
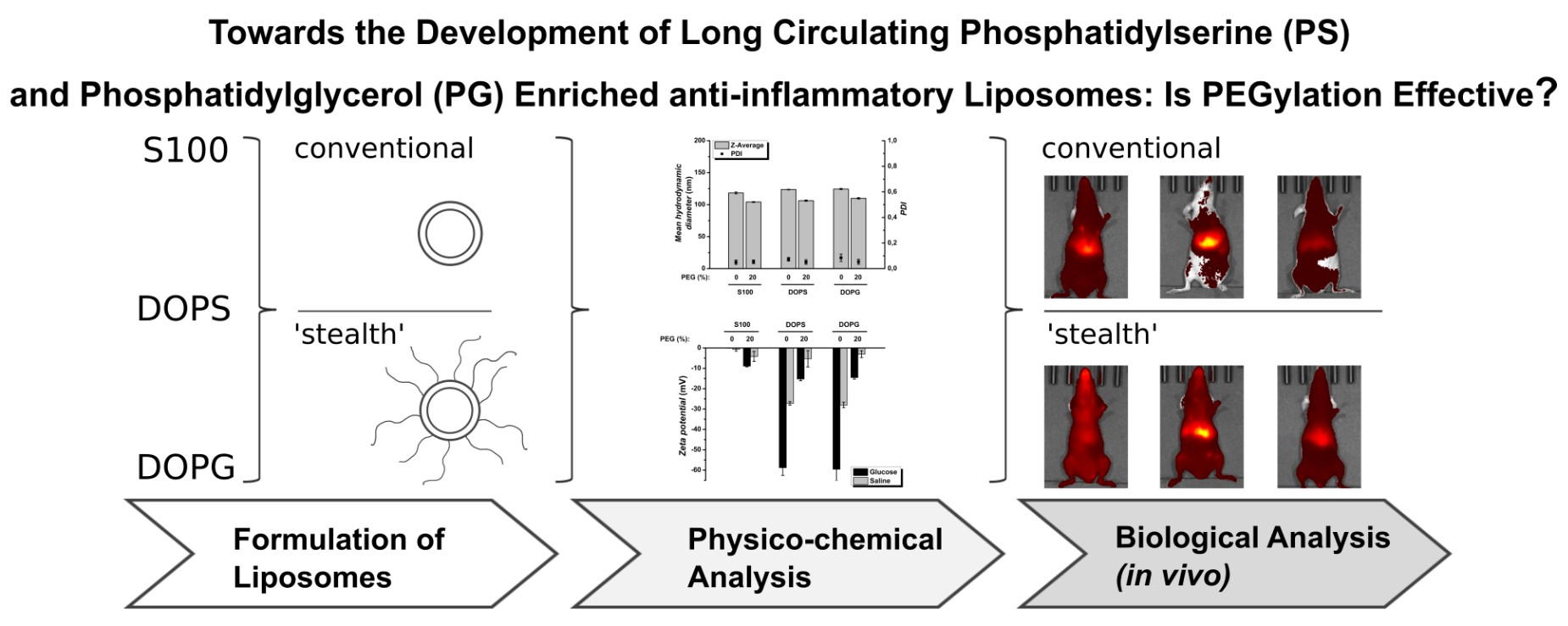
Towards the Development of Long Circulating Phosphatidylserine (PS)- and Phosphatidylglycerol (PG)-Enriched Anti-Inflammatory Liposomes: Is PEGylation Effective?

 , ,
, ,  and
and
Abstract
:
1. Introduction
- Are the biodistribution profiles of PS- and PG-enriched nanodispersions in healthy mice comparable?
- Does PEGylation of PS- and PG-enriched nanodispersions effect their biodistribution in healthy mice?
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Liposome Preparation
2.2.2. Dynamic Light Scattering (DLS)
2.2.3. Cryo-Transmission Electron Microscopy (Cryo-TEM)
2.2.4. Zeta Potential Measurements
2.2.5. Mice
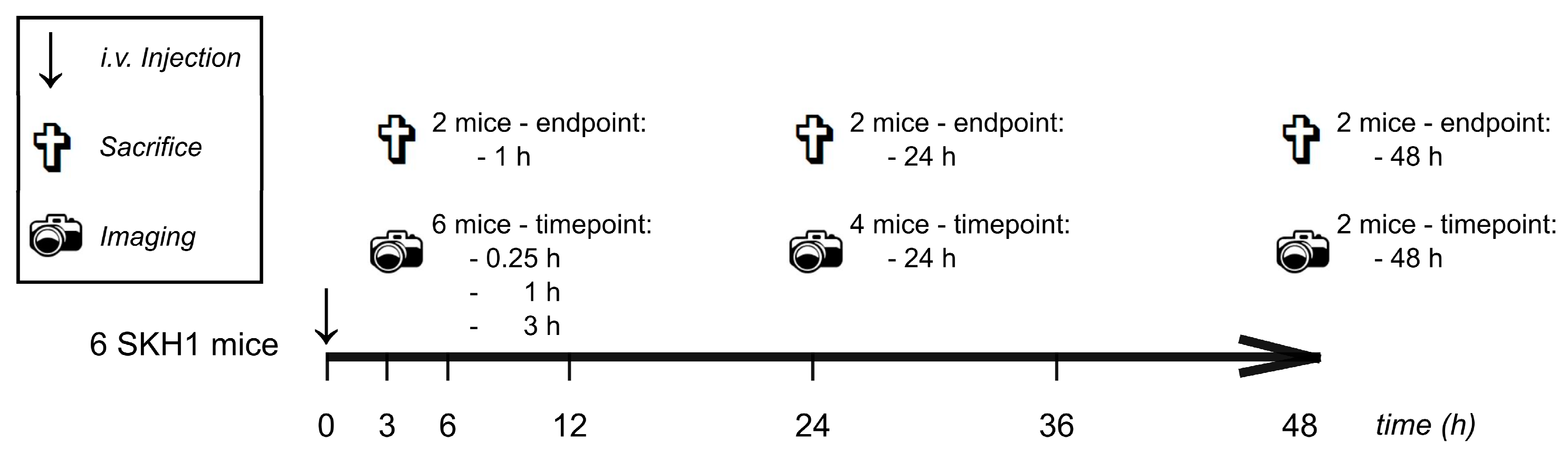
2.2.6. Experimental Design for the Evaluation of In Vivo and Ex Vivo Biodistribution
2.2.7. Fluorescence Imaging (FI)
2.2.8. Statistical Analysis
3. Results
3.1. Physico-Chemical Characterization Showed Defined Size and Charge of DiR-Loaded Liposomes
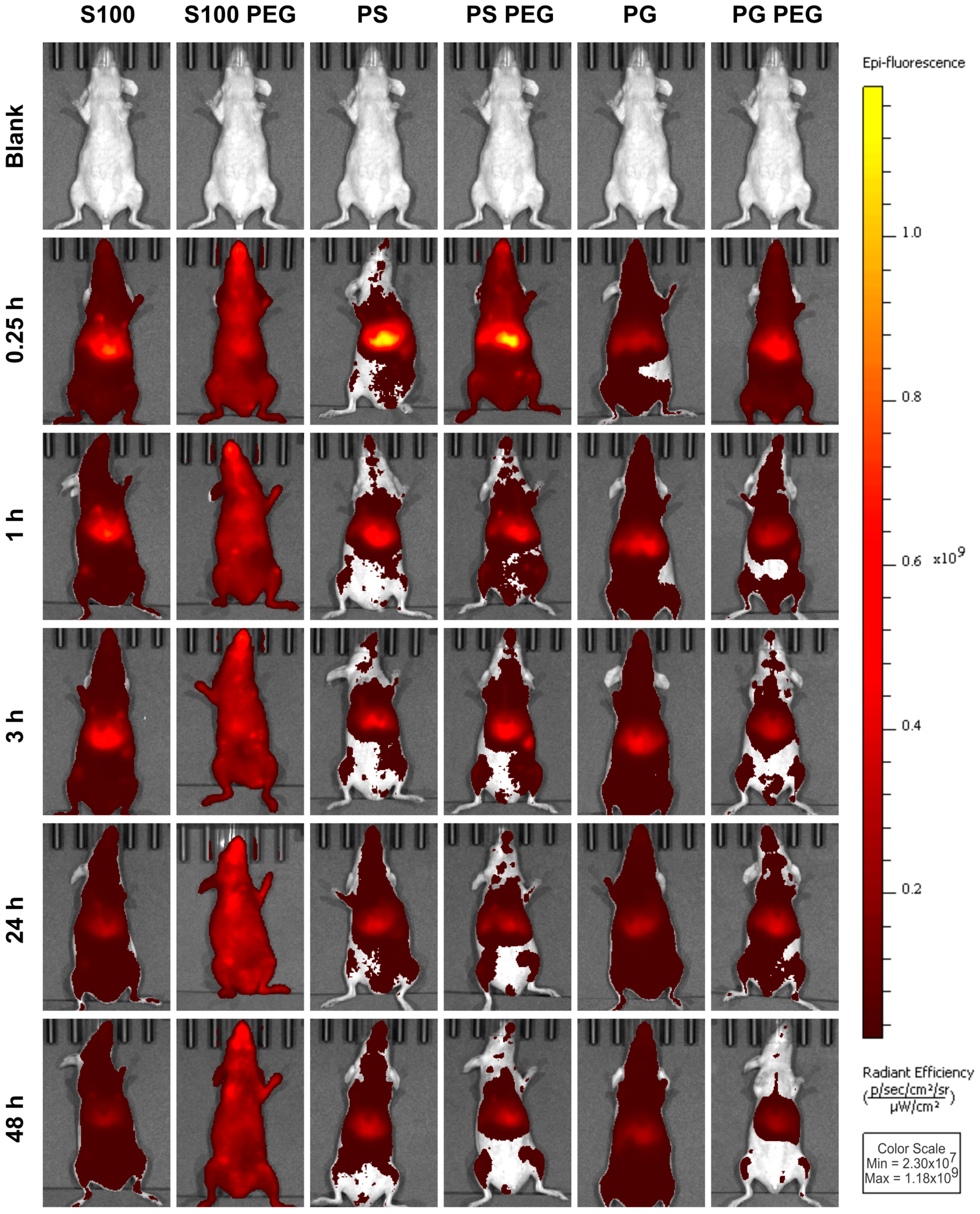
3.2. In Vivo Liposome Biodistribution Differed in Biodistribution and Abdominal Accumulation, Depending on Both Formulation and PEGylation
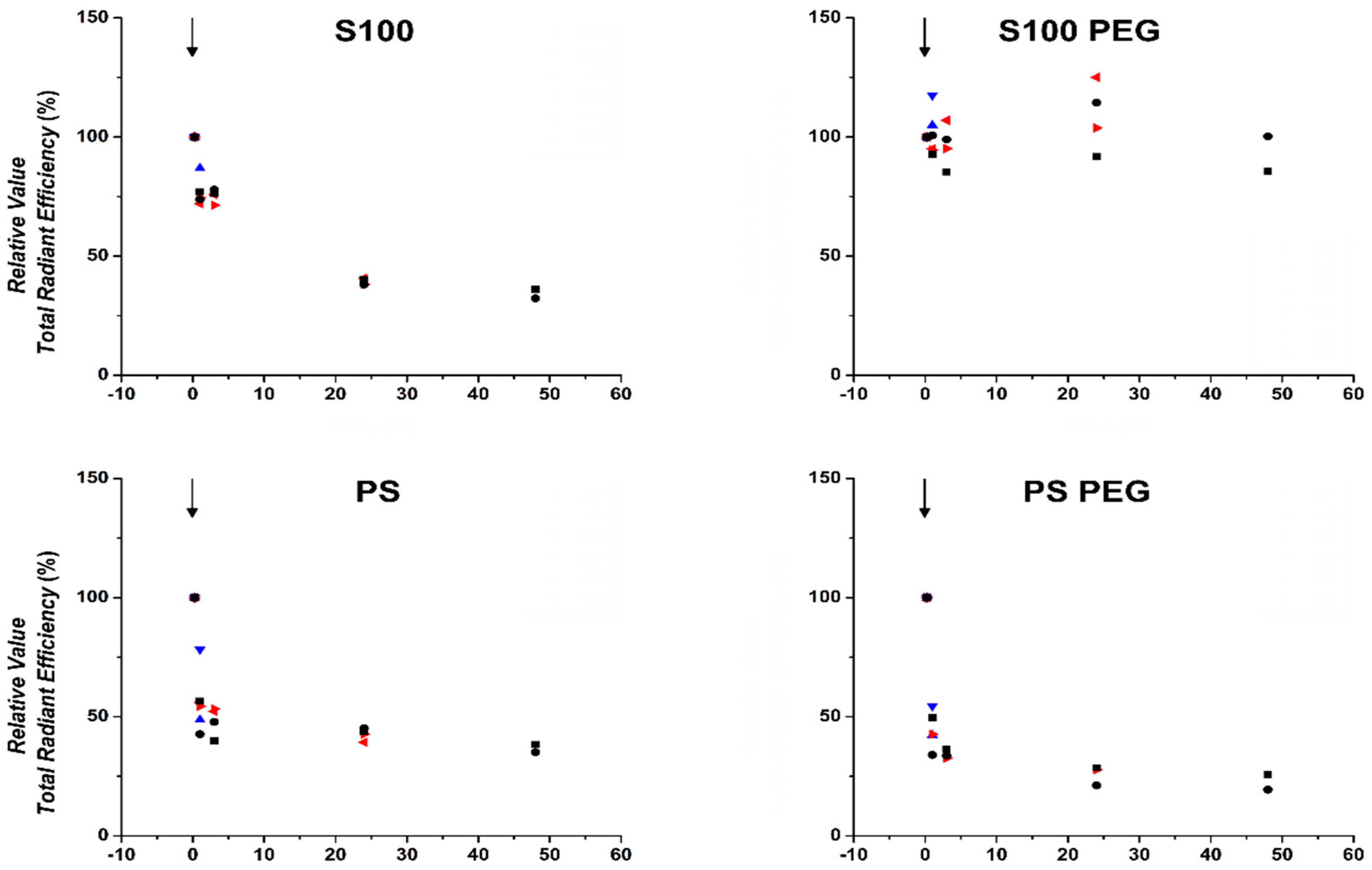
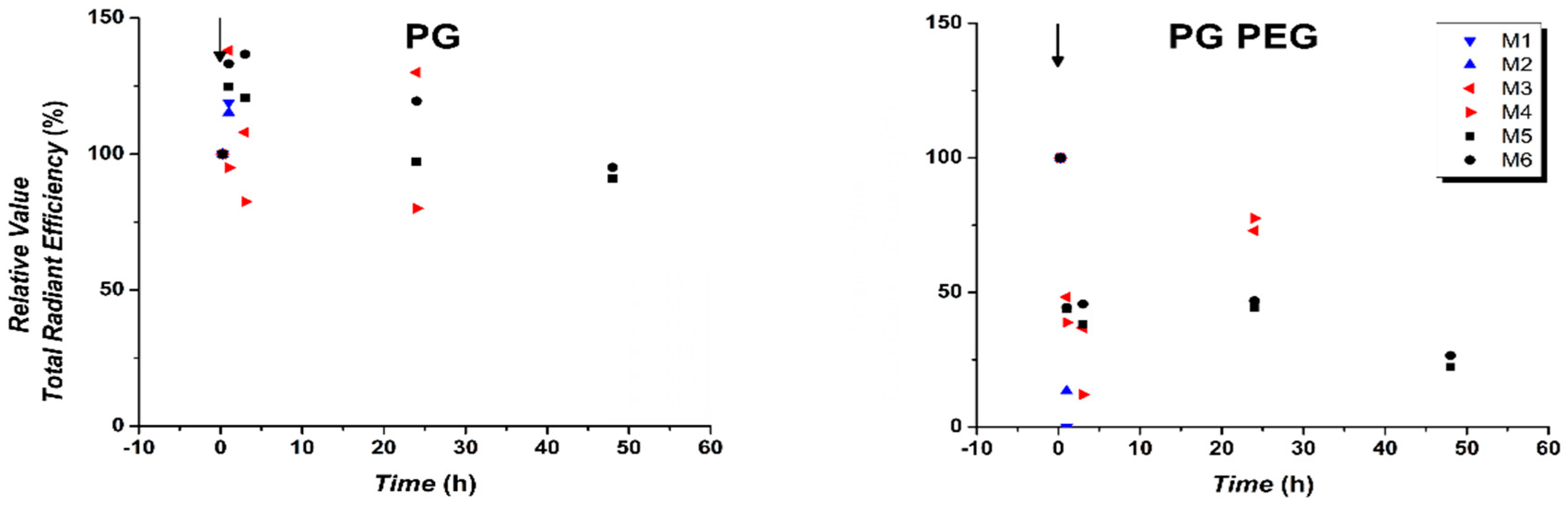
3.3. Quantification of In Vivo Fluorescence Signal Showed Prolonged Circulation of S100PEG and Fast Clearance of Other Formulations
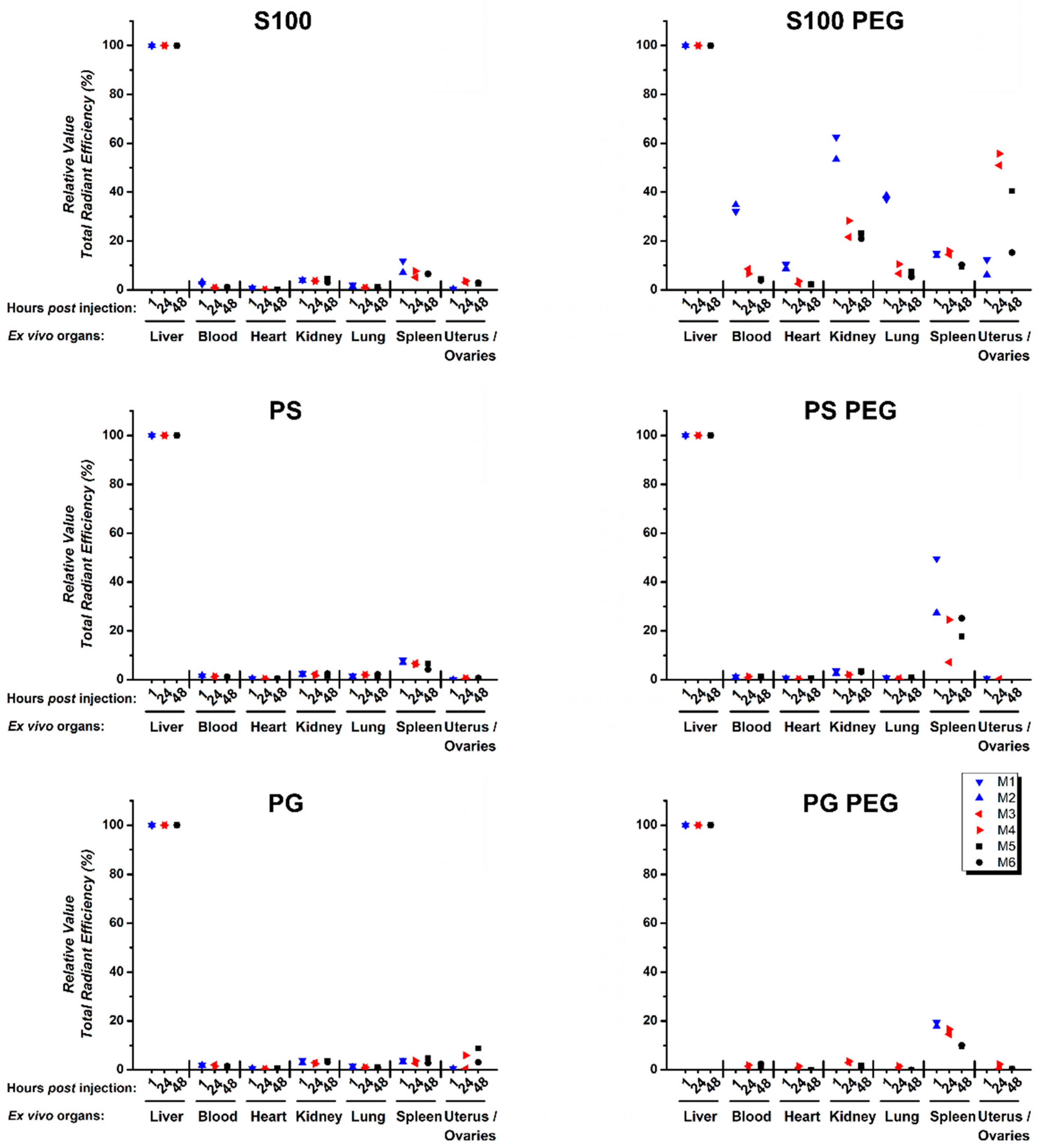
3.4. Quantification of Ex Vivo Single Organ TREs Shows Systemic Accumulation of S100 PEG and Fast Clearance of Other Formulations via Liver and Spleen
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weiss, V.M.; Lucas, H.; Mueller, T.; Chytil, P.; Etrych, T.; Naolou, T.; Kressler, J.; Mäder, K. Intended and Unintended Targeting of Polymeric Nanocarriers: The Case of Modified Poly(Glycerol Adipate) Nanoparticles. Macromol. Biosci. 2018, 18. [Google Scholar] [CrossRef] [Green Version]
- Allen, T.M.; Cullis, P.R. Liposomal Drug Delivery Systems: From Concept to Clinical Applications. Adv. Drug Deliv. Rev. 2013. [Google Scholar] [CrossRef]
- Crommelin, D.J.A.; van Hoogevest, P.; Storm, G. The Role of Liposomes in Clinical Nanomedicine Development. What Now? Now What? J. Control. Release 2020, 318, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.S. The Origins and Evolution of “Controlled” Drug Delivery Systems. J. Control. Release 2008, 132, 153–163. [Google Scholar] [CrossRef]
