
Tumor Activated Cell Penetrating Peptides to Selectively Deliver Immune Modulatory Drugs

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Synthesis of ACPP-Resiquimod Conjugate
2.2. Synthesis of Ratiometric ACPP, ACPP-Cy5 Conjugate and CPP-Cy5 Conjugate
2.3. Cell culture Cy5 Imaging
2.4. In Vivo Murine Fluorescent Imaging Experiments
2.5. Confocal Imaging Of Murine Tissue Sections
2.6. Tissue Drug Concentration
2.7. In Vivo Murine Therapy Experiments
3. Results
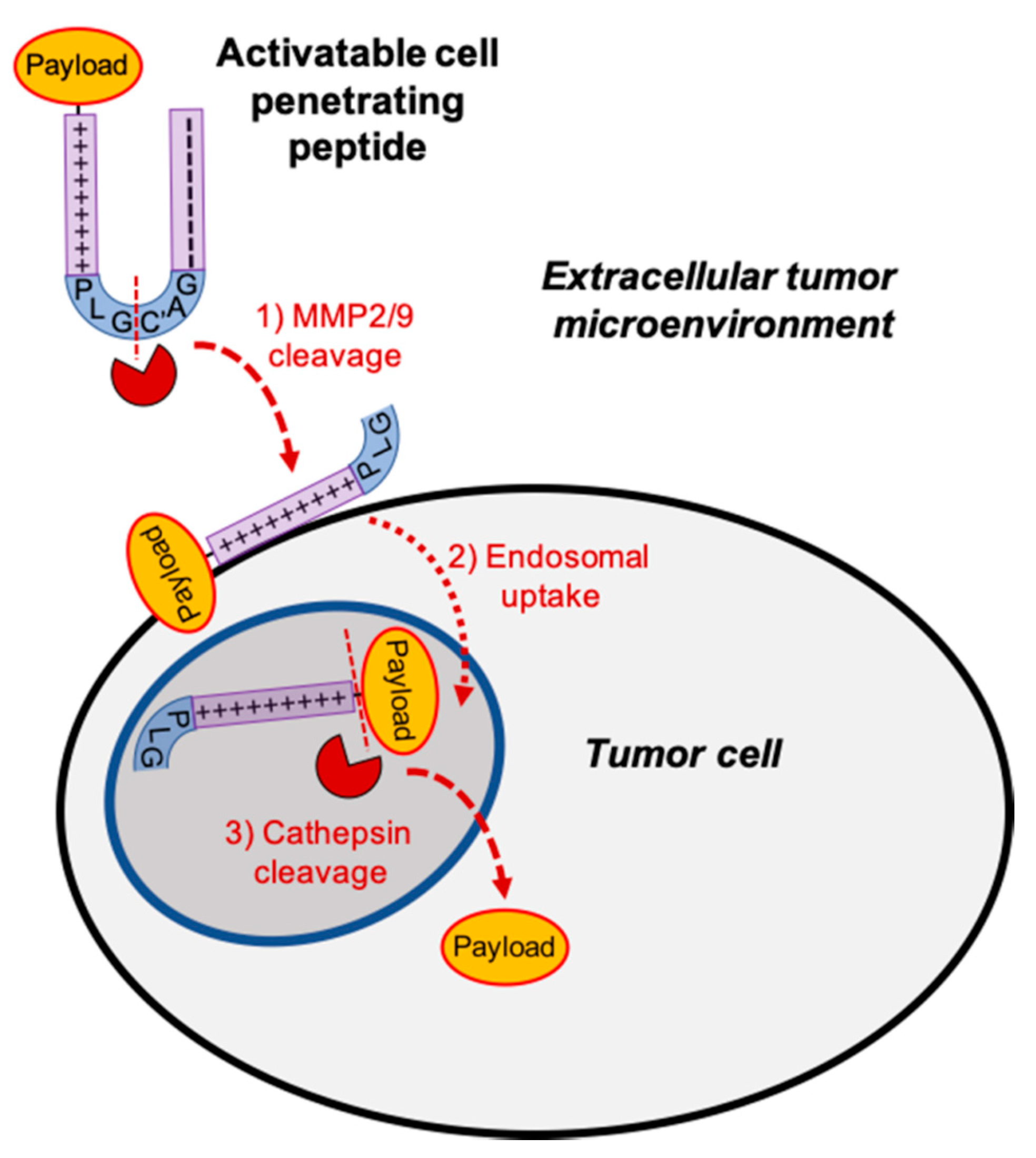
3.1. Tumor Targeted Activatable Cell Penetrating Peptides
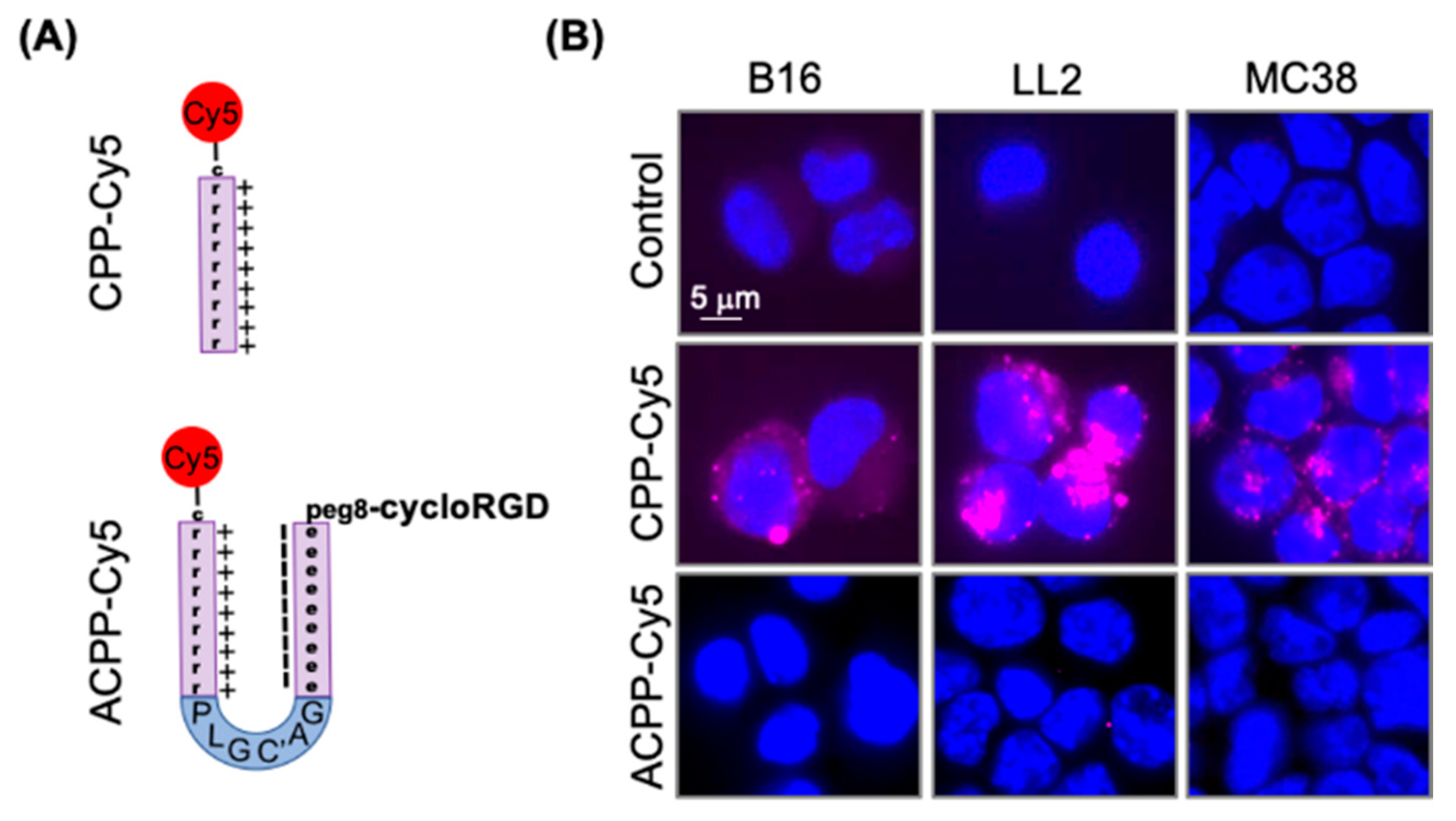
3.2. Intact ACPP Cloak Polycationic Cell Penetrating Peptides
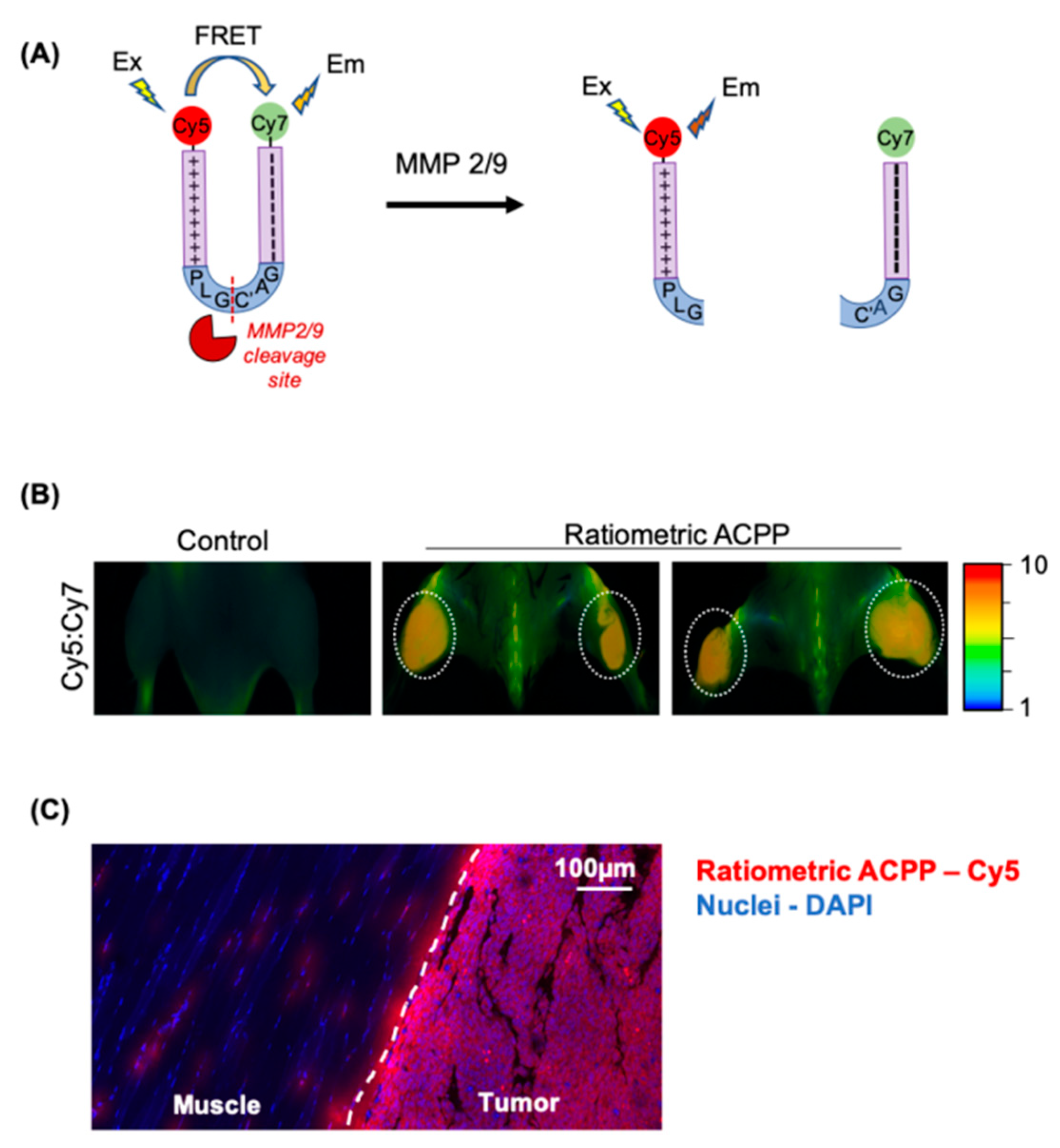
3.3. Cleavage of PLGC(Me)AG Targeted ACPPs in Live Mice
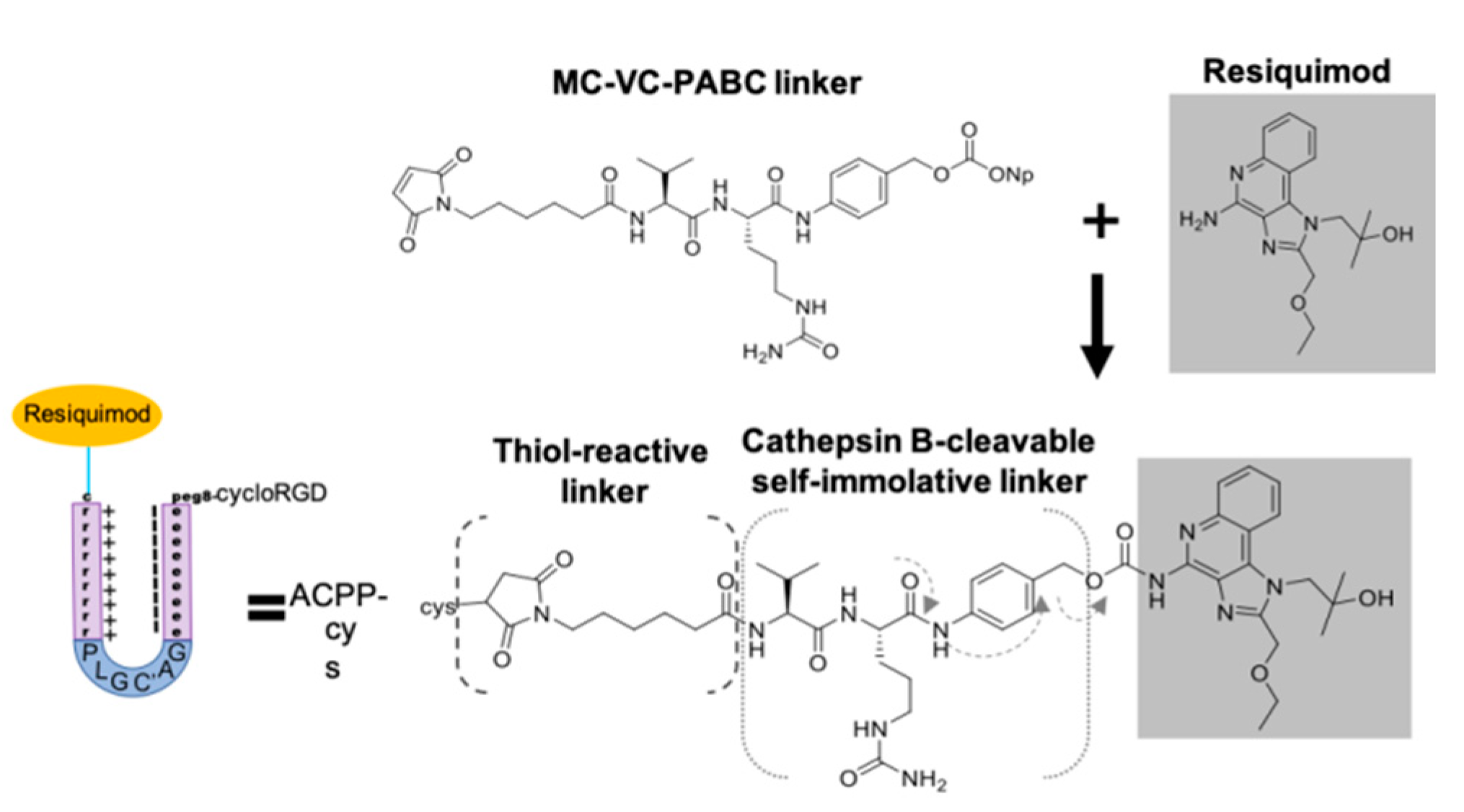
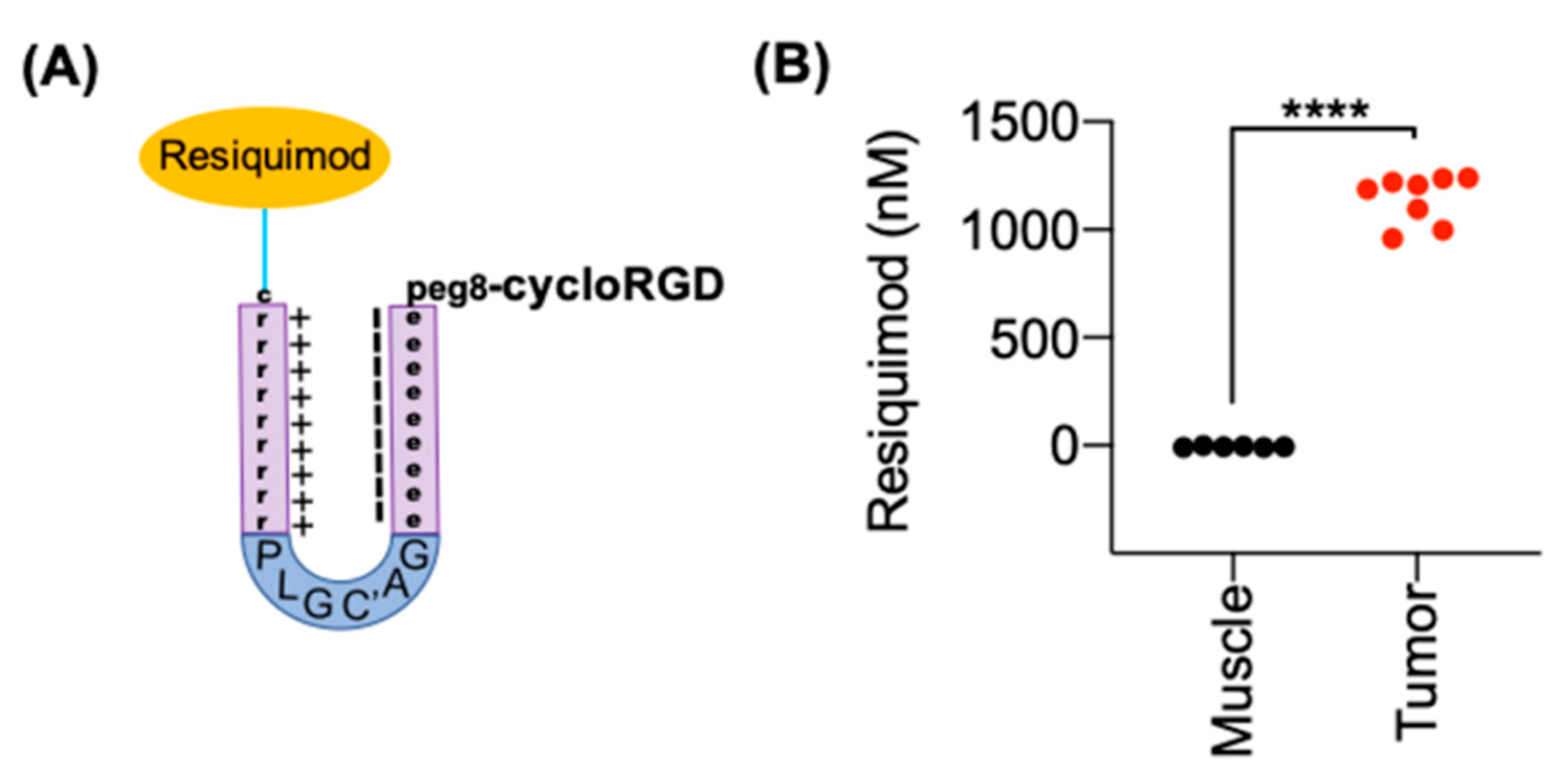
3.4. Synthesis of ACPP Resiquimod Conjugate
3.5. ACPP-Resiquimod Biodistribution
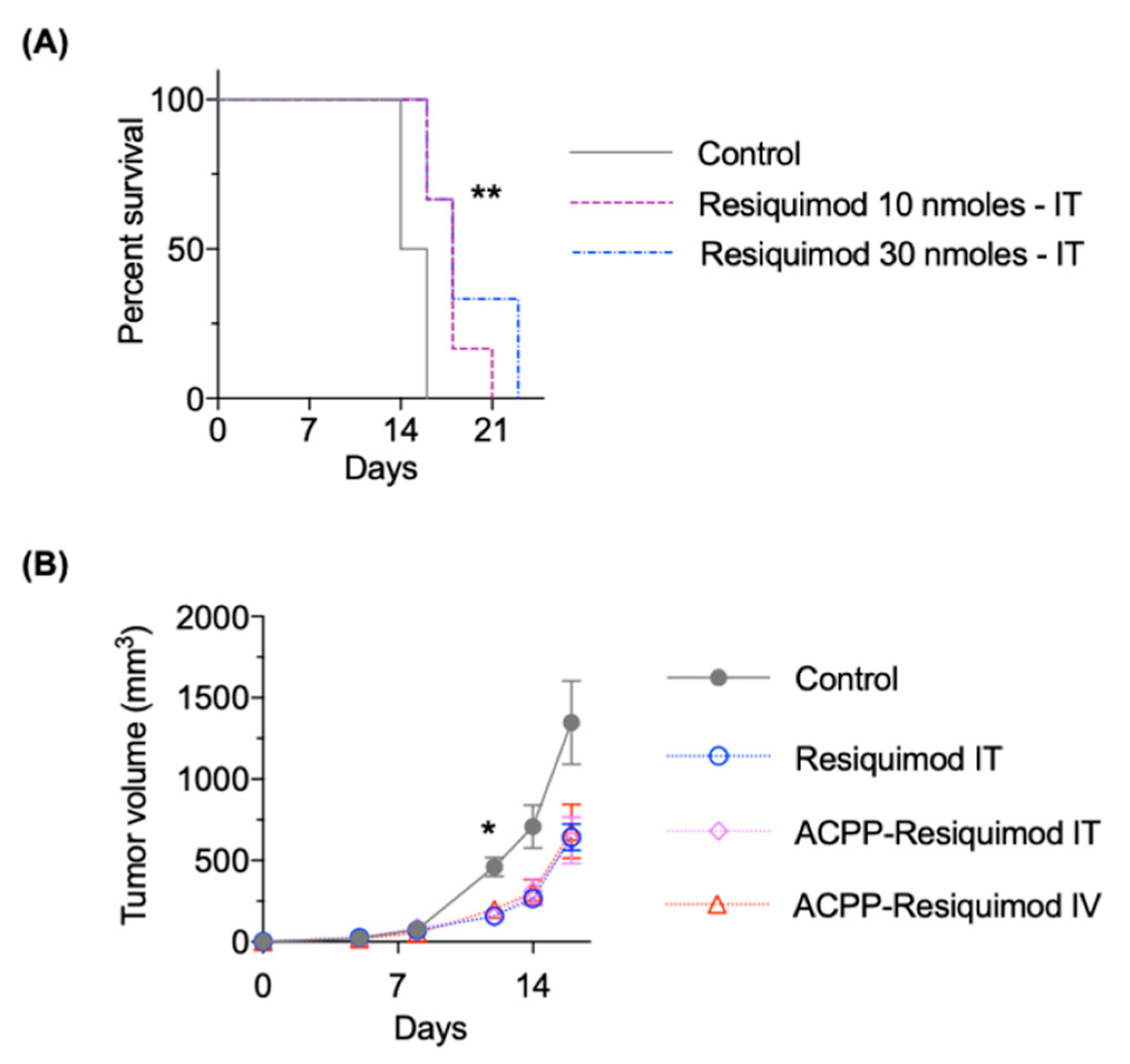
3.6. Antitumor Efficacy of ACPP-Resiquimod
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gameiro, G.R.; Sinkunas, V.; Liguori, G.R.; Auler-Júnior, J.O.C. Precision Medicine: Changing the way we think about healthcare. Clinics 2018, 73, e723. [Google Scholar] [CrossRef]
