
On Absorption Modeling and Food Effect Prediction of Rivaroxaban, a BCS II Drug Orally Administered as an Immediate-Release Tablet


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical and Reagents
2.2. Software
2.3. Chromatographic Quantitative Analysis
2.4. Biopharmaceutical Properties of Riva
2.4.1. Biorelevant Solubility Determination
2.4.2. In Vitro Dynamic Biorelevant Dissolution
2.4.3. In Vitro Caco-2 Permeability Determination
2.4.4. Determination of Systemic Disposition Parameters of Riva
2.5. Physiologically Based Gastrointestinal Absorption Modeling
2.5.1. Model Compound Parameters
2.5.2. Development of In-Silico Physiology Based Gastrointestinal Absorption Model
Model Verification
Parameter Sensitivity Analysis (PSA)
2.6. IVIVC Studies
2.7. Food Effect (FE) Studies of Riva in Simulated Healthy Population
3. Results and Discussion
3.1. Biopharmaceutical Properties of Riva
3.1.1. Equilibrium solubility in simulated media
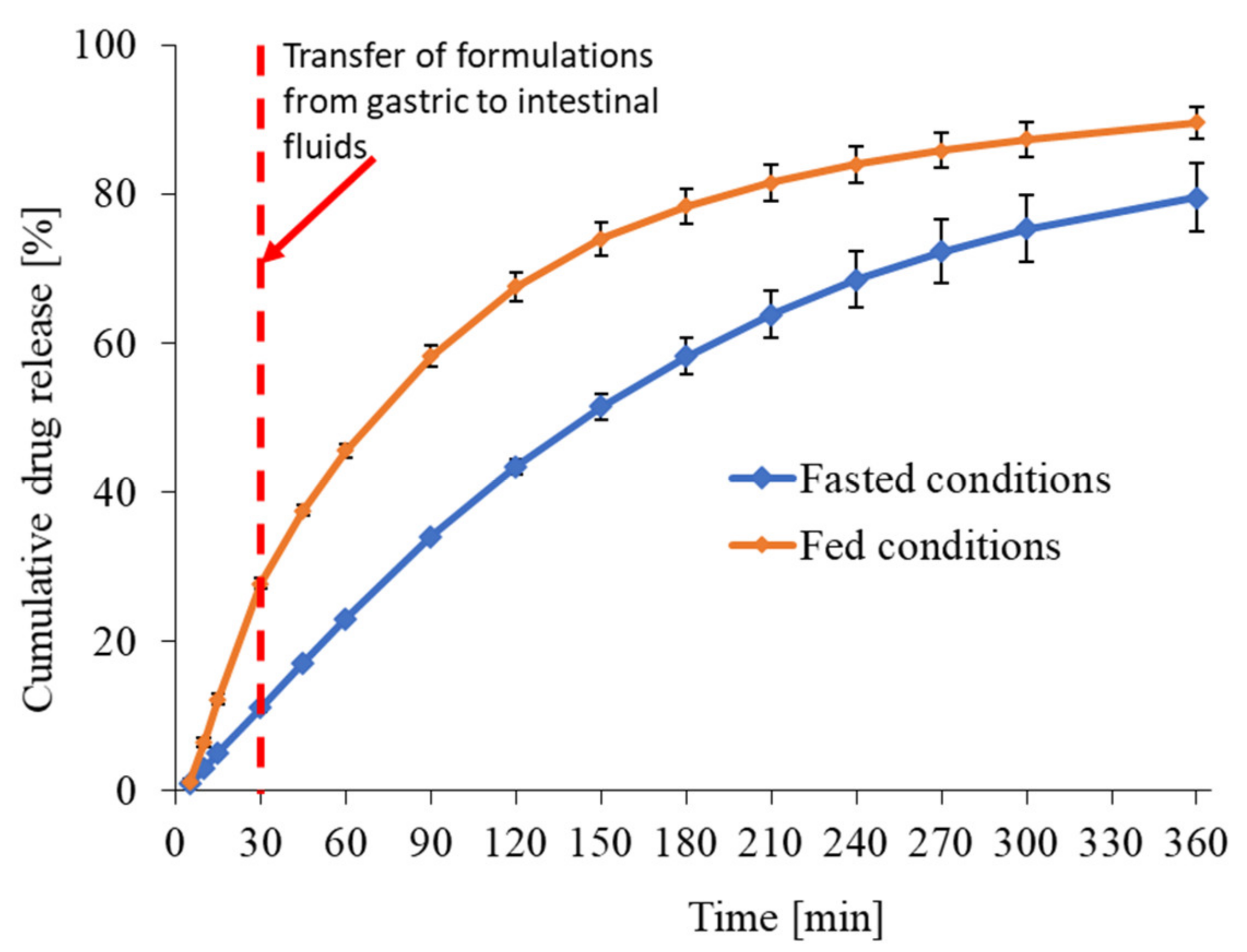
3.1.2. In Vitro Release Profile of Riva in Fasted and Fed Conditions
3.1.3. In Vitro Caco-2 Permeability
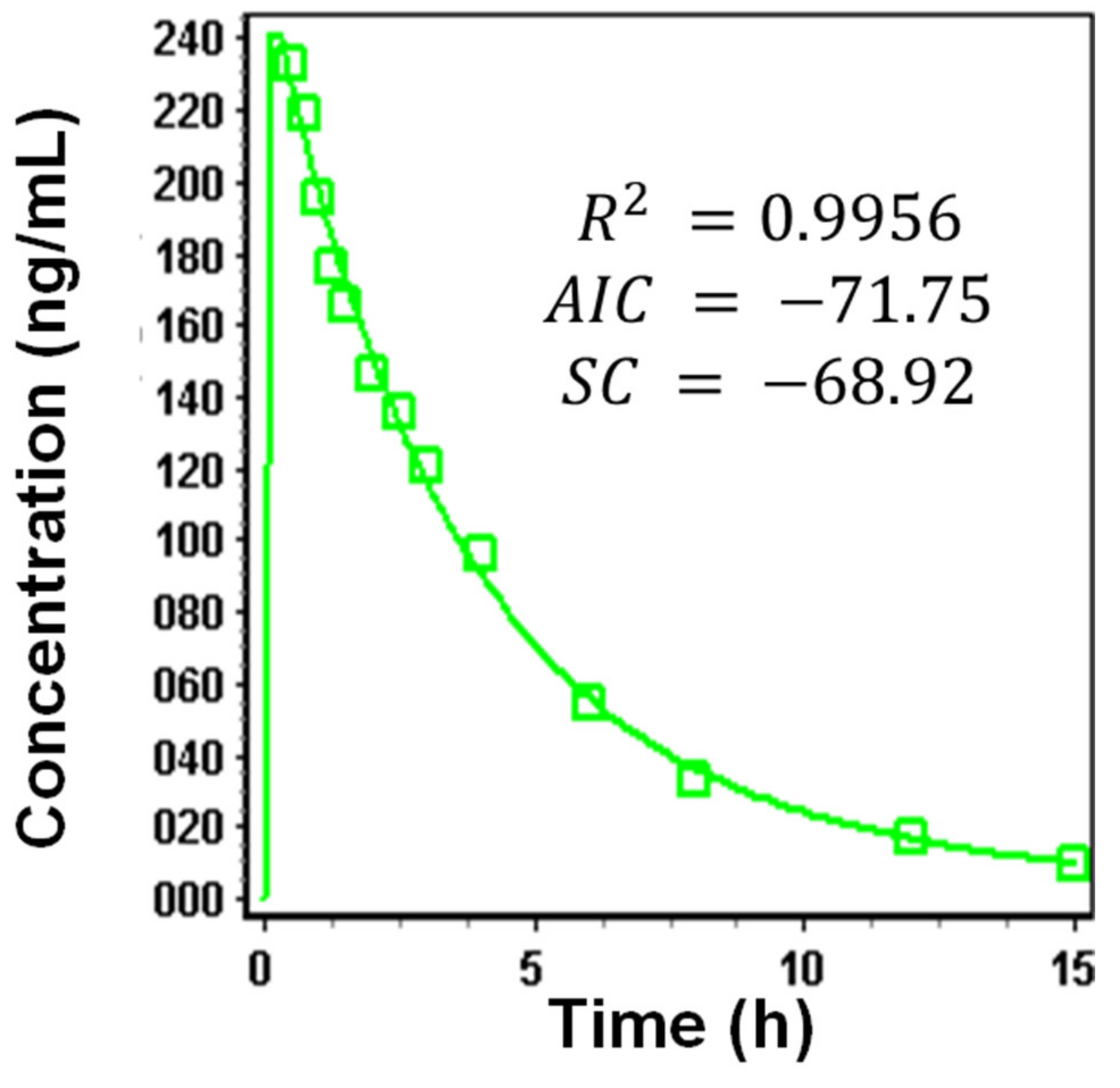
3.1.4. Systemic Disposition Parameters of Riva
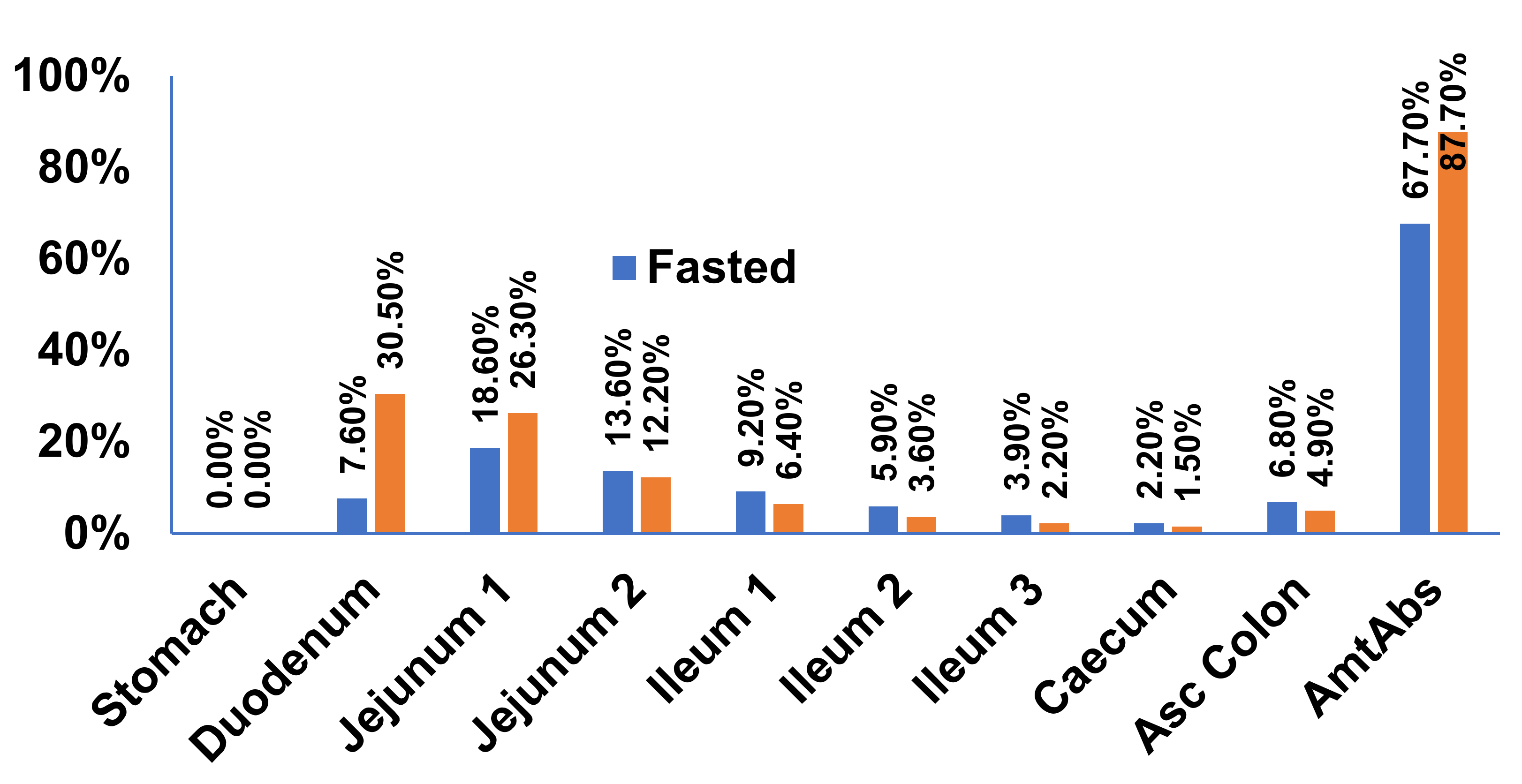
3.2. Physiology Based Gastrointestinal Absorption Model of Riva Formulation
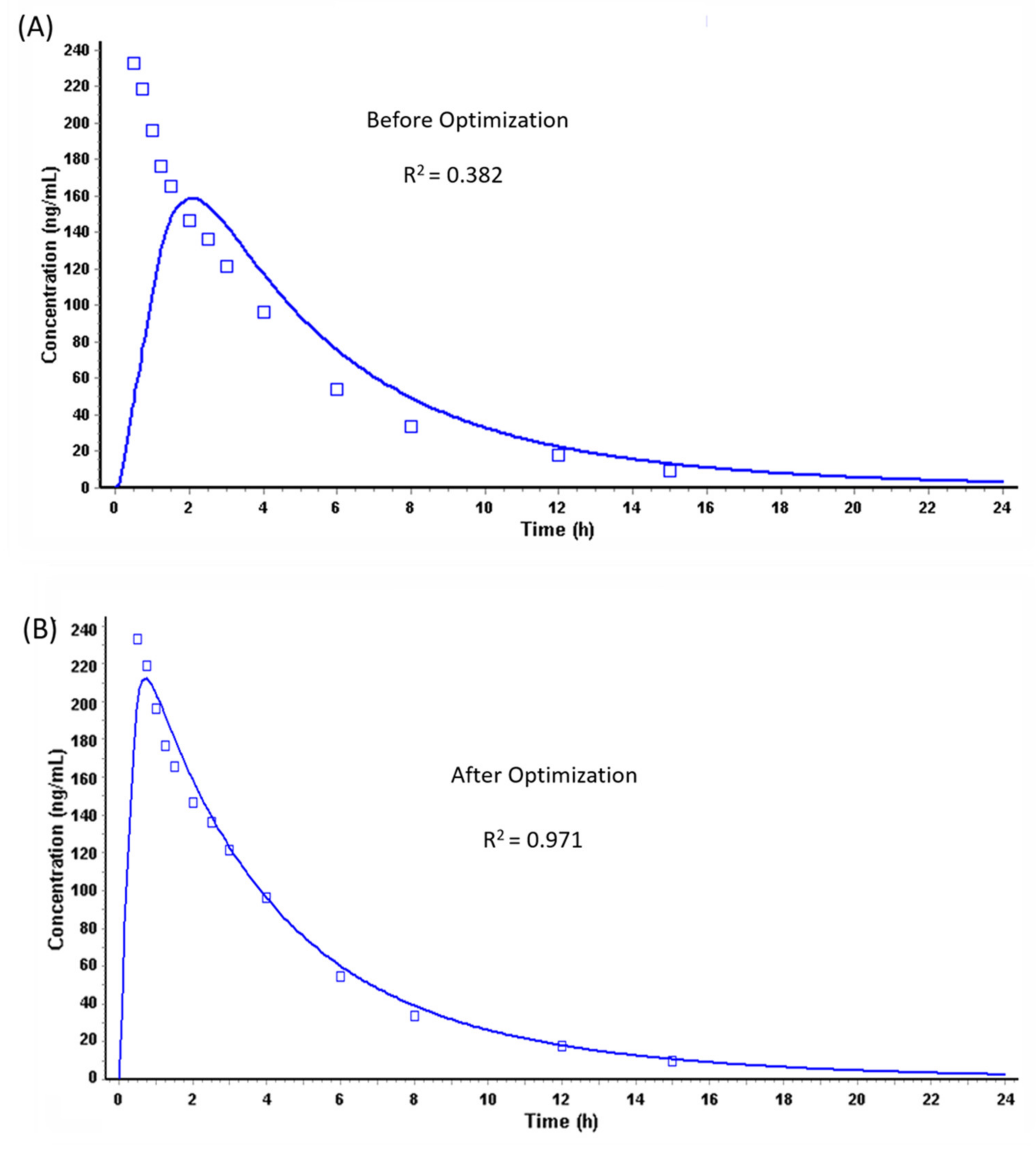
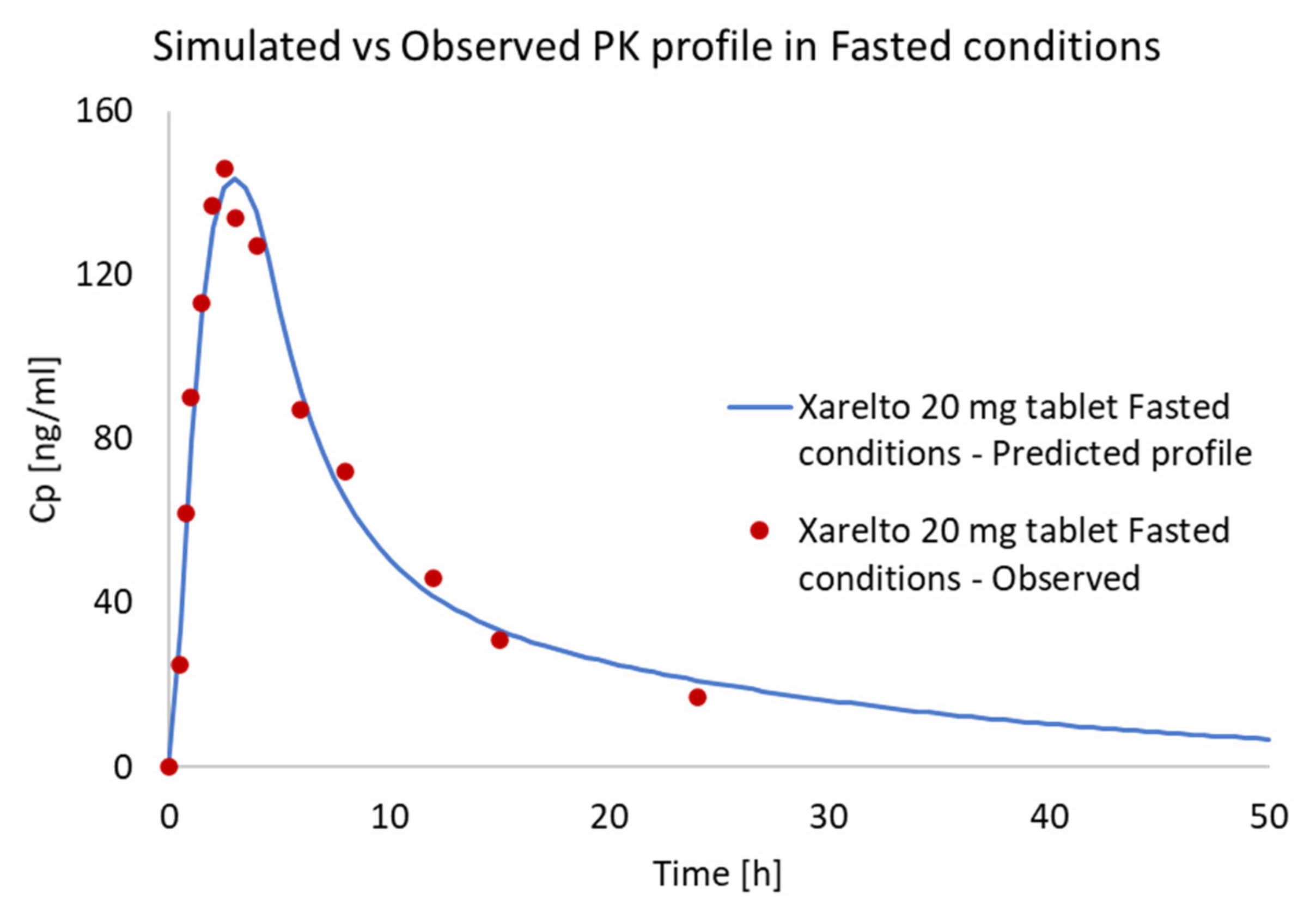
