
Quinuclidine-Based Carbamates as Potential CNS Active Compounds

 ,
,  and
and
Abstract
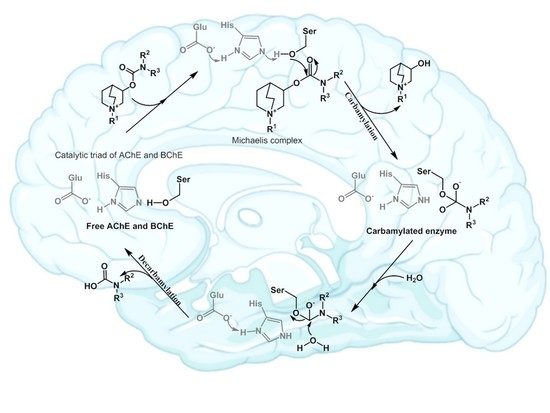
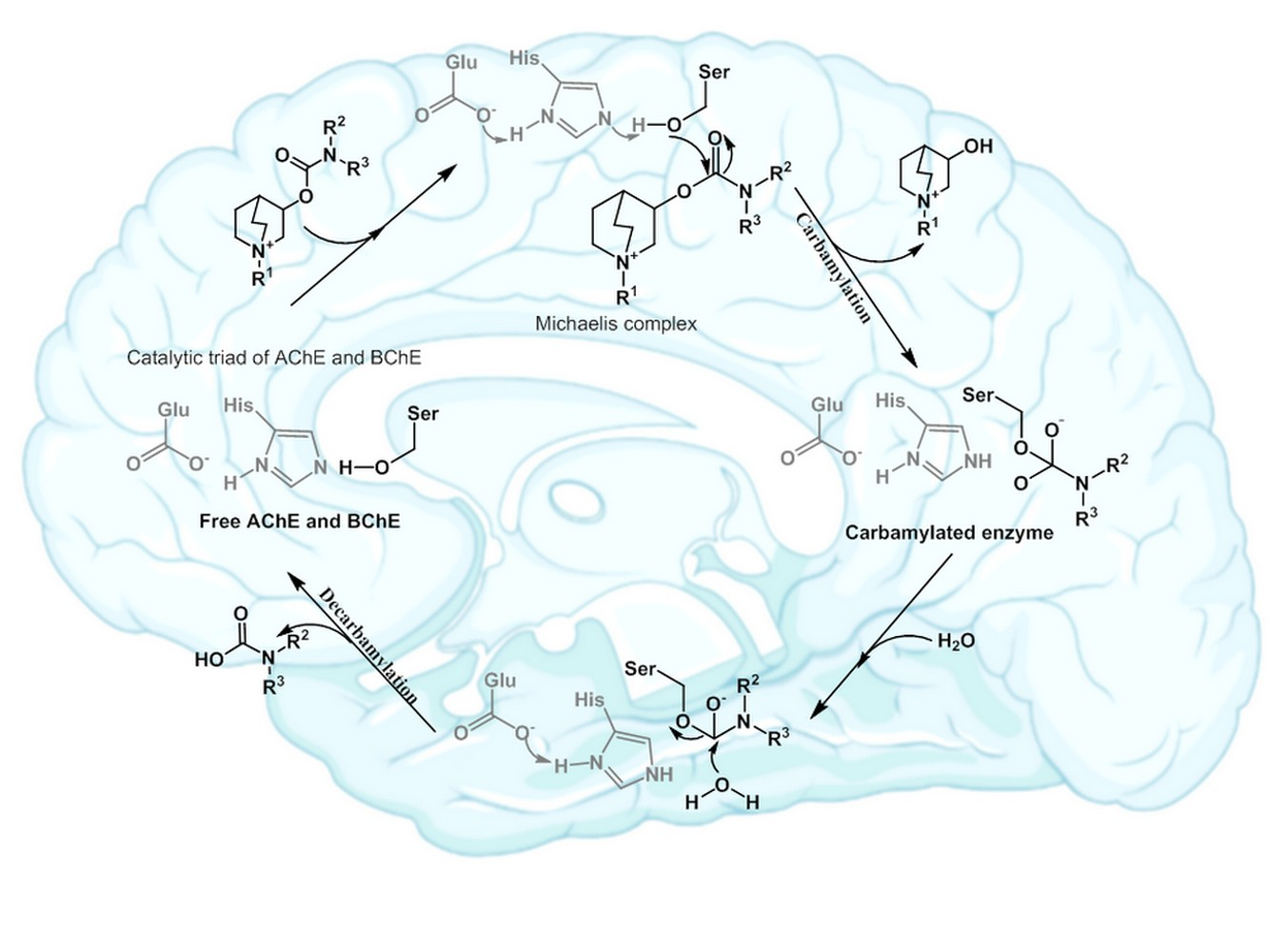
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.1.1. Enzyme Sources
2.1.2. Cell Culture
2.2. Synthesis of Compounds
2.3. Cholinesterase Inhibition
2.3.1. Enzyme Activity Measurement
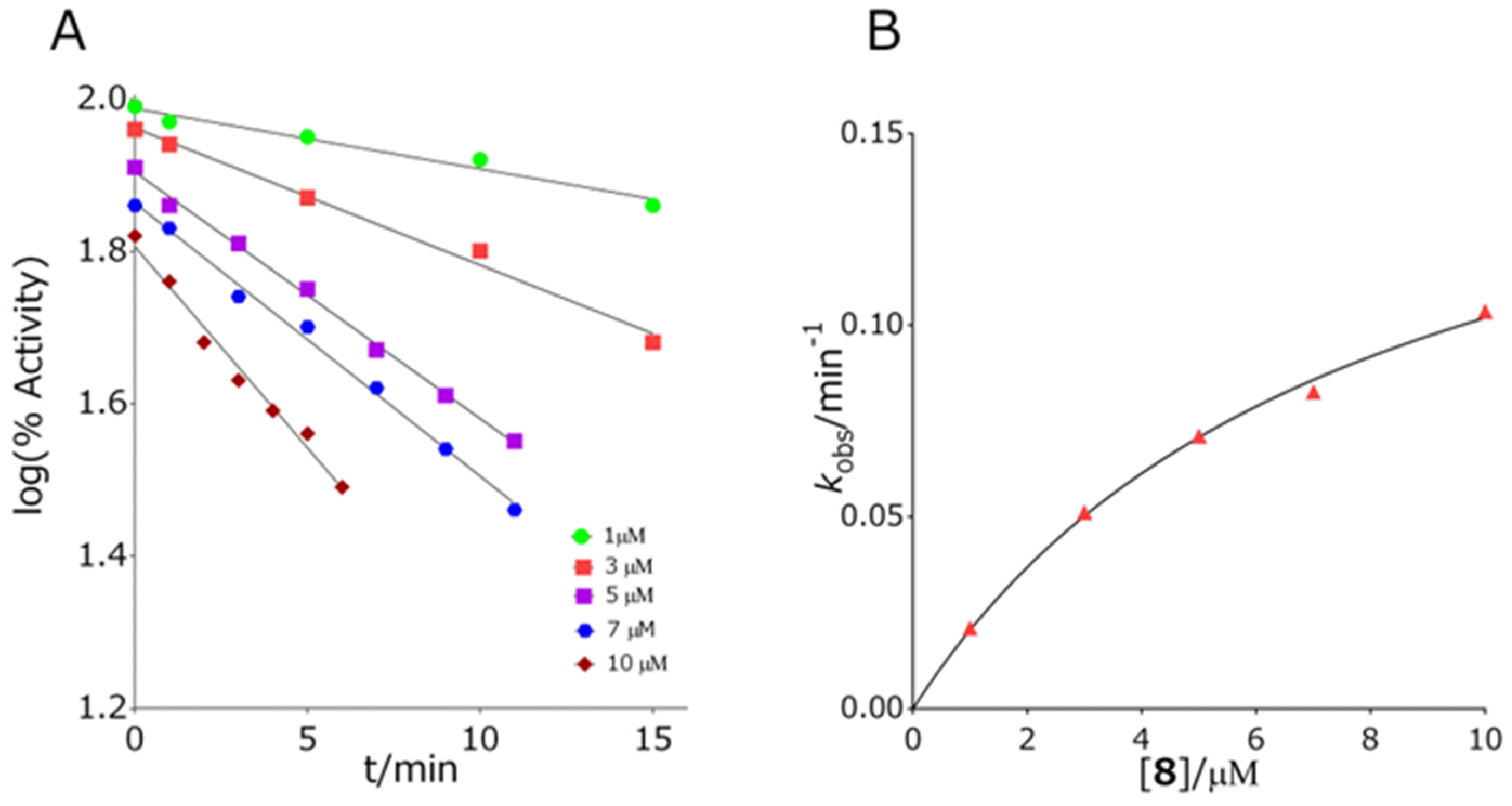
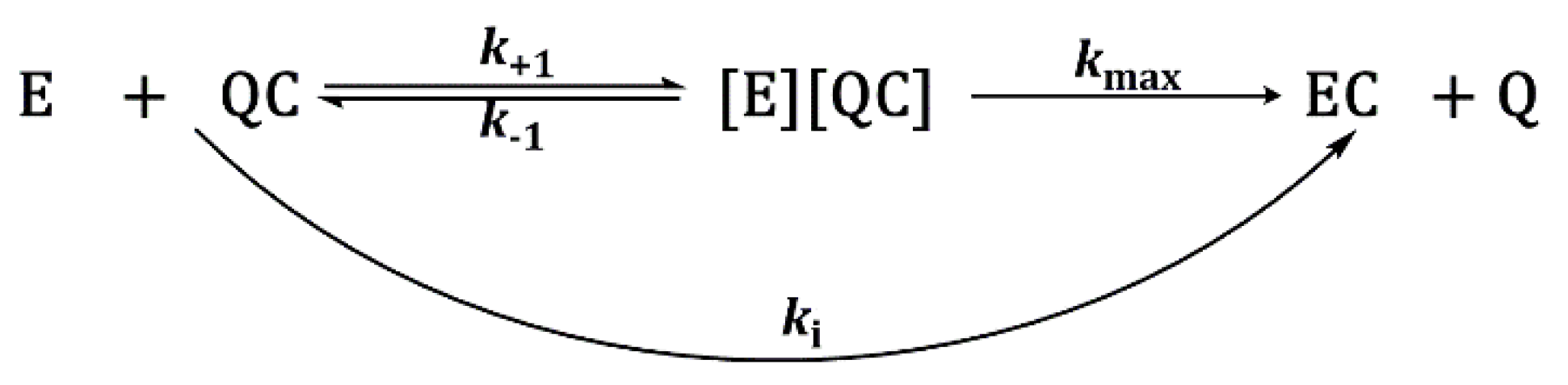
2.3.2. Inhibition Constants Determination
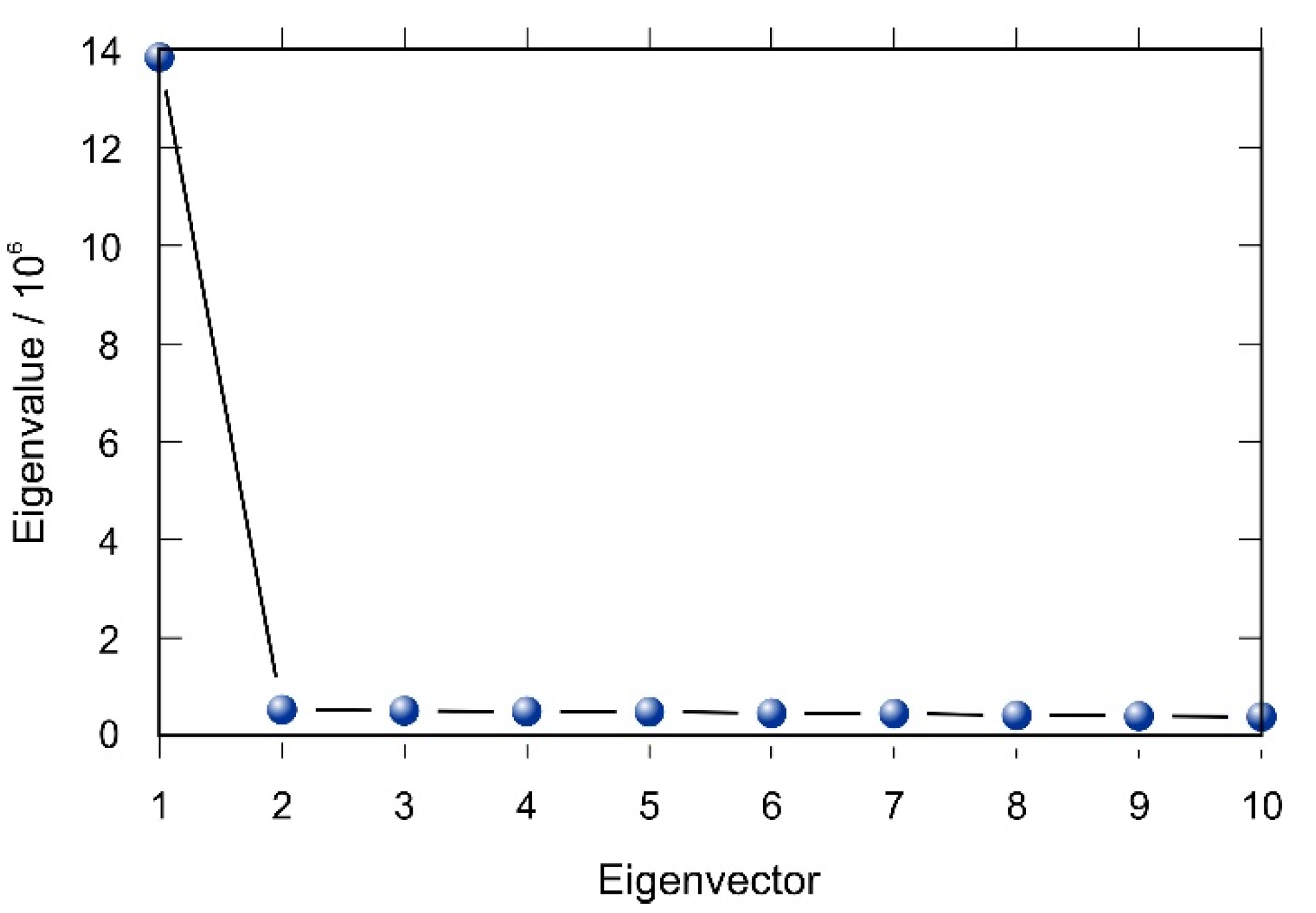
2.4. Multivariate Analysis
2.5. Machine Learning Procedure
2.6. In Silico Prediction of Blood–Brain Barrier (BBB) Penetration
2.7. Cytotoxicity of Carbamates
3. Results and Discussion
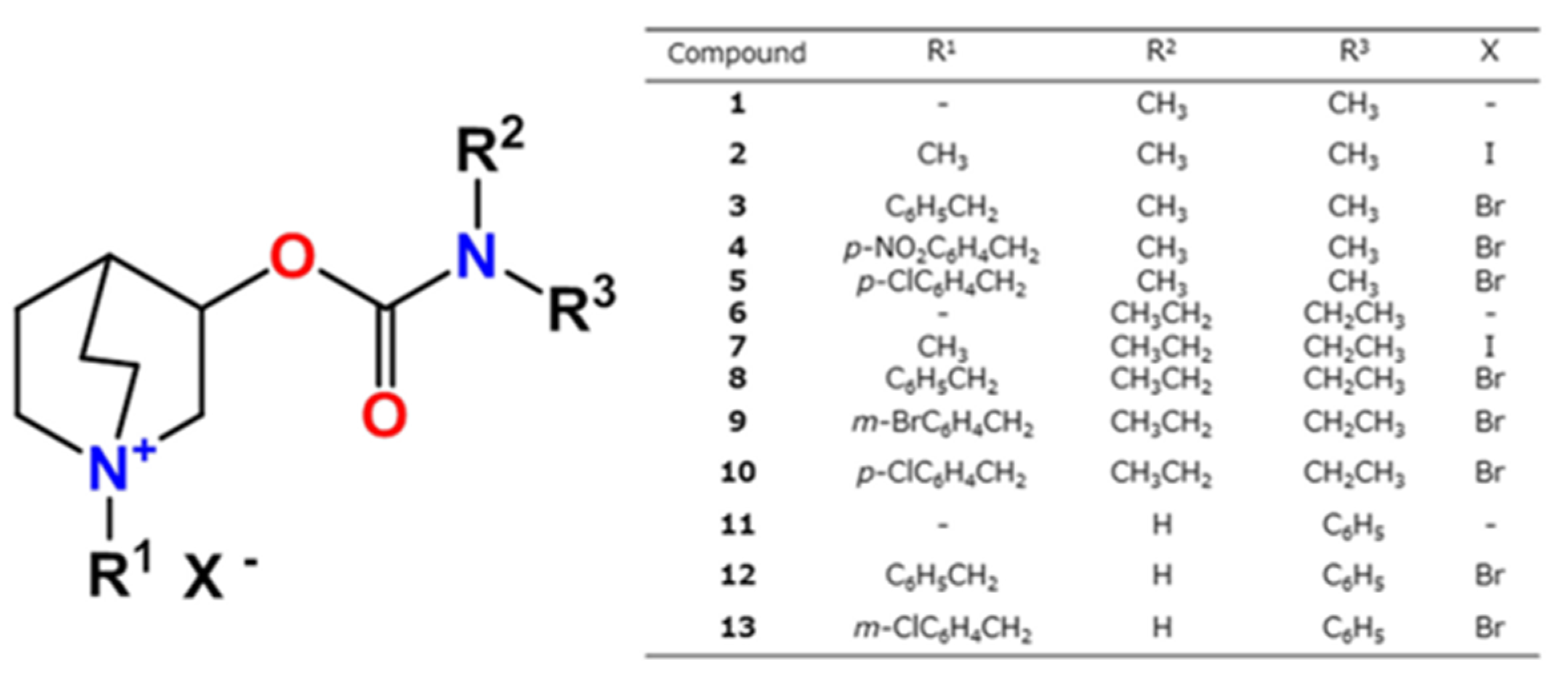
3.1. Synthesis of Compounds
3.2. Inhibition of Cholinesterases
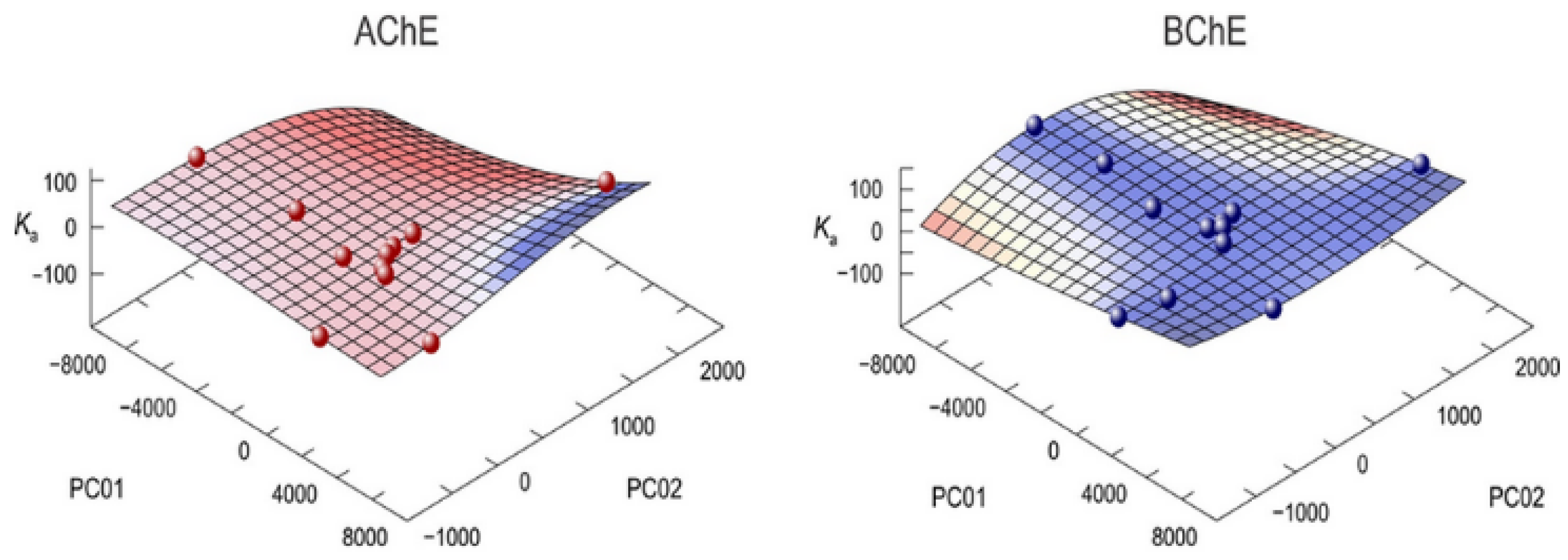
3.3. Multivariate Analysis and Activity Models
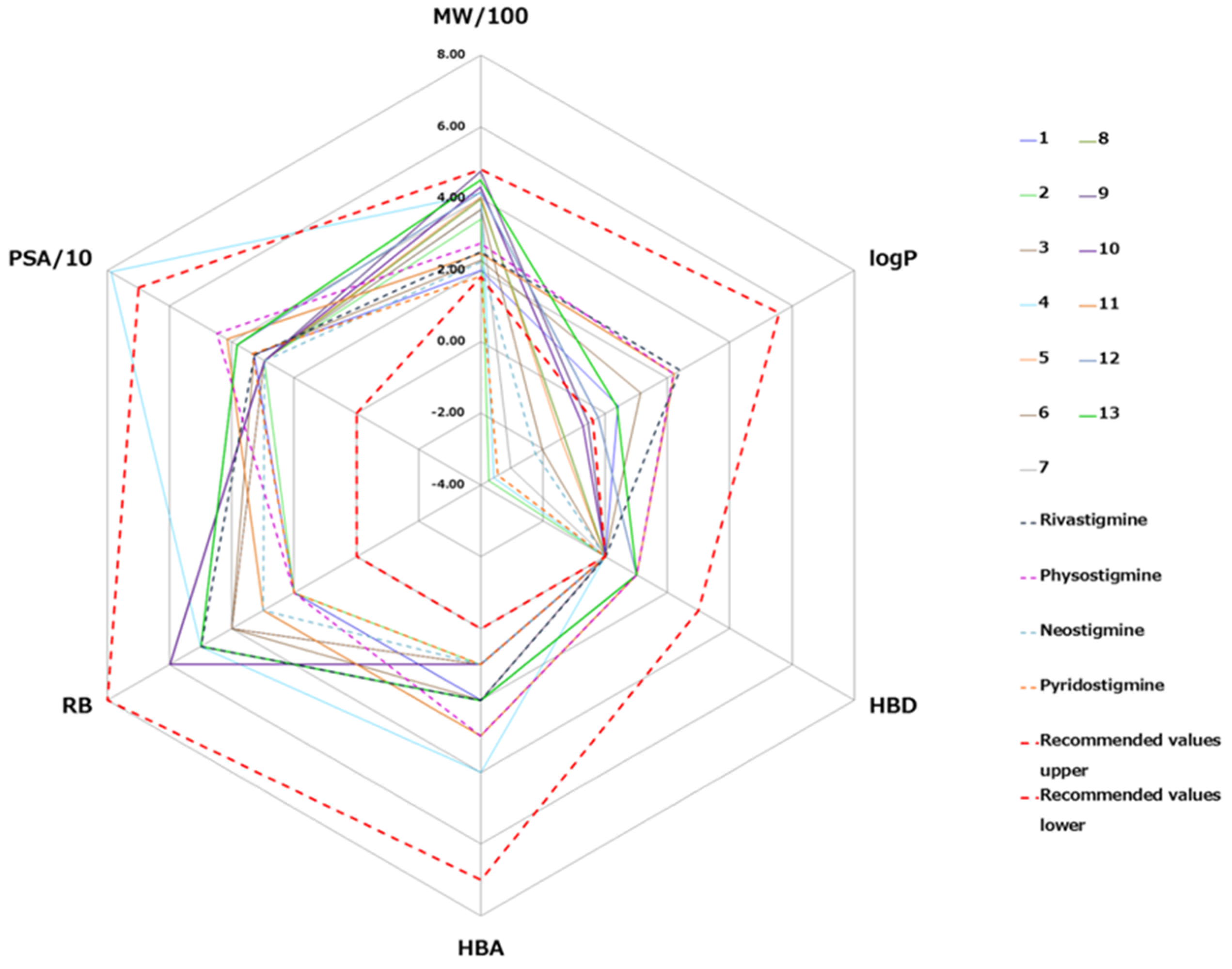
3.4. The BBB Penetration Ability of Tested Quinuclidinium Carbamates
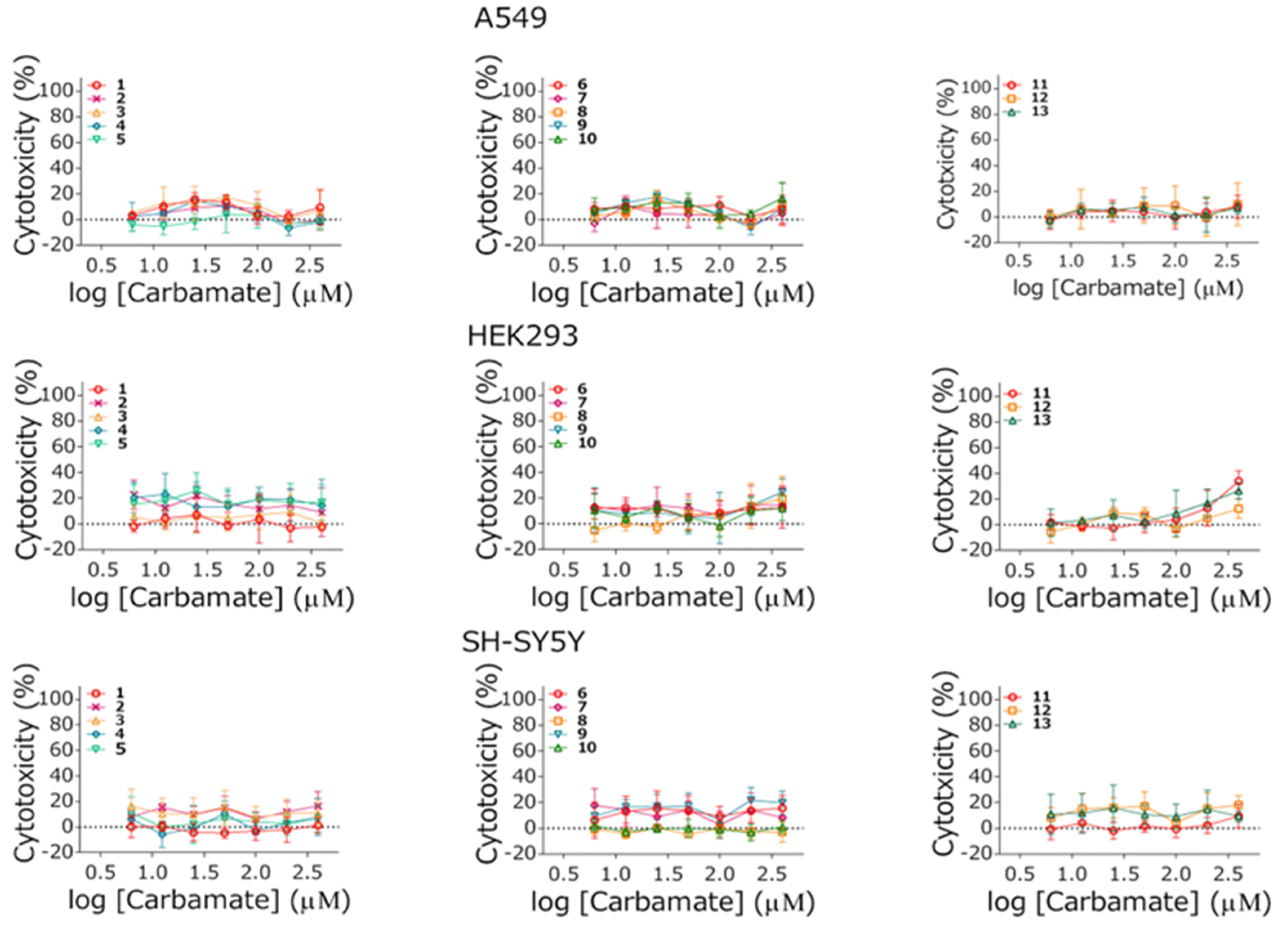