- Gindy, M.E.; Prud’Homme, R.K. Multifunctional nanoparticles for imaging, delivery and targeting in cancer therapy. Expert Opin. Drug Deliv. 2009, 6, 865–878. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T. EPR-effect: Utilizing size-dependent nanoparticle delivery to solid tumors. Ther. Deliv. 2013, 4, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lin, Y.; Gillies, R.J. Tumor pH and Its Measurement. J. Nucl. Med. 2010, 51, 1167–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodell, C.B.; Ahmed, M.S.; Garris, C.S.; Pittet, M.J.; Weissleder, R. Development of adamantane-conjugated TLR7/8 agonists for supramolecular delivery and cancer immunotherapy. Theranostics 2019, 9, 8426. [Google Scholar] [CrossRef]
- Thauvin, C.; Widmer, J.; Mottas, I.; Hocevar, S.; Allémann, E.; Bourquin, C.; Delie, F. Development of resiquimod-loaded modified PLA-based nanoparticles for cancer immunotherapy: A kinetic study. Eur. J. Pharm. Biopharm. 2019, 139, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Tang, W.-L.; Kheirolomoom, A.; Fite, B.Z.; Wu, B.; Lau, K.; Baikoghli, M.; Raie, M.N.; Tumbale, S.K.; Foiret, J.; et al. Development of thermosensitive resiquimod-loaded liposomes for enhanced cancer immunotherapy. J. Control. Release 2021, 330, 1080–1094. [Google Scholar] [CrossRef]
- Denny, W.A. Prodrug strategies in cancer therapy. Eur. J. Med. Chem. 2001, 36, 577–595. [Google Scholar] [CrossRef]
- Mahato, R.; Tai, W.; Cheng, K. Prodrugs for improving tumor targetability and efficiency. Adv. Drug Deliv. Rev. 2011, 63, 659–670. [Google Scholar] [CrossRef] [Green Version]