- Crommelin, D.J.A.; Metselaar, J.M.; Storm, G. Liposomes: The Science and the Regulatory Landscape. In Non-Biological Complex Drugs–The Science and the Regulatory Landscape; Crommelin, D.J.A., Mühlebach, S., Eds.; Springer: Cham, Switzerland, 2015; Volume 20, pp. 77–106. [Google Scholar]
- Barenholz, Y. Doxil®–The First FDA-Approved Nano-Drug: Lessons Learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- van Hoogevest, P.; Wendel, A. The Use of Natural and Synthetic Phospholipids as Pharmaceutical Excipients. Eur. J. Lipid Sci. Technol. 2014, 116, 1088–1107. [Google Scholar] [CrossRef] [Green Version]
- Daleke, D.L. Regulation of Phospholipid Asymmetry in the Erythrocyte Membrane. Curr. Opin. Hematol. 2008, 15, 191–195. [Google Scholar] [CrossRef]
- Fadok, V.A.; Voelker, D.R.; Campbell, P.A.; Cohen, J.J.; Bratton, D.L.; Henson, P.M. Exposure of Phosphatidylserine on the Surface of Apoptotic Lymphocytes Triggers Specific Recognition and Removal by Macrophages. J. Immunol. 1992, 148, 2207–2216. [Google Scholar] [PubMed]
- Hoffmann, P.R.; Kench, J.A.; Vondracek, A.; Kruk, E.; Daleke, D.L.; Jordan, M.; Marrack, P.; Henson, P.M.; Fadok, V.A. Interaction between Phosphatidylserine and the Phosphatidylserine Receptor Inhibits Immune Responses in Vivo. J. Immunol. 2005, 174, 1393–1404. [Google Scholar] [CrossRef]
- Bevers, E.M.; Williamson, P.L. Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiol. Rev. 2016, 96, 605–645. [Google Scholar] [CrossRef]
- Bagalkot, V.; Deiuliis, J.A.; Rajagopalan, S.; Maiseyeu, A. “Eat Me” Imaging and Therapy. Adv. Drug Deliv. Rev. 2016, 2–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szondy, Z.; Sarang, Z.; Kiss, B.; Garabuczi, É.; Köröskényi, K. Anti-Inflammatory Mechanisms Triggered by Apoptotic Cells during Their Clearance. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Hashioka, S.; Han, Y.H.; Fujii, S.; Kato, T.; Monji, A.; Utsumi, H.; Sawada, M.; Nakanishi, H.; Kanba, S. Phosphatidylserine and Phosphatidylcholine-Containing Liposomes Inhibit Amyloid β and Interferon-γ-Induced Microglial Activation. Free Radic. Biol. Med. 2007, 42, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Dvoriantchikova, G.; Agudelo, C.; Hernandez, E.; Shestopalov, V.I.; Ivanov, D. Phosphatidylserine-Containing Liposomes Promote Maximal Survival of Retinal Neurons after Ischemic Injury. J. Cereb. Blood Flow Metab. 