3.5. Cytotoxicity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Neurological Disorders: Public Health Challenges. WHO Library Cataloguing in Publication Data; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Peterson, B. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar]
- Sharma, P.; Srivastava, P.; Seth, A.; Nath Tripathi, P.; Banerjee, A.G.; Shrivastava, S.K. Comprehensive review of mechanisms of pathogenesis involved in Alzheimer’s disease and potential therapeutic strategies. Prog. Neurobiol. 2019, 174, 53–89. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Mobely, W.C. Alzheimer disease pathogenesis: Insights from molecular and cellular biology studies of oligomeric Aβ and Tau species. Front. Neurosci. 2019, 13, 1–21. [Google Scholar] [CrossRef]
- Rizek, P.; Kumar, N.; Jog, M.S. An update on the diagnosis and treatment of Parkinson disease. Can. Med. Assoc. J. 2016, 188, 1157–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szeto, J.Y.Y.; Lewis, S.J.G. Current treatment options for Alzheimer’s disease and Parkinson’s disease dementia. Curr. Neuropharmacol. 2016, 14, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2012, 6, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Goetzl, E.J.; Kapogiannis, D.; Lista, S.; Vergallo, A. Biomarker-Drug and Liquid Biopsy Co-development for Disease Staging and Targeted Therapy: Cornerstones for Alzheimer’s Precision Medicine and Pharmacology. Front. Pharmacol. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Contestabile, A. The history of the cholinergic hypothesis. Behav. Brain Res. 2011, 221, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E. Cholinesterases and Cholinesterases Inhibitors, 3rd ed.; Informa Healthcare: London, UK, 2000. [Google Scholar]
- Giacobini, E.; Pepeu, G. The Brain Cholinergic System in Health and Disease, 1st ed.; Informa Healthcare: London, UK, 2006. [Google Scholar]
- Giacobini, E. Butyrylcholinesterase: Its Role in Brain Function, 1st ed.; Informa Healthcare: London, UK, 2003. [Google Scholar]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [Green Version]