- Sievers, E.L.; Senter, P.D. Antibody-Drug Conjugates in Cancer Therapy. Annu. Rev. Med. 2013, 64, 15–29. [Google Scholar] [CrossRef]
- Sagi, A.; Weinstain, R.; Karton, N.; Shabat, D. Self-Immolative Polymers. J. Am. Chem. Soc. 2008, 130, 5434–5435. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, P.; Lepland, A.; Scodeller, P.; Fontana, F.; Torrieri, G.; Tiboni, M.; Shahbazi, M.; Casettari, L.; Kostiainen, M.A.; Hirvonen, J.; et al. Peptide-guided resiquimod-loaded lignin nanoparticles convert tumor-associated macrophages from M2 to M1 phenotype for enhanced chemotherapy. Acta Biomater. 2020. [Google Scholar] [CrossRef] [PubMed]
- Shadidi, M.; Sioud, M. Selective targeting of cancer cells using synthetic peptides. Drug Resist. Updat. 2003, 6, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Callmann, C.E.; Barback, C.V.; Thompson, M.P.; Hall, D.J.; Mattrey, R.F.; Gianneschi, N.C. Therapeutic Enzyme-Responsive Na-noparticles for Targeted Delivery and Accumulation in Tumors. Adv. Mater. 2015, 27, 4611–4615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collier, M.A.; Junkins, R.D.; Gallovic, M.D.; Johnson, B.M.; Johnson, M.M.; Macintyre, A.N. Acetalated dextran microparticles for codelivery of STING and TLR7/8 agonists. Mol. Pharm. 2018, 15, 4933–4946. [Google Scholar] [CrossRef] [PubMed]
