprotein is sufficient to mediate pH-dependent membrane fusion. J. Virol. 1992, 66, 6829–6835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.; Pan, X.; Kormelink, R.; Vlak, J.M. Functional entry of baculovirus into insect and mammalian cells is dependent on clathrin-mediated endocytosis. J. Virol. 2006, 80, 8830–8833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, S.; Wang, M.; Qiu, Z.; Deng, F.; Vlak, J.M.; Hu, Z.; Wang, H. Autographa californica Multicapsid Nucleopolyhedrovirus Efficiently Infects Sf9 Cells and Transduces Mammalian Cells via Direct Fusion with the Plasma Membrane at Low pH. J. Virol. 2010. [Google Scholar] [CrossRef] [Green Version]
- Leikina, E.; Onaran, H.O.; Zimmerberg, J. Acidic pH induces fusion of cells infected with baculovirus to form syncytia. FEBS Lett. 1992, 304, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Au, S.; Pante, N. Nuclear transport of baculovirus: Revealing the nuclear pore complex passage. J. Struct. Biol. 2012, 177, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, T.; Volkman, L.E.; Welch, M.D. Actin-based motility drives baculovirus transit to the nucleus and cell surface. J. Cell Biol. 2010, 190, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Sinn, P.L.; Hwang, B.-Y.; Li, N.; Ortiz, J.L.S.; Shirazi, E.; Parekh, K.R.; Cooney, A.L.; Schaffer, D.V.; McCray, P.B. Novel GP64 envelope variants for improved delivery to human airway epithelial cells. Gene Ther. 2017, 24, 674–679. [Google Scholar] [CrossRef]
- Kim, Y.K.; Choi, J.Y.; Yoo, M.K.; Jiang, H.L.; Arote, R.; Je, Y.H.; Cho, M.H.; Cho, C.S. Receptor-mediated gene delivery by folate-PEG-baculovirus in vitro. J. Biotechnol. 2007, 131, 353–361. [Google Scholar] [CrossRef]
- Yang, Y.; Lo, S.L.; Yang, J.; Goh, S.S.; Wu, C.; Feng, S.S.; Wang, S. Polyethylenimine coating to produce serum-resistant baculoviral vectors for in vivo gene delivery. Biomaterials 2009, 30, 5767–5774. [Google Scholar] [CrossRef]
- Kim, Y.K.; Choi, J.Y.; Jiang, H.L.; Arote, R.; Jere, D.; Cho, M.H.; Je, Y.H.; Cho, C.S. Hybrid of baculovirus and galactosylated PEI for efficient gene carrier. Virology 2009, 387, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-K.; Park, I.-K.; Jiang, H.-L.; Choi, J.-Y.; Je, Y.-H.; Jin, H.; Kim, H.-W.; Cho, M.-H.; Cho, C.-S. Regulation of transduction efficiency by pegylation of baculovirus vector in vitro and in vivo. J. Biotechnol. 2006, 125, 104–109. [Google Scholar] [CrossRef]
- Yang, Q.; Lai, S.K. Anti-PEG immunity: Emergence, characteristics, and unaddressed questions. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 655–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regnström, K.; Ragnarsson, E.G.E.; Köping-Höggård, M.; Torstensson, E.; Nyblom, H.; Artursson, P. PEI – a potent, but not harmless, mucosal immuno-stimulator of mixed T-helper cell response and FasL-mediated cell death in mice. Gene Ther. 2003, 10, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Jorio, H.; Tran, R.; Meghrous, J.; Bourget, L.; Kamen, A. Analysis of baculovirus aggregates using flow cytometry. J. Virol. Methods 2006, 134, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Kaikkonen, M.U.; Räty, J.K.; Airenne, K.J.; Wirth, T.; Heikura, T.; Ylä-Herttuala, S. Truncated vesicular stomatitis virus G protein improves baculovirus transduction efficiency in vitro and in vivo. Gene Ther. 2005, 13, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Barsoum, J.; Brown, R.; McKee, M.; Boyce, F.M. Efficient transduction of mammalian cells by a recombinant baculovirus having the vesicular stomatitis virus G glycoprotein. Hum. Gene 1997, 8, 2011–2018. [Google Scholar] [CrossRef]