- Bosak, A.; Gazić, I.; Vinković, V.; Kovarik, Z. Amino acid residues involved in stereoselective inhibition of cholinesterases with bambuterol. Arch. Biochem. Biophys. 2008, 471, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Bosak, A.; Smilovic, I.G.; Šinko, G.; Vinković, V.; Kovarik, Z. Metaproterenol, isoproternol and their bisdimethyl-carbaamte derivates as human cholinesterase inhibitors. J. Med. Chem. 2012, 55, 6716–6723. [Google Scholar] [CrossRef]
- Matošević, A.; Bosak, A. Carbamate group as structural motif in drugs: A review of carbamate derivatives used as therapeutic agents. Arch. Hig. Rada Toksikol. 2020, 71, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Brindisi, M. Organic Carbamates in Drug Design and Medicinal Chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiner, E.; Radić, Z. Mechanism of Action of Cholinesterase Inhibitors. In Cholinesterase’s and Cholinesterase Inhibitors, 3rd ed.; Giaccobini, E., Dunitz, M., Eds.; Informa Healthcare: London, UK, 2000; pp. 103–144. [Google Scholar]
- Aldrige, W.N.; Reiner, E. Enzyme Inhibitors as Substrates, 1st ed.; Northoland Publishing Company: Amsterdam, The Netherlands, 1972. [Google Scholar]
- Gold, R.; Hohlfeld, R.; Toyka, K.V. Review: Progress in the treatment of myasthenia gravis. Ther. Adv. Neurol. Disord. 2008, 1, 99–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, M.; Adem, A.; Sabbagh, M. New acetylcholinesterase inhibitors for Alzheimer’s disease. Int. J. Alzheimers Dis. 2012, 2012. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Cavalli, A.; Valgimigli, L.; Bartolini, M.; Rosini, M.; Andrisano, V.; Recanatini, M.; Melchiorre, C. Multi-Target-Directed Drug Design Strategy: From a Dual Binding Site Acetylcholinesterase Inhibitor to a Trifunctional Compound against Alzheimer’s Disease. J. Med. Chem. 2007, 50, 6446–6449. [Google Scholar] [CrossRef]
- Wu, M.; Ma, J.; Ji, L.; Wang, M.; Han, J.; Li, Z. Design, synthesis, and biological evaluation of rutacecarpine derivatives as multitarget-directed ligands for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 177, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimer’s Dement. 2020, 6, e12050. [Google Scholar] [CrossRef] [PubMed]