2009, 29, 1755–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Ma, H.M.; Kukita, T.; Nakanishi, Y.; Nakanishi, H. Phosphatidylserine-Containing Liposomes Inhibit the Differentiation of Osteoclasts and Trabecular Bone Loss. J. Immunol. 2010, 184, 3191–3201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.M.; Wu, Z.; Nakanishi, H. Phosphatidylserine-Containing Liposomes Suppress Inflammatory Bone Loss by Ameliorating the Cytokine Imbalance Provoked by Infiltrated Macrophages. Lab. Investig. 2011, 91, 921–931. [Google Scholar] [CrossRef]
- Yeom, M.; Hahm, D.H.; Sur, B.J.; Han, J.J.; Lee, H.J.; Yang, H.I.; Kim, K.S. Phosphatidylserine Inhibits Inflammatory Responses in Interleukin-1β-Stimulated Fibroblast-like Synoviocytes and Alleviates Carrageenan-Induced Arthritis in Rat. Nutr. Res. 2013, 33, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Urbano, P.C.M.; Soccol, V.T.; Teixeira, V.N.; Oliveira, P.G.; Filippin, L.I.; Bonat, W.H.; de Oliveira, C.; Rossi, G.R.; Xavier, R.M.; Azevedo, V.F. Effect of Pegylated Phosphatidylserine-Containing Liposomes in Experimental Chronic Arthritis. BMC Pharmacol. Toxicol. 2015, 16, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Harel-Adar, T.; Ben Mordechai, T.; Amsalem, Y.; Feinberg, M.S.; Leor, J.; Cohen, S. Modulation of Cardiac Macrophages by Phosphatidylserine-Presenting Liposomes Improves Infarct Repair. Proc. Natl. Acad. Sci. USA 2011, 108, 1827–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Yang, J.; Liu, R.; Qiao, C.; Lu, Z.; Shi, Y.; Fan, Z.; Zhang, Z.; Zhang, X. Dual-Targeting Theranostic System with Mimicking Apoptosis to Promote Myocardial Infarction Repair via Modulation of Macrophages. Theranostics 2017, 7, 4149–4167. [Google Scholar] [CrossRef]