- Neutsch, L.; Plattner, V.E.; Polster-Wildhofen, S.; Zidar, A.; Chott, A.; Borchard, G.; Zechner, O.; Gabor, F.; Wirth, M. Lectin Mediated Biorecognition as a Novel Strategy for Targeted Delivery to Bladder Cancer. J. Urol. 2011, 186, 1481–1488. [Google Scholar] [CrossRef]
- Jiang, T.; Olson, E.S.; Nguyen, Q.T.; Roy, M.; Jennings, P.A.; Tsien, R.Y. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 17867–17872. [Google Scholar] [CrossRef] [Green Version]
- Olson, E.S.; Aguilera, T.A.; Jiang, T.; Ellies, L.G.; Nguyen, Q.T.; Wong, E.H. In vivo characterization of activatable cell pene-trating peptides for targeting protease activity in cancer. Integr. Biol. 2009, 1, 382–393. [Google Scholar] [CrossRef] [Green Version]
- Crisp, J.L.; Savariar, E.N.; Glasgow, H.L.; Ellies, L.G.; Whitney, M.A.; Tsien, R.Y. Dual targeting of integrin alphavbeta3 and matrix metalloproteinase-2 for optical imaging of tumors and chemotherapeutic delivery. Mol. Cancer Ther. 2014, 13, 1514–1525. [Google Scholar] [CrossRef] [Green Version]
- Buckel, L.; Savariar, E.N.; Crisp, J.L.; Jones, K.A.; Hicks, A.M.; Scanderbeg, D.J.; Nguyen, Q.T.; Sicklick, J.K.; Lowy, A.M.; Tsien, R.Y.; et al. Tumor radiosensitization by monomethyl auristatin E: Mechanism of action and targeted delivery. Cancer Res. 2015, 75, 1376–1387. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, D.V.; Crisp, J.L.; Doan, M.K.; Camargo, M.F.; Quraishi, M.A.; Aguilera, J.; Gilardi, M.; Gross, L.A.; Jiang, T.; Li, W.T.; et al. Redirecting extracellular proteases to molecularly guide radiosensitizing drugs to tumors. Biomaterials 2020, 248, 120032. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Dissecting the mechanisms of immune checkpoint therapy. Nat. Rev. Immunol. 2020, 20, 75–76. [Google Scholar] [CrossRef] [PubMed]
- Geisse, J.; Caro, I.; Lindholm, J.; Golitz, L.; Stampone, P.; Owens, M. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: Results from two phase III, randomized, vehicle-controlled studies. J. Am. Acad. Dermatol. 2004, 50, 722–733. [Google Scholar] [CrossRef]
- Meyer, T.; Stockfleth, E. Clinical investigations of Toll-like receptor agonists. Expert Opin. Investig. Drugs 2008, 17, 1051–1065. [Google Scholar] [CrossRef]
- Fife, K.H.; Meng, T.-C.; Ferris, D.G.; Liu, P. Effect of Resiquimod 0.01% Gel on Lesion Healing and Viral Shedding When Applied to Genital Herpes Lesions. Antimicrob. Agents Chemother. 2007, 52, 477–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzavon, Y.M.; Zhao, P.; Lv, B.; Liu, M.; Zhang, X.; Xie, F.; Yang, L.; Shang, L.; Zhang, M.; Li, Q.; et al. TLR7 and TLR8 agonist resiquimod (R848) differently regulates MIF expression in cells and organs. Cytokine 2017, 97, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Park, C.-S.; Lee, Y.-R.; Im, S.-A.; Song, S.; Lee, C.-K. Resiquimod, a TLR7/8 agonist, promotes differentiation of mye-loid-derived suppressor cells into macrophages and dendritic cells. Arch. Pharm. Res. 2014, 37, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.L.; Holt, G.E.; Lu, H. The pharmacokinetics of Toll-like receptor agonists and the impact on the immune system. Expert Rev. Clin. Pharmacol. 