- Simeon-Rudolf, V.; Reiner, E.; Škrinjarić-Špoljar, M.; Radić, B.; Lucić, A.; Primožič, I.; Tomić, S. Quinuclidini-um-imidazolium compounds: Synthesis, mode of interaction with acetylcholinesterase and effect upon soman intoxicated mice. Arch. Toxicol. 1998, 72, 289–295. [Google Scholar] [CrossRef]
- Lučić, A.; Radić, B.; Peraica, M.; Mesic, M.; Primožič, I.; Binenfeld, Z. Antidotal efficacy of quinuclidinium oximes against soman poisoning. Arch. Toxicol. 1997, 71, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Zandona, A.; Katalinić, M.; Šinko, G.; Kastelic, A.R.; Primožič, I.; Kovarik, Z. Targeting organophosphorus com-pounds poisoning by novel quinuclidine-3 oximes: Development of butyrylcholinesterase-based bioscavengers. Arch. Toxicol. 2020, 94, 3157–3171. [Google Scholar] [CrossRef] [PubMed]
- Sterling, G.H.; Doukas, P.H.; Jackson, C.; Caccese, R.; O’Neill, K.J.; O’Neill, J.J. 3-carbamyl-N-allylquinuclidinium bromide. Biochem. Pharmacol. 1993, 45, 465–472. [Google Scholar] [CrossRef]
- Primožič, I.; Hrenar, T.; Tomić, S. Binding Modes of Quinuclidinium Esters to Butyrylcholinesterase. Croat. Chem. Acta 2012, 85, 77–83. [Google Scholar] [CrossRef]
- Bosak, A.; Primožič, I.; Oršulić, M.; Tomić, S.; Simeon-Rudolf, V. Enantiomers of quinuclidin-3-ol derivates: Resolution and interactions with human cholinesterases. Croat Chem. Acta 2005, 78, 121–128. [Google Scholar]
- Reiner, E.; Škrinjarić-Špoljar, M.; Dunaj, S.; Simeon-Rudolf, V.; Primožič, I. 3-hydroxyquinuclidinium derivatives: Synthesis of compounds and inhibition of acetylcholinesterase. Chem. Biol. Interact. 1999, 120, 173–181. [Google Scholar] [CrossRef]
- Bosak, A.; Ramić, A.; Šmidlehner, T.; Hrenar, T.; Primožič, I.; Kovarik, Z. Design and evaluation of selective butyryl-cholinesterase inhibitors based on Cinchona alkaloid scaffold. PLoS ONE 2018, 13, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Simeon-Rudolf, V.; Evans, T. Interlaboratory