- Stokes, C.; Kaur, R.; Edwards, M.R.; Mondhe, M.; Robinson, D.; Prestwich, E.C.; Hume, R.D.; Marshall, C.A.; Perrie, Y.; O’Donnell, V.B.; et al. Human Rhinovirus-Induced Inflammatory Responses Are Inhibited by Phosphatidylserine Containing Liposomes. Mucosal Immunol. 2016, 9, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, A.; Fisch, A.; Seuwen, K.; Baumgarten, B.; Ruffner, H.; Aebi, A.; Rausch, M.; Kiessling, F.; Bartneck, M.; Weiskirchen, R.; et al. Glucocorticoid-Loaded Liposomes Induce a pro-Resolution Phenotype in Human Primary Macrophages to Support Chronic Wound Healing. Biomaterials 2018, 1–15. [Google Scholar] [CrossRef]
- King, R.J.; MacBeth, M.C. Interaction of the Lipid and Protein Components of Pulmonary Surfactant Role of Phosphatidylglycerol and Calcium. Biochim. Biophys. Acta 1981, 647, 159–168. [Google Scholar] [CrossRef]
- Numata, M.; Chu, H.W.; Dakhama, A.; Voelker, D.R. Pulmonary Surfactant Phosphatidylglycerol Inhibits Respiratory Syncytial Virus-Induced Inflammation and Infection. Proc. Natl. Acad. Sci. USA 2010, 107, 320–325. [Google Scholar] [CrossRef] [Green Version]
- Kandasamy, P.; Zarini, S.; Chan, E.D.; Leslie, C.C.; Murphy, R.C.; Voelker, D.R. Pulmonary Surfactant Phosphatidylglycerol Inhibits Mycoplasma Pneumoniae-Stimulated Eicosanoid Production from Human and Mouse Macrophages. J. Biol. Chem. 2011, 286, 7841–7853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numata, M.; Kandasamy, P.; Nagashima, Y.; Posey, J.; Hartshorn, K.; Woodland, D.; Voelker, D.R. Phosphatidylglycerol Suppresses Influenza A Virus Infection. Am. J. Respir. Cell Mol. Biol. 2012, 46, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Bollag, W.B.; Olala, L.O.; Xie, D.; Lu, X.; Qin, H.; Choudhary, V.; Patel, R.; Bogorad, D.; Estes, A.; Watsky, M. Dioleoylphosphatidylglycerol Accelerates Corneal Epithelial Wound Healing. Investig. Ophthalmol. Vis. Sci. 2020, 61, 29. [Google Scholar] [CrossRef]
- Choudhary, V.; Uaratanawong, R.; Patel, R.R.; Patel, H.; Bao, W.; Hartney, B.; Cohen, E.; Chen, X.; Zhong, Q.; Isales, C.M.; et al. Phosphatidylglycerol Inhibits Toll-Like Receptor–Mediated Inflammation by Danger-Associated Molecular Patterns. J. Investig. Dermatol. 2019, 139, 868–877. [Google Scholar] [CrossRef] [Green Version]