Prediction of PK Profiles and Optimization of ACAT Model
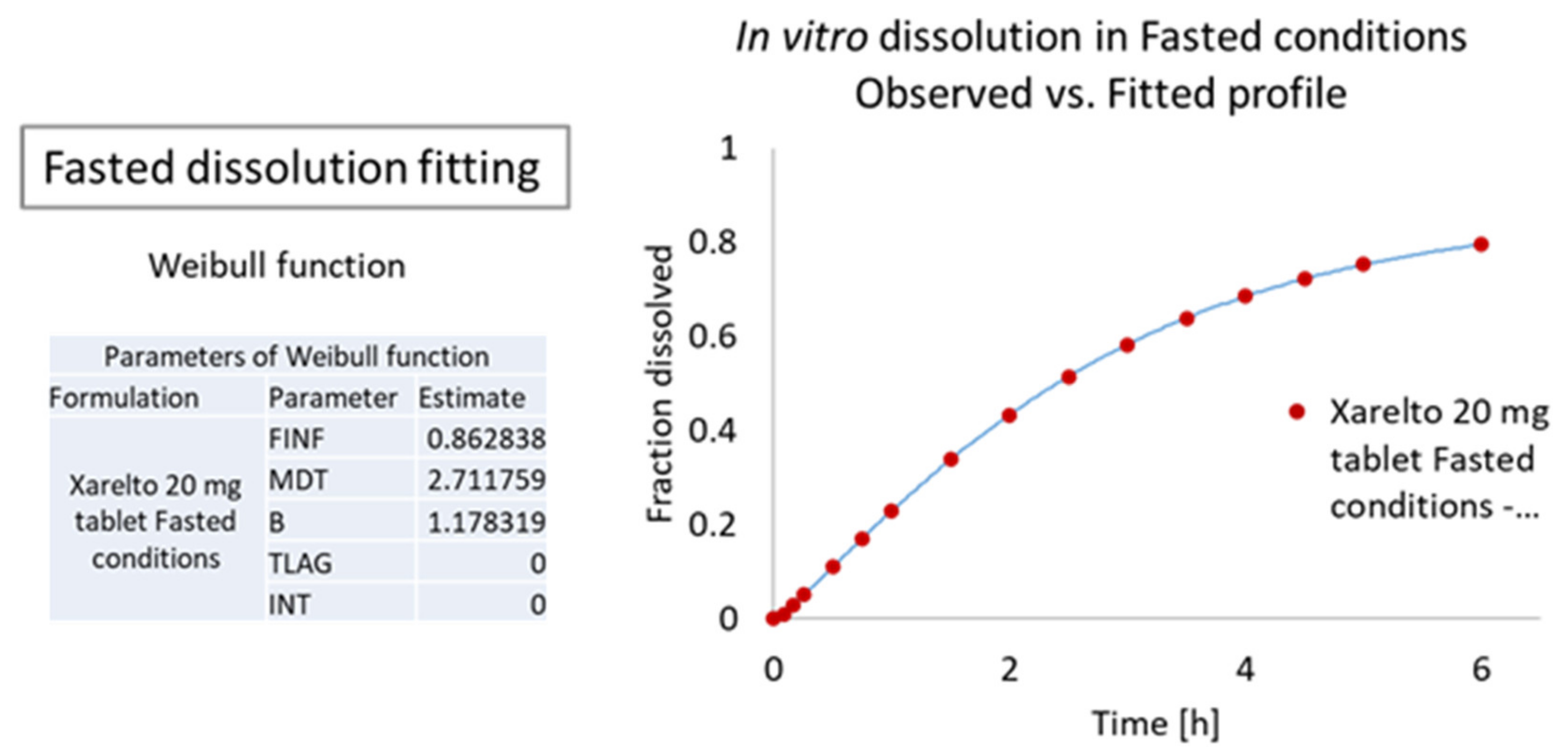
3.3. IVIVC Studies
3.3.1. Modelling
In Vitro and In Vivo Raw Data
Dissolution Curve Fitting
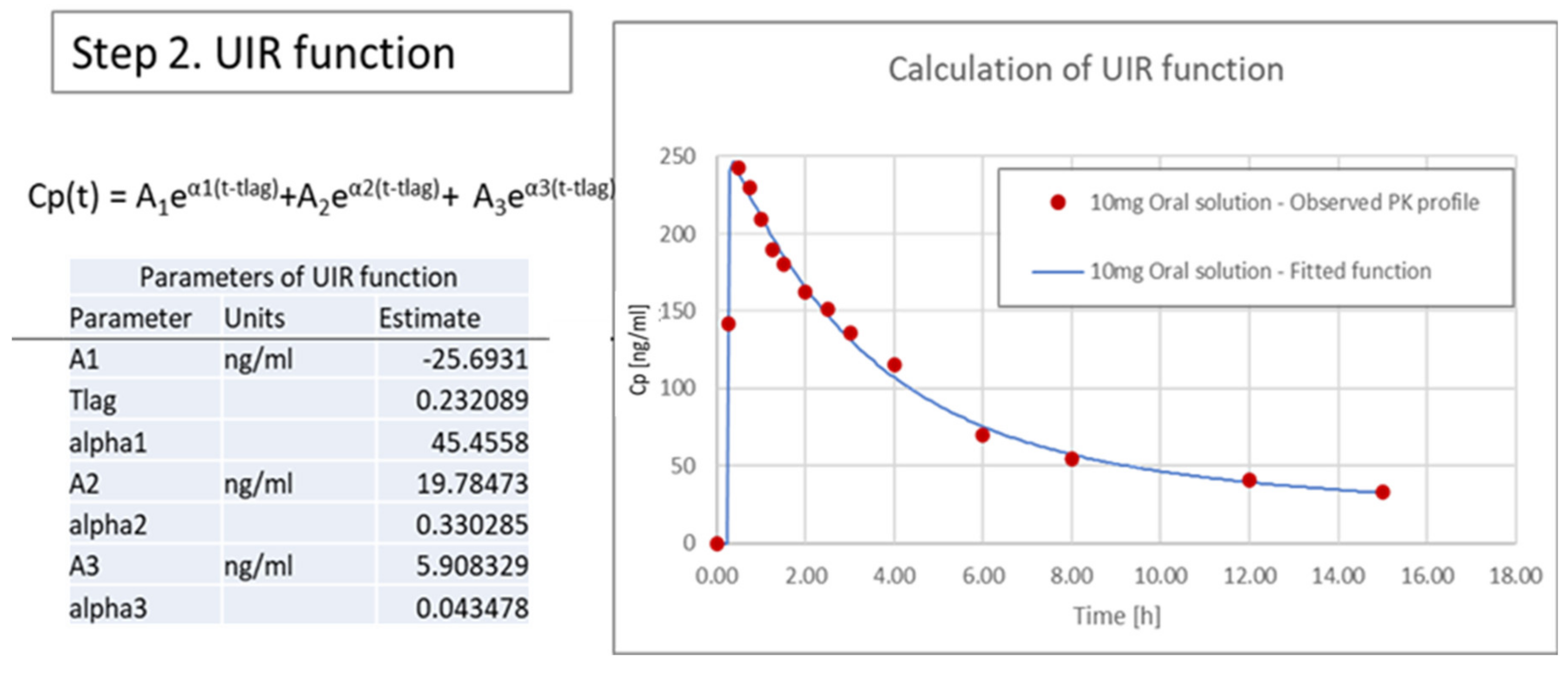
3.3.2. Calculation of Unit Impulse Response (UIR) Function
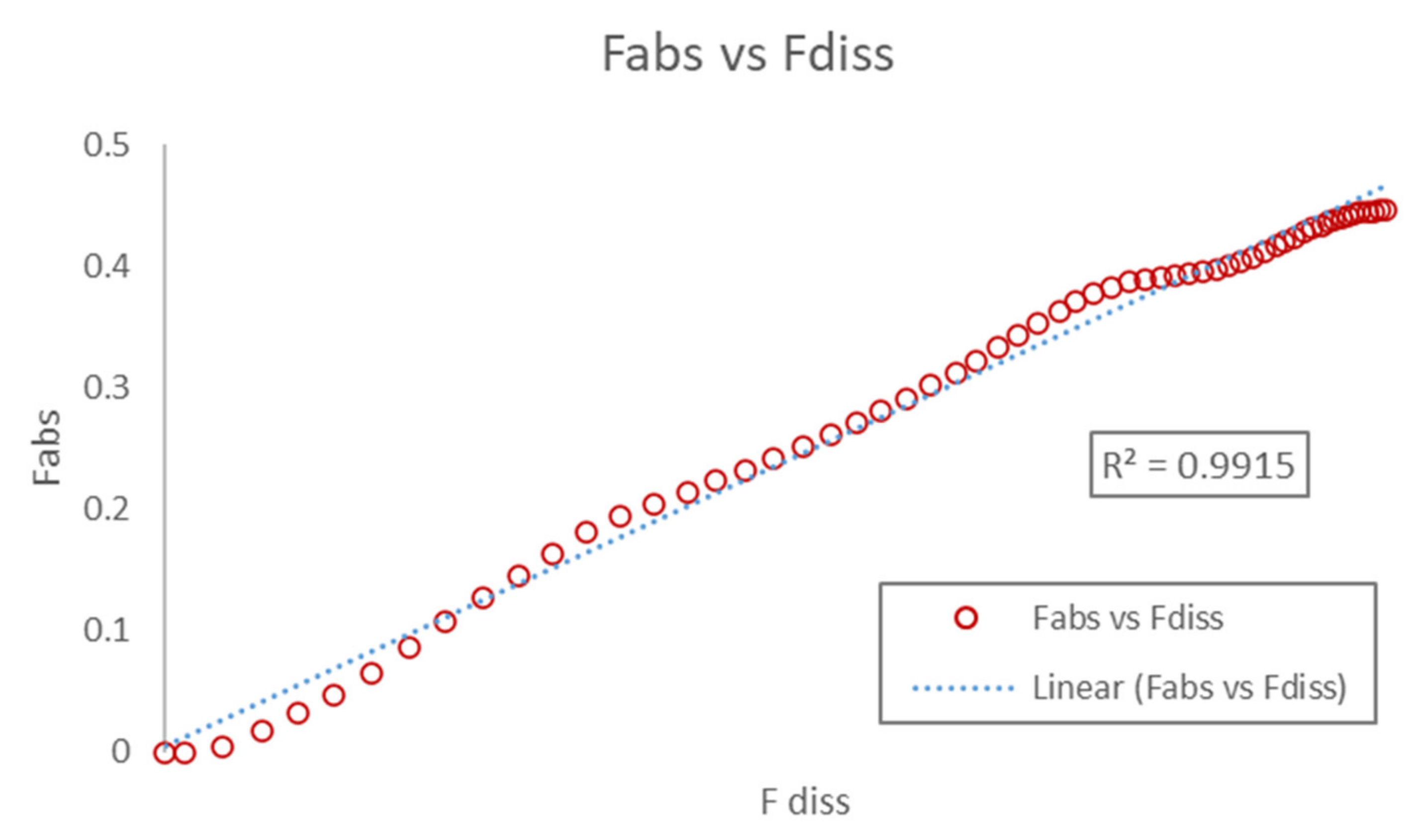
3.3.3. Correlation
3.3.4. Internal Validation and Prediction
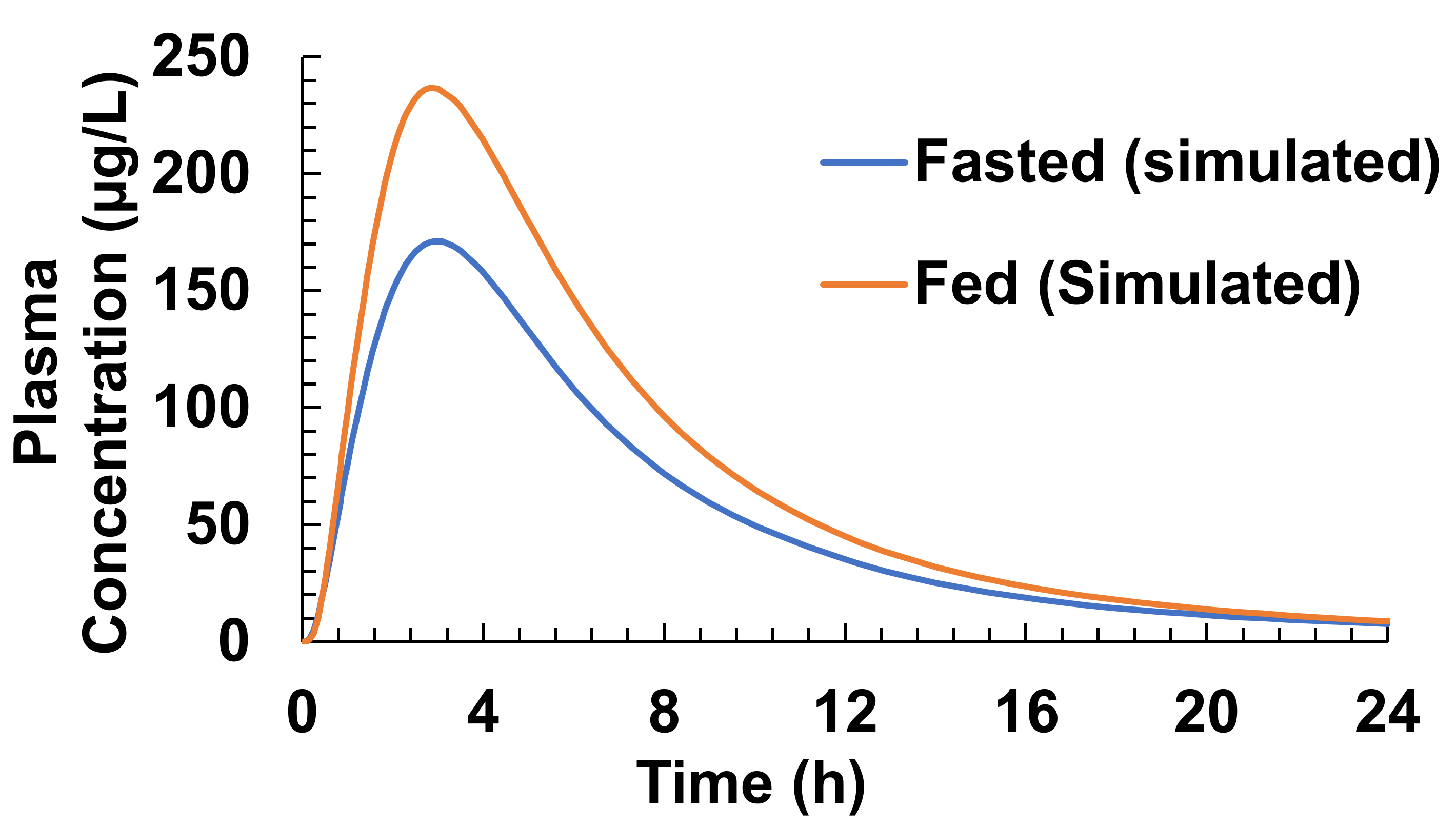
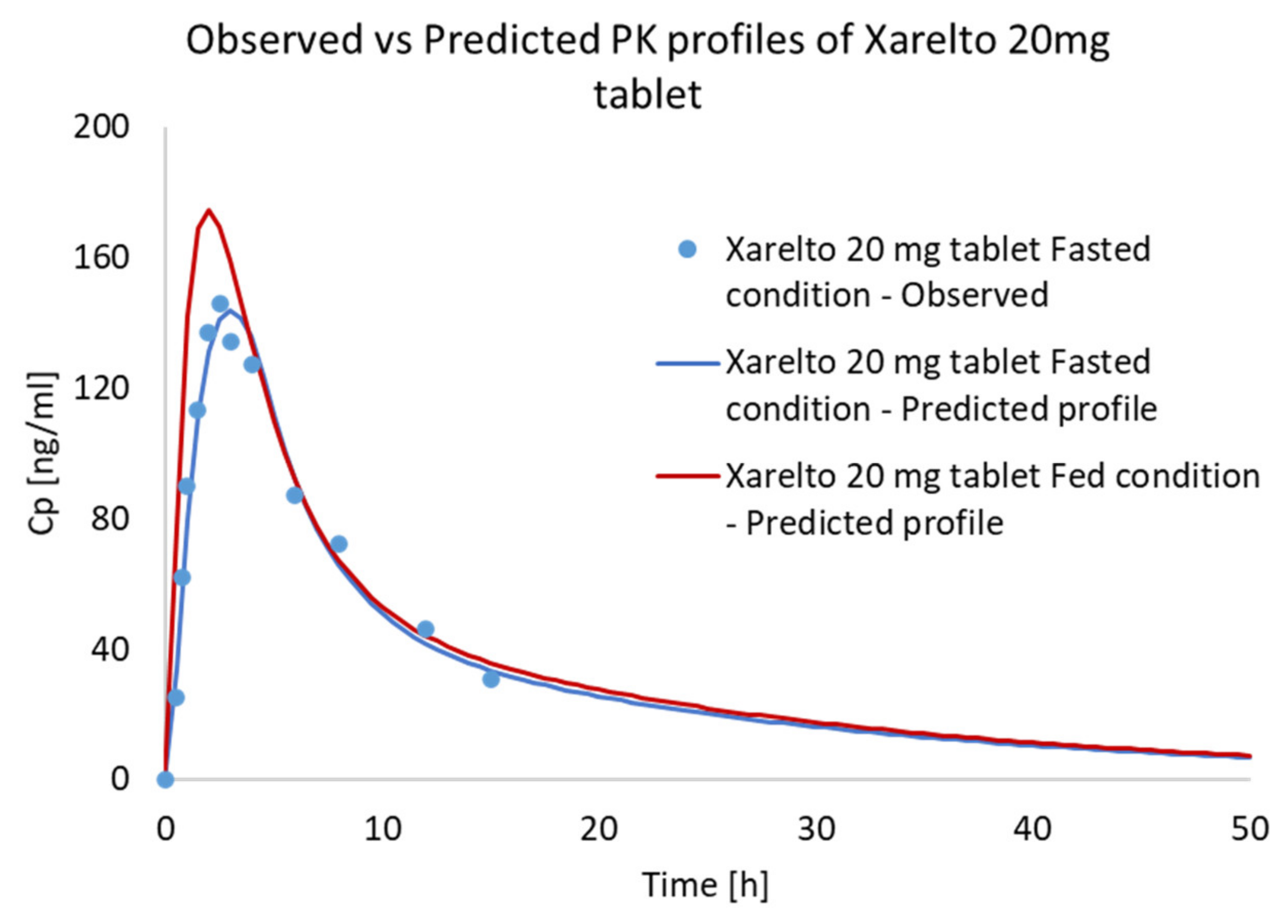
3.4. Food Effect (FE) Studies of Riva in Simulated Healthy Subjects
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations:
| Absorption Scale Factors | ASF |
| Active pharmaceutical ingredients | APIs |