- Mangor, J.T.; Monsma, S.A.; Johnson, M.C.; Blissard, G.W. A GP64-Null Baculovirus Pseudotyped with Vesicular Stomatitis Virus G Protein. J. Virol. 2001, 75, 2544. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Yu, F.; Xu, J.; Li, Y.; Chen, H.; Xiao, S.; Fu, Z.F.; Fang, L. Rabies-virus-glycoprotein-pseudotyped recombinant baculovirus vaccine confers complete protection against lethal rabies virus challenge in a mouse model. Vet. Microbiol. 2014, 171, 93–101. [Google Scholar] [CrossRef]
- Buchholz, C.J.; Friedel, T.; Büning, H. Surface-Engineered Viral Vectors for Selective and Cell Type-Specific Gene Delivery. Trends Biotechnol. 2015, 33, 777–790. [Google Scholar] [CrossRef]
- Hu, L.; Li, Y.; Deng, F.; Hu, Z.; Wang, H.; Wang, M. Improving Baculovirus Transduction of Mammalian Cells by Incorporation of Thogotovirus Glycoproteins. Virol. Sin. 2019, 34, 454–466. [Google Scholar] [CrossRef]
- Huser, A.; Rudolph, M.; Hofmann, C. Incorporation of decay-accelerating factor into the baculovirus envelope generates complement-resistant gene transfer vectors. Nat. Biotechnol. 2001, 19, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y.; Kawabata, C.; Sakaguchi, M.; Tamura, T. Protection of Baculovirus Vectors Expressing Complement Regulatory Proteins against Serum Complement Attack. Biol. Pharm. Bull. 2018, 41, 1600–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condreay, J.P.; Witherspoon, S.M.; Clay, W.C.; Kost, T.A. Transient and stable gene expression in mammalian cells transduced with a recombinant baculovirus vector. Proc. Natl. Acad. Sci. USA 1999, 96, 127–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.C.; Tsai, C.T.; Chang, Y.J.; Huang, J.H. Enhancement and prolongation of baculovirus-mediated expression in mammalian cells: Focuses on strategic infection and feeding. Biotechnol. Prog. 2003, 19, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Ono, C.; Ninomiya, A.; Yamamoto, S.; Abe, T.; Wen, X.; Fukuhara, T.; Sasai, M.; Yamamoto, M.; Saitoh, T.; Satoh, T.; et al. Innate immune response induced by baculovirus attenuates transgene expression in mammalian cells. J. Virol. 2014, 88, 2157–2167. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-H.; Naik, N.G.; Liao, L.-L.; Wei, S.-C.; Chao, Y.-C. Global Screening of Antiviral Genes that Suppress Baculovirus Transgene Expression in Mammalian Cells. Mol. Ther. Methods Clin. Dev. 2017, 6, 194–206. [Google Scholar] [CrossRef] [Green Version]
- Makela, A.R.; Enback, J.; Laakkonen, J.P.; Vihinen-Ranta, M.; Laakkonen, P.; Oker-Blom, C. Tumor targeting of baculovirus displaying a lymphatic homing peptide. J. Gene Med. 2008, 10, 1019–1031. [Google Scholar] [CrossRef]
- Ojala, K.; Mottershead, D.G.; Suokko, A.; Oker-Blom, C. Specific binding of baculoviruses displaying gp64 fusion proteins to mammalian cells. Biochem. Biophys. Res. Commun. 2001, 284, 777–784. [Google Scholar] [CrossRef]
- Raty, J.K.; Airenne, K.J.; Marttila, A.T.; Marjomaki, V.; Hytonen, V.P.; Lehtolainen, P.; Laitinen, O.H.; Mahonen, A.J.; Kulomaa, M.S.; Yla-Herttuala, S. Enhanced gene delivery by avidin-displaying baculovirus. Mol. Ther. 2004, 9, 282–291. [Google Scholar] [CrossRef]
- Matilainen, H.; Makela, A.R.; Riikonen, R.; Saloniemi, T.; Korhonen, E.; Hyypia, T.; Heino, J.; Grabherr, R.; Oker-Blom, C. RGD motifs on the surface of baculovirus enhance transduction of human lung carcinoma cells. J. Biotechnol. 2006, 125, 114–126. [Google Scholar] [CrossRef]