- Kuronuma, K.; Mitsuzawa, H.; Takeda, K.; Nishitani, C.; Chan, E.D.; Kuroki, Y.; Nakamura, M.; Voelker, D.R. Anionic Pulmonary Surfactant Phospholipids Inhibit Inflammatory Responses from Alveolar Macrophages and U937 Cells by Binding the Lipopolysaccharide-Interacting Proteins CD14 and MD-2. J. Biol. Chem. 2009, 284, 25488–25500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollag, W.B.; Gonzales, J.N. Phosphatidylglycerol and Surfactant: A Potential Treatment for COVID-19? Med. Hypotheses 2020, 144. [Google Scholar] [CrossRef]
- Kunjachan, S.; Jose, S.; Thomas, C.A.; Joseph, E.; Kiessling, F.; Lammers, T. Physicochemical and Biological Aspects of Macrophage-Mediated Drug Targeting in Anti-Microbial Therapy. Fundam. Clin. Pharmacol. 2012, 26, 63–71. [Google Scholar] [CrossRef]
- Asano, T.; Kleinerman, E.S. Liposome-Encapsulated MTP-PE: A Novel Biologic Agent for Cancer Therapy. J. Immunother. 1993, 14, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Nardin, A.; Lefebvre, M.; Labroquere, K.; Faure, O.; Abastado, J. Liposomal Muramyl Tripeptide Phosphatidylethanolamine: Targeting and Activating Macrophages for Adjuvant Treatment of Osteosarcoma. Curr. Cancer Drug Targets 2006, 6, 123–133. [Google Scholar] [CrossRef]
- Adler-Moore, J.P.; Proffitt, R.T. Development, Characterization, Efficacy and Mode of Action of Ambisome, a Unilamellar Liposomal Formulation of Amphotericin B. J. Liposome Res. 1993, 3, 429–450. [Google Scholar] [CrossRef]
- Adler-Moore, J.; Proffitt, R.T. AmBisome: Liposomal Formulation, Structure, Mechanism of Action and Pre-Clinical Experience. J. Antimicrob. Chemother. 2002, 49, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Hunter, A.C.; Andresen, T.L. Factors Controlling Nanoparticle Pharmacokinetics: An Integrated Analysis and Perspective. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 481–503. [Google Scholar] [CrossRef] [PubMed]
- Koster, V.S.; Kuks, P.F.M.; Lange, R.; Talsma, H. Particle Size in Parenteral Fat Emulsions, What Are the True Limitations? Int. J. Pharm. 1996, 134, 235–238. [Google Scholar] [CrossRef]
- Woodle, M.C.; Lasic, D.D. Sterically Stabilized Liposomes. Biochim. Biophys. Acta Gen. Subj. 1992, 1113, 171–199. [Google Scholar] [CrossRef]
- Klein, M.E.; Mauch, S.; Rieckmann, M.; Martínez, D.G.; Hause, G.; Noutsias, M.; Hofmann, U.; Lucas, H.; Meister, A.; Ramos, G.; et al. Phosphatidylserine (PS) and Phosphatidylglycerol (PG) Nanodispersions as Potential Anti-Inflammatory Therapeutics: Comparison of in Vitro Activity and Impact of Pegylation. Nanomed. Nanotechnol. Biol. Med. 2020, 23. [Google Scholar] [CrossRef] [PubMed]