| Advanced compartmental absorption and transit | ACAT |
| Akaike information criterion | AIC |
| Apparent permeability coefficient | Papp |
| American Type Culture Collection | ATCC |
| Bioavailability | BA |
| Bioequivalence | BE |
| Biopharmaceutical Classification System | BCS |
| Immediate release | IR |
| In Vitro–In Vivo correlation | IVIVC |
| Minimal Essential Medium | MEM |
| Fasted state simulated gastric fluid | FaSSGF |
| Fasted state simulated intestinal fluid | FaSSIF |
| Fed state simulated gastric fluid | FeSSGF |
| Fed state simulated intestinal fluid | FeSSIF |
| Fetal bovine serum | FBS |
| Parameter Sensitivity Analysis | PSA |
| Pharmacokinetics | PK |
| Physiologically based pharmacokinetic | PBPK |
| Rivaroxaban | Riva |
| Schwarz criterion | SC |
| Sodium lauryl sulfate | SLS |
| Transepithelial electrical resistance | TEER |
| Ultra-High-Performance Liquid Chromatography | UHPLC |
| Unit impulse response | UIR |
| United States Pharmacopeia | USP |
References
- Jamei, M.; Abrahamsson, B.; Brown, J.; Bevernage, J.; Bolger, M.B.; Heimbach, T.; Karlsson, E.; Kotzagiorgis, E.; Lindahl, A.; McAllister, M. Current status and future opportunities for incorporation of dissolution data in PBPK modeling for pharmaceutical development and regulatory applications: OrBiTo consortium commentary. Eur. J. Pharm. Biopharm. 2020, 155, 55–68. [Google Scholar] [CrossRef]
- European Medicines Agency. ICH M9 on biopharmaceutics classification system based biowaivers. Available online: https://www.ema.europa.eu/en/ich-m9-biopharmaceutics-classification-system-based-biowaivers#current-version-section (accessed on 18 January 2021).
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of In Vitro Drug Product Dissolution and In Vivo Bioavailability. Pharm. Res. An Off. J. Am. Assoc. Pharm. Sci. 1995, 12, 413–420. [Google Scholar] [CrossRef] [Green Version]
- FDA. Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/extended-release-oral-dosage-forms-development-evaluation-and-application-vitroin-vivo-correlations (accessed on 18 January 2021).