2011, 4, 275–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geller, M.A.; Cooley, S.; Argenta, P.A.; Downs, L.S.; Carson, L.F.; Judson, P.L. Toll-like receptor-7 agonist administered sub-cutaneously in a prolonged dosing schedule in heavily pretreated recurrent breast, ovarian, and cervix cancers. Cancer Immunol. Immunother. 2010, 59, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Vascotto, F.; Petschenka, J.; Walzer, K.C.; Vormehr, M.; Brkic, M.; Strobl, S.; Rösemann, R.; Diken, M.; Kreiter, S.; Türeci, Ö.; et al. Intravenous delivery of the toll-like receptor 7 agonist SC1 confers tumor control by inducing a CD8+ T cell response. OncoImmunology 2019, 8, e1601480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigel, B.J.; Cooley, S.; DeFor, T.; Weisdorf, D.J.; Panoskaltsis-Mortari, A.; Chen, W. Prolonged subcutaneous administration of 852A, a novel systemic toll-like receptor 7 agonist, to activate innate immune responses in patients with advanced hema-tologic malignancies. Am. J. Hematol. 2012, 87, 953–956. [Google Scholar] [CrossRef] [Green Version]
- Meyer, T.; Surber, C.; E French, L.; Stockfleth, E. Resiquimod, a topical drug for viral skin lesions and skin cancer. Expert Opin. Investig. Drugs 2012, 22, 149–159. [Google Scholar] [CrossRef]