- Partain, B.D.; Unni, M.; Rinaldi, C.; Allen, K.D. The Clearance and Biodistribution of Magnetic Composite Nanoparticles in Healthy and Osteoarthritic Rat Knees. J. Control. Release 2020, 321, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.E.; Rieckmann, M.; Lucas, H.; Meister, A.; Loppnow, H.; Mäder, K. Phosphatidylserine (PS) and Phosphatidylglycerol (PG) Enriched Mixed Micelles (MM): A New Nano-Drug Delivery System with Anti-Inflammatory Potential? Eur. J. Pharm. Sci. 2020, 152, 105451. [Google Scholar] [CrossRef] [PubMed]
- Voipio, H.-M.; Baneux, P.; De Segura, I.A.G.; Hau, J.; Wolfensohn, S. Guidelines for the Veterinary Care of Laboratory Animals: Report of the FELASA/ECLAM/ESLAV Joint Working Group on Veterinary Care. Lab. Anim. 2008, 42, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillen, J. FELASA Guidelines and Recommendations. J. Am. Assoc. Lab. Anim. Sci. 2012, 51, 311–321. [Google Scholar]
- Mertens, S.; Vogt, M.A.; Gass, P.; Palme, R.; Hiebl, B.; Chourbaji, S. Effect of Three Different Forms of Handling on the Variation of Aggression-Associated Parameters in Individually and Group-Housed Male C57BL/6NCrl Mice. PLoS ONE 2019, 14. [Google Scholar] [CrossRef] [Green Version]
- Bayne, K. Environmental Enrichment and Mouse Models: Current Perspectives. Anim. Model. Exp. Med. 2018, 1, 82–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloomsmith, M.A.; Perlman, J.E.; Hutchinson, E.; Sharpless, M. Behavioral Management Programs to Promote Laboratory Animal Welfare; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Bhattacharjee, S. DLS and Zeta Potential—What They Are and What They Are Not? J. Control. Release 2016, 235, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Schädlich, A.; Hoffmann, S.; Mueller, T.; Caysa, H.; Rose, C.; Göpferich, A.; Li, J.; Kuntsche, J.; Mäder, K. Accumulation of Nanocarriers in the Ovary: A Neglected Toxicity Risk? J. Control. Release 2012, 160, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Mérian, J.; Boisgard, R.; Decleves, X.; Thezé, B.; Texier, I.; Tavitian, B. Synthetic Lipid Nanoparticles Targeting Steroid Organs. J. Nucl. Med. 2013, 54, 1996–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, T.M.; Hansen, C. Pharmacokinetics of Stealth versus Conventional Liposomes: Effect of Dose. BBA Biomembr. 1991, 1068, 133–141. [Google Scholar] [CrossRef]