- Stillhart, C.; Pepin, X.; Tistaert, C.; Good, D.; Bergh, A.; Van Den Parrott, N.; Kesisoglou, F. PBPK Absorption Modeling: Establishing the In Vitro–In Vivo Link—Industry Perspective. AAPS J. 2019, 21, 1–13. [Google Scholar] [CrossRef]
- Kesisoglou, F.; Xia, B.; Agrawal, N.G.B. Comparison of Deconvolution-Based and Absorption Modeling IVIVC for Extended Release Formulations of a BCS III Drug Development Candidate. AAPS J. 2015, 17, 1492–1500. [Google Scholar] [CrossRef] [Green Version]
- Kaur, N.; Narang, A.; Bansal, A.K. Use of biorelevant dissolution and PBPK modeling to predict oral drug absorption. Eur. J. Pharm. Biopharm. 2018, 129, 222–246. [Google Scholar] [CrossRef]
- Pepin, X.J.H.; Flanagan, T.R.; Holt, D.J.; Eidelman, A.; Treacy, D.; Rowlings, C.E. Justification of drug product dissolution rate and drug substance particle size specifications based on absorption PBPK modeling for lesinurad immediate release tablets. Mol. Pharm. 2016, 13, 3256–3269. [Google Scholar] [CrossRef] [PubMed]
- Willmann, S.; Thelen, K.; Becker, C.; Dressman, J.B.; Lippert, J. Mechanism-based prediction of particle size-dependent dissolution and absorption: Cilostazol pharmacokinetics in dogs. Eur. J. Pharm. Biopharm. 2010, 76, 83–94. [Google Scholar] [CrossRef]
- Pepin, X.J.H.; Huckle, J.E.; Alluri, R.V.; Basu, S.; Dodd, S.; Parrott, N.; Emami Riedmaier, A. Understanding Mechanisms of Food Effect and Developing Reliable PBPK Models Using a Middle-out Approach. AAPS J. 2021, 23, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Davit, B.M.; Kanfer, I.; Tsang, Y.C.; Cardot, J.M. BCS biowaivers: Similarities and differences among EMA, FDA, and WHO requirements. AAPS J. 2016, 18, 612–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Services, H. Vladimir Nikolaevich Chernigovski. Acta Physiol. Pharmacol. Bulg. 1977, 3, 3–5. [Google Scholar]
- Jain, S.; Jain, R.; Das, M.; Agrawal, A.K.; Thanki, K.; Kushwah, V. Combinatorial bio-conjugation of gemcitabine and curcumin enables dual drug delivery with synergistic anticancer efficacy and reduced toxicity. RSC Adv. 2014, 4, 29193–29201. [Google Scholar] [CrossRef]
- Arora, R.; Katiyar, S.S.; Kushwah, V.; Jain, S. Solid lipid nanoparticles and nanostructured lipid carrier-based nanotherapeutics in treatment of psoriasis: A comparative study. Expert Opin. Drug Deliv. 2017, 14, 165–177. [Google Scholar] [CrossRef]
- Shilpi, D.; Kushwah, V.; Agrawal, A.K.; Jain, S. Improved Stability and Enhanced Oral Bioavailability of Atorvastatin Loaded Stearic Acid Modified Gelatin Nanoparticles. Pharm. Res. 2017, 34, 1505–1516. [Google Scholar] [CrossRef]
- Tripathi, S.; Kushwah, V.; Thanki, K.; Jain, S. Triple antioxidant SNEDDS formulation with enhanced oral bioavailability: Implication of chemoprevention of breast cancer. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1431–1443. [Google Scholar] [CrossRef]
- Kubitza, D.; Becka, M.; Zuehlsdorf, M.; Mueck, W. Effect of food, an antacid, and the H2 antagonist ranitidine on the absorption of BAY 59-7939 (rivaroxaban), an oral, direct Factor Xa inhibitor, in healthy subjects. J. Clin. Pharmacol. 2006, 46, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Kubitza, D.; Becka, M.; Voith, B.; Zuehlsdorf, M.; Wensing, G. Safety, pharmacodynamics, and pharmacokinetics of single doses of BAY 59-7939, an oral, direct factor Xa inhibitor. Clin. Pharmacol. Ther. 2005, 78, 412–421. [Google Scholar] [CrossRef]
- Takács-Novák, K.; Szőke, V.; Völgyi, G.; Horváth, P.; Ambrus, R.; Szabó-Révész, P. Biorelevant solubility of poorly soluble drugs: Rivaroxaban, furosemide, papaverine and niflumic acid. J. Pharm. Biomed. Anal. 2013, 83, 279–285. [Google Scholar] [CrossRef]
- EMEA. Note for Guidance on Quality of Modified Release Products: A: Oral Dosage Forms B: Transdermal Dosage Forms Section I (Quality). Guidance 1999, 96, 6–7. [Google Scholar]
- Malinowski, H.; Marroum, P.; Uppoor, V.R.; Gillespie, W.; Ahn, H.Y.; Lockwood, P.; Henderson, J.; Baweja, R.; Hossain, M.; Fleischer, N.; et al. FDA guidance for industry extended release solid oral dosage forms: Development, evaluation, and application of In Vitro/In Vivo correlations. Dissolution Technol. 1997, 4, 23–32. [Google Scholar] [CrossRef]
- Stampfuss, J.; Kubitza, D.; Becka, M.; Mueck, W. The effect of food on the absorption and pharmacokinetics of rivaroxaban. Int. J. Clin. Pharmacol. Ther. 2013, 51, 549–561. [Google Scholar] [CrossRef]
- EMA, C. Guideline on the conduct of bioequivalence studies for veterinary medicinal products. 2010, 44, 1–25. [Google Scholar]
- Shah, V.P.; Lesko, L.J.; Fan, J.; Fleischer, N.; Handerson, J.; Malinowski, H.; Makary, M.; Ouderkirk, L.; Bay, S.; Sathe, P.; et al. FDA guidance for industry 1 dissolution testing of immediate release solid oral dosage forms. Dissolution Technol. 1997, 4, 15–22. [Google Scholar] [CrossRef]
- Gnoth, M.J.; Buetehorn, U.; Muenster, U.; Schwarz, T.; Sandmann, S. In Vitro and In Vivo P-glycoprotein transport characteristics of rivaroxaban. J. Pharmacol. Exp. Ther. 2011, 338, 372–380. [Google Scholar] [CrossRef]
- Thanki, K.; Kushwah, V.; Jain, S. Recent Advances in Tumor Targeting Approaches; Springer: Cham, Germany, 2015; pp. 41–112. [Google Scholar]
- Wilcox, M.D.; Van Rooij, L.K.; Chater, P.I.; Pereira De Sousa, I.; Pearson, J.P. The effect of nanoparticle permeation on the bulk rheological properties of mucus from the small intestine. Eur. J. Pharm. Biopharm. 2015, 96, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Pontier, C.; Pachot, J.; Botham, R.; Lenfant, B.; Arnaud, P. HT29-MTX and Caco-2/TC7 monolayers as predictive models for human intestinal absorption: Role of the mucus layer. J. Pharm. Sci. 2001, 90, 1608–1619. [Google Scholar] [CrossRef]
- Wharf, C.; Kingdom, U. Keppra CHMP Assessment Report for Paediatric Use Studies Submitted According to Article 46 of the RegulationEC No. 1901/. 2006, 2013, 44. [Google Scholar]
- Weinz, C.; Schwarz, T.; Kubitza, D.; Mueck, W.; Lang, D. Metabolism and excretion of rivaroxaban, an oral, direct factor Xa inhibitor, in rats, dogs, and humans. Drug Metab. Dispos. 2009, 37, 1056–1064. [Google Scholar] [CrossRef]
- Michailidou, A.; Trenz, H.-J.; de Wilde, P.; Annex, I. The Internet and European Integration. JSTOR 2019, 167–172. [Google Scholar] [CrossRef]
- Mueck, W.; Schwers, S.; Stampfuss, J. Rivaroxaban and other novel oral anticoagulants: Pharmacokinetics in healthy subjects, specific patient populations and relevance of coagulation monitoring. Thromb. J. 2013, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- European Medicines Agency (EMA) Assessment report Xarelto (Rivaroxaban) Procedure No. EMEA/H/C/000944/X/0017. 2013; 44, 1–75.
- Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/022406s028lbl.pdf (accessed on 15 February 2021).
- Available online: https://www.ema.europa.eu/en/documents/product-information/rivaroxaban-accord-epar-product-information_en.pdf (accessed on 15 February 2021).
- Pepin, X.J.H.; Moir, A.J.; Mann, J.C.; Sanderson, N.J.; Barker, R.; Meehan, E.; Plumb, A.P.; Bailey, G.R.; Murphy, D.S.; Krejsa, C.M.; et al. Bridging In Vitro dissolution and In Vivo exposure for acalabrutinib. Part II. A mechanistic PBPK model for IR formulation comparison, proton pump inhibitor drug interactions, and administration with acidic juices. Eur. J. Pharm. Biopharm. 2019, 142, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Committee for Medicinal Products for Human Use (CHMP) Assessment report: Xarelto; EMA/CHMP/301607/2011. 2011; 44.