- Savariar, E.N.; Felsen, C.N.; Nashi, N.; Jiang, T.; Ellies, L.G.; Steinbach, P.; Tsien, R.Y.; Nguyen, Q.T. Real-time In Vivo Molecular Detection of Primary Tumors and Metastases with Ratiometric Activatable Cell-Penetrating Peptides. Cancer Res. 2013, 73, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Genchi, G. An overview on D-amino acids. Amino Acids. 2017, 49, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.C.; Strömblad, S.; Sanders, L.C.; von Schalscha, T.L.; Aimes, R.T.; Stetler-Stevenson, W.G.; Quigley, J.P.; A Cheresh, D. Localization of Matrix Metalloproteinase MMP-2 to the Surface of Invasive Cells by Interaction with Integrin αvβ3. Cell 1996, 85, 683–693. [Google Scholar] [CrossRef] [Green Version]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.F.; Matrisian, L.M. Changing views of the role of matrix metalloproteinases in metastasis. J. Natl. Cancer Inst. 1997, 89, 1260–1270. [Google Scholar] [CrossRef] [Green Version]
- Overall, C.M.; Kleifeld, O. Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 2006, 6, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.; Winer, E.P. Trastuzumab Emtansine: A Novel Antibody–Drug Conjugate for HER2-Positive Breast Cancer. Clin. Cancer Res. 2014, 20, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Dubowchik, G.M.; Firestone, R.A. Cathepsin B-sensitive dipeptide prodrugs. 1. A model study of structural requirements for efficient release of doxorubicin. Bioorganic Med. Chem. Lett. 1998, 8, 3341–3346. [Google Scholar] [CrossRef]