- Boerman, O.C.; Oyen, W.J.; van Bloois, L.; Koenders, E.B.; van der Meer, J.W.; Corstens, F.H.; Storm, G. Optimization of Technetium-99m-Labeled PEG Liposomes to Image Focal Infection: Effects of Particle Size and Circulation Time. J. Nucl. Med. 1997, 38, 489–493. [Google Scholar] [PubMed]
- Semple, S.C.; Chonn, A.; Cullis, P.R. Influence of Cholesterol on the Association of Plasma Proteins with Liposomes. Biochemistry 1996, 35, 2521–2525. [Google Scholar] [CrossRef] [PubMed]
- Sieber, S.; Grossen, P.; Detampel, P.; Siegfried, S.; Witzigmann, D.; Huwyler, J. Zebrafish as an Early Stage Screening Tool to Study the Systemic Circulation of Nanoparticulate Drug Delivery Systems in Vivo. J. Control. Release 2017, 264, 180–191. [Google Scholar] [CrossRef]
- Gabizon, A.; Papahadjopoulos, D. The Role of Surface Charge and Hydrophilic Groups on Liposome Clearance in Vivo. BBA Biomembr. 1992, 1103, 94–100. [Google Scholar] [CrossRef]
- Daemen, T.; Velinova, M.; Regts, J.; De Jager, M.; Kalicharan, R.; Donga, J.; Van Der Want, J.J.L.; Scherphof, G.L. Different Intrahepatic Distribution of Phosphatidylglycerol and Phosphatidylserine Liposomes in the Rat. Hepatology 1997, 26, 416–423. [Google Scholar] [CrossRef]
- Levchenko, T.S.; Rammohan, R.; Lukyanov, A.N.; Whiteman, K.R.; Torchilin, V.P. Liposome Clearance in Mice: The Effect of a Separate and Combined Presence of Surface Charge and Polymer Coating. Int. J. Pharm. 2002, 240, 95–102. [Google Scholar] [CrossRef]
- Chiu, G.N.C.; Bally, M.B.; Mayer, L.D. Selective Protein Interactions with Phosphatidylserine Containing Liposomes Alter the Steric Stabilization Properties of Poly(Ethylene Glycol). Biochim. Biophys. Acta Biomembr. 2001, 1510, 56–69. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Suzuki, Y.; Hihara, T.; Kubara, K.; Kondo, K.; Hyodo, K.; Yamazaki, K.; Ishida, T.; Ishihara, H. PEG Shedding-Rate-Dependent Blood Clearance of PEGylated Lipid Nanoparticles in Mice: Faster PEG Shedding Attenuates Anti-PEG IgM Production. Int. J. Pharm. 2020, 588, 119792. [Google Scholar] [CrossRef]