- Parrott, N.; Lukacova, V.; Fraczkiewicz, G.; Bolger, M.B. Predicting pharmacokinetics of drugs using physiologically based modeling—application to food effects. AAPS J. 2009, 11, 45–53. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physiochemical Parameter | Values |
|---|---|
| Molecular Weight (g/mol) | 435.89 |
| logP | 1.36 |
| pKa | strongest acidic: 13.6strongest basic: 1.6 |
| Solubility vs. pH | water solubility (pH = 7) = 10 µg/mL pH 1.2, FaSSGF = 11 µg/mL pH 6.5, FaSSIF = 9.9 µg/ml pH 5.0, FeSSIF =16.8 µg/ml |
| Particle Size (Radius) | 7.5 µm (Xarelto tablet, 20 mg) d90 = 9.4 µm; d50 =3.8 µm; d10 =0.7 µm |
| Caco-2 Permeability Dissolution Profiles (USP 4) | 2.69 ± 0.72 × 10−6 cm/s (Xarelto) Xarelto IR tablet (20 mg) |
| Solubility in | pH | Values (µg/mL) |
|---|---|---|
| Unbuffered water | 7.0 | 10.0 |
| FaSSGF | 1.6 | 11.0 |
| FaSSIF | 6.5 | 9.9 |
| FeSSGF | 4.5 | 24.0 |
| FeSSIF | 5.0 | 16.8 |
| Parameter | Values |
|---|---|
| Clearance (L/h) | 9.43 |
| Vc (L/kg) | 0.47 |
| T1/2 (h) | 4.62 |
| K12 (1/h) | 0.04 |
| K21 (1/h) | 0.21 |
| V2 (L/kg) | 0.09 |
| Compartment | Default (GastroPlus) ASF | Optimized ASF |
|---|---|---|
| Stomach | 0 | 0 |
| Duodenum | 2.673 | 36.44 |
| Jejunum 1 | 2.658 | 36.25 |
| Jejunum 2 | 2.629 | 35.85 |
| Ileum 1 | 2.592 | 35.35 |
| Ileum 2 | 2.568 | 35.02 |
| Ileum 3 | 2.505 | 34.16 |
| Caecum | 0.535 | 0.535 |
| Asc Colon | 1.038 | 1.038 |
| Parameter | Actual (Reported) a | Predicted (Before Optimization) | Predicted (After Optimization) |
|---|---|---|---|
| PK parameters obtained from simulated PK profile of solution oral dose (10 mg) of Riva before and after optimization | |||
| Cmax (µg/L) | 266/25.1 (187–412) | 159.27 | 212.23 |
| Tmax (h) | 0.50 (0.25–1.00) | 1.92 | 0.72 |
| AUC (µg·h/L) | 997/25.1 (613–1383) | 1056 | 1058 |
| F% | >90% | 99.54 | 99.79 |
| Pharmacokinetic parameters for 5 mg oral (solution) dose of Riva in fasted conditions | |||
| Cmax (µg/L) | 119/18.5 (97.2–158) | 80.13 | 107.06 |
| Tmax (h) | 0.63 (0.5–0.75) | 1.92 | 0.66 |
| AUC (µg·h/L) | 461/17.2 (348–587) | 528.24 | 529.37 |
| F% | >90% | 99.58 | 99.80 |
| Parameter | Actual (Reported) a | Predicted (Optimized ASF) |
|---|---|---|
| Pharmacokinetic parameters for 5 mg oral (IR Tablet) dose of Riva in fasted conditions | ||
| Cmax (µg/L) | 72/19.7 (55–96) | 76.13 |
| Tmax (h) | 1.88 (0.5–4.00) | 2.1 |
| AUC (µg·h/L) | 466/23.0 (348–677) | 524.42 |
| F% | 80–100% | 98.86 |
| Pharmacokinetic parameters for 10 mg oral (IR Tablet) dose of Riva in fasted conditions | ||
| Cmax (µg/L) | 141/15.5 (112–184) | 149 |
| Tmax (h) | 2.00 (0.5–2.50) | 2.20 |
| AUC (µg·h/L) | 1020/14.9 (797–1217) | 1037 |
| F% | 80–100% | 97.75 |
| Parameters | Fasted State | Fed State | ||
|---|---|---|---|---|
| Actual (Reported) a | Predicted (Optimized ASF) | Actual(Reported) b | Predicted (Optimized ASF) | |
| Cmax (µg/L) | 173/35.6 (111–294) | 171.15 | 294.4/15 (225.4–360.6) | 236.64 |
| Tmax (h) | 1.50 (0.5–4.00) | 3 | 3.00 (0.5–6.00) | 2.9 |
| AUC (µg·h/L) | 1612/36.1 (859–2193) | 1433.8 | 2294/19 (1464–3227) | 1857.3 |
| F% | 66% | 67.57 | 80–100% | 87.54 |
| Formulation | Parameter | Predicted | Observed a | %PE | Ratio |
|---|---|---|---|---|---|
| Xarelto 20 mg tablet Fasted condition | AUClast (µg·h/L) | 1381.946 | 1361.125 | 1.52966 | 1.015297 |
| Xarelto 20 mg tablet Fasted condition | Cmax (µg/L) | 143.567 | 146.000 | −1.66657 | 0.983334 |
| Xarelto 20 mg tablet Fasted condition | Tmax (h) | 3.0 | 1.5 | N/A | N/A |
| Xarelto 20 mg tablet Fed condition | AUClast (µg·h/L) | 1543.120 | 1750.175 | 13.41790 | 1.134179 |
| Xarelto 20 mg tablet Fed condition | Cmax (µg/L) | 174.566 | 241.000 | 38.05670 | 1.380567 |
| Xarelto 20 mg tablet Fed condition | Tmax (h) | 2.0 | 3.0 | N/A | N/A |
| Parameters | Population A | Population B | Population C |
|---|---|---|---|
| Cmax (ng/mL) | 238 | 236 | 237 |
| AUC0–∞ (ng·h/mL) | 1856 | 1888 | 1988 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kushwah, V.; Arora, S.; Tamás Katona, M.; Modhave, D.; Fröhlich, E.; Paudel, A. On Absorption Modeling and Food Effect Prediction of Rivaroxaban, a BCS II Drug Orally Administered as an Immediate-Release Tablet. Pharmaceutics 2021, 13, 283. https://doi.org/10.3390/pharmaceutics13020283
Kushwah V, Arora S, Tamás Katona M, Modhave D, Fröhlich E, Paudel A. On Absorption Modeling and Food Effect Prediction of Rivaroxaban, a BCS II Drug Orally Administered as an Immediate-Release Tablet. Pharmaceutics. 2021; 13(2):283. https://doi.org/10.3390/pharmaceutics13020283
Chicago/Turabian StyleKushwah, Varun, Sumit Arora, Miklós Tamás Katona, Dattatray Modhave, Eleonore Fröhlich, and Amrit Paudel. 2021. "On Absorption Modeling and Food Effect Prediction of Rivaroxaban, a BCS II Drug Orally Administered as an Immediate-Release Tablet" Pharmaceutics 13, no. 2: 283. https://doi.org/10.3390/pharmaceutics13020283
APA StyleKushwah, V., Arora, S., Tamás Katona, M., Modhave, D., Fröhlich, E., & Paudel, A. (2021). On Absorption Modeling and Food Effect Prediction of Rivaroxaban, a BCS II Drug Orally Administered as an Immediate-Release Tablet. Pharmaceutics, 13(2), 283. https://doi.org/10.3390/pharmaceutics13020283