- Hingorani, D.V.; Lemieux, A.J.; Acevedo, J.R.; Glasgow, H.L.; Kedarisetty, S.; Whitney, M.A. Early detection of squamous cell carcinoma in carcinogen induced oral cancer rodent model by ratiometric activatable cell penetrating peptides. Oral oncol. 2017, 71, 156–162. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hingorani, D.V.; Camargo, M.F.; Quraishi, M.A.; Adams, S.R.; Advani, S.J. Tumor Activated Cell Penetrating Peptides to Selectively Deliver Immune Modulatory Drugs. Pharmaceutics 2021, 13, 365. https://doi.org/10.3390/pharmaceutics13030365
Hingorani DV, Camargo MF, Quraishi MA, Adams SR, Advani SJ. Tumor Activated Cell Penetrating Peptides to Selectively Deliver Immune Modulatory Drugs. Pharmaceutics. 2021; 13(3):365. https://doi.org/10.3390/pharmaceutics13030365
Chicago/Turabian StyleHingorani, Dina V., Maria F. Camargo, Maryam A. Quraishi, Stephen R. Adams, and Sunil J. Advani. 2021. "Tumor Activated Cell Penetrating Peptides to Selectively Deliver Immune Modulatory Drugs" Pharmaceutics 13, no. 3: 365. https://doi.org/10.3390/pharmaceutics13030365
APA StyleHingorani, D. V., Camargo, M. F., Quraishi, M. A., Adams, S. R., & Advani, S. J. (2021). Tumor Activated Cell Penetrating Peptides to Selectively Deliver Immune Modulatory Drugs. Pharmaceutics, 13(3), 365. https://doi.org/10.3390/pharmaceutics13030365