- Shimizu, T.; Awata, M.; Abu Lila, A.S.; Yoshioka, C.; Kawaguchi, Y.; Ando, H.; Ishima, Y.; Ishida, T. Complement Activation Induced by PEG Enhances Humoral Immune Responses against Antigens Encapsulated in PEG-Modified Liposomes. J. Control. Release 2020. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Liposomal Formulation | Z-Average (nm) | PDI | Zeta Potential (mV) in Saline | Zeta Potential (mV) in Glucose 5% |
|---|---|---|---|---|
| S100 | 125 ± 6 | 0.06 ± 0.01 | −0.9 ± 1.4 | 0.3 ± 0.3 |
| S100 PEG | 116 ± 13 | 0.06 ± 0.01 | −2.5 ± 1.5 | −9.1 ± 0.8 |
| PS | 125 ± 11 | 0.07 ± 0.01 | −26.9 ± 1.3 | −60.0 ± 3.9 |
| PS PEG | 115 ± 10 | 0.06 ± 0.01 | −4.2 ± 2.0 | −16.0 ± 1.4 |
| PG | 120 ± 4 | 0.08 ± 0.02 | −27.1 ± 2.1 | −57.8 ± 1.5 |
| PG PEG | 116 ± 13 | 0.06 ± 0.01 | −3.7 ± 1.2 | −16.1 ± 2.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, M.E.; Rieckmann, M.; Sedding, D.; Hause, G.; Meister, A.; Mäder, K.; Lucas, H. Towards the Development of Long Circulating Phosphatidylserine (PS)- and Phosphatidylglycerol (PG)-Enriched Anti-Inflammatory Liposomes: Is PEGylation Effective? Pharmaceutics 2021, 13, 282. https://doi.org/10.3390/pharmaceutics13020282
Klein ME, Rieckmann M, Sedding D, Hause G, Meister A, Mäder K, Lucas H. Towards the Development of Long Circulating Phosphatidylserine (PS)- and Phosphatidylglycerol (PG)-Enriched Anti-Inflammatory Liposomes: Is PEGylation Effective? Pharmaceutics. 2021; 13(2):282. https://doi.org/10.3390/pharmaceutics13020282
Chicago/Turabian StyleKlein, Miriam E., Max Rieckmann, Daniel Sedding, Gerd Hause, Annette Meister, Karsten Mäder, and Henrike Lucas. 2021. "Towards the Development of Long Circulating Phosphatidylserine (PS)- and Phosphatidylglycerol (PG)-Enriched Anti-Inflammatory Liposomes: Is PEGylation Effective?" Pharmaceutics 13, no. 2: 282. https://doi.org/10.3390/pharmaceutics13020282
APA StyleKlein, M. E., Rieckmann, M., Sedding, D., Hause, G., Meister, A., Mäder, K., & Lucas, H. (2021). Towards the Development of Long Circulating Phosphatidylserine (PS)- and Phosphatidylglycerol (PG)-Enriched Anti-Inflammatory Liposomes: Is PEGylation Effective? Pharmaceutics, 13(2), 282. https://doi.org/10.3390/pharmaceutics13020282