- Hofmann, C.; Strauss, M. Baculovirus-mediated gene transfer in the presence of human serum or blood facilitated by inhibition of the complement system. Gene Ther. 1998, 5, 531–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, C.; Sandig, V.; Jennings, G.; Rudolph, M.; Schlag, P.; Strauss, M. Efficient gene transfer into human hepatocytes by baculovirus vectors. Proc. Natl. Acad. Sci. USA 1995, 92, 10099–10103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaikkonen, M.U.; Ylä-Herttuala, S.; Airenne, K.J. How to avoid complement attack in baculovirus-mediated gene delivery. J. Invertebr. Pathol. 2011, 107, S71–S79. [Google Scholar] [CrossRef] [PubMed]
- Wickham, T.J.; Davis, T.; Granados, R.R.; Hammer, D.A.; Shuler, M.L.; Wood, H.A. Baculovirus defective interfering particles are responsible for variations in recombinant protein production as a function of multiplicity of infection. Biotechnol. Lett. 1991, 13, 483–488. [Google Scholar] [CrossRef]
- Vijayachandran, L.S.; Thimiri Govinda Raj, D.B.; Edelweiss, E.; Gupta, K.; Maier, J.; Gordeliy, V.; Fitzgerald, D.J.; Berger, I. Gene gymnastics: Synthetic biology for baculovirus expression vector system engineering. Bioengineered 2013, 4, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Berger, I.; Fitzgerald, D.J.; Richmond, T.J. Baculovirus expression system for heterologous multiprotein complexes. Nat. Biotechnol. 2004, 22, 1583–1587. [Google Scholar] [CrossRef]
- Koehler, C.; Lemke, E.A. MultiBacTAG-Genetic Code Expansion Using the Baculovirus Expression System in Sf21 Cells. Methods Mol. Biol. 2018, 1728, 297–311. [Google Scholar] [CrossRef]
- Palmberger, D.; Klausberger, M.; Berger, I.; Grabherr, R. MultiBac turns sweet. Bioengineered 2013, 4, 78–83. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baculovirus in Vivo Delivery Challenges | Approaches to Overcome Issue |
|---|---|
| Narrow tropism (e.g., target cells or tissue are not efficiently transduced) | Pseudotyping can be used to change or expand cell tropism: |
| Serum complement-mediated inactivation Baculovirus is inactivated by human serum complement-cascade | Pseudotyping with complement shielding factors can enhance viral stability in the bloodstream:
Chemical modifications have been reported to enhance serum resistance:
|
| Intracellular immune response inactivation Baculovirus efficiently reaches target cells but is rapidly inactivated and silenced by intracellular immune response pathways. | Histone deacetylases (HDACs) inhibitors can be used ex vivo to counteract silencing. Due to broad spectra and high toxicity they cannot be used systemically: •Valproic acid (VPA) or sodium butyrate (NaBu) [74,75]. Genetically encoded intracellular immune suppression strategies increase transgene expression: |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aulicino, F.; Capin, J.; Berger, I. Synthetic Virus-Derived Nanosystems (SVNs) for Delivery and Precision Docking of Large Multifunctional DNA Circuitry in Mammalian Cells. Pharmaceutics 2020, 12, 759. https://doi.org/10.3390/pharmaceutics12080759
Aulicino F, Capin J, Berger I. Synthetic Virus-Derived Nanosystems (SVNs) for Delivery and Precision Docking of Large Multifunctional DNA Circuitry in Mammalian Cells. Pharmaceutics. 2020; 12(8):759. https://doi.org/10.3390/pharmaceutics12080759
Chicago/Turabian StyleAulicino, Francesco, Julien Capin, and Imre Berger. 2020. "Synthetic Virus-Derived Nanosystems (SVNs) for Delivery and Precision Docking of Large Multifunctional DNA Circuitry in Mammalian Cells" Pharmaceutics 12, no. 8: 759. https://doi.org/10.3390/pharmaceutics12080759
APA StyleAulicino, F., Capin, J., & Berger, I. (2020). Synthetic Virus-Derived Nanosystems (SVNs) for Delivery and Precision Docking of Large Multifunctional DNA Circuitry in Mammalian Cells. Pharmaceutics, 12(8), 759. https://doi.org/10.3390/pharmaceutics12080759