study into the proficiency of attribution of human serum butyrylcholin-esterase phenotypes: Reference values of activities and inhibitor numbers. Acta Pharm. 2001, 51, 289–296. [Google Scholar]
- ECACC. Fundamental Techniques in Cell Culture Laboratory Handbook, 4th ed.; Merck KGaA: Darmstadt, Germany, 2018. [Google Scholar]
- Dulbecco, R.; Vogt, M. Plaque Formation and Isolation of Pure Lines with Poliomyelitis Viruses. J. Exp. Med. 1954, 99, 167–182. [Google Scholar] [CrossRef] [Green Version]
- Annadurai, S.; Zhang, M.; Gabriel, J.L.; Bencheriff, M.; Canney, D.J. Ether and Carbamate Derivatives of 3-quinuclidinol and 3- hydroxymethylquinuclidine: Synthesis and Evaluation as Nicotinic Ligands. Med. Chem. 2016, 12, 574–584. [Google Scholar] [CrossRef] [Green Version]
- Kastelic, A.R.; Odžak, R.; Pezdirc, I.; Sović, K.; Hrenar, T.; Gašparović, A.Č.; Skočibušić, M.; Primožič, I. New and potent quinuclidine-based antimicrobial agents. Molecules 2019, 24, 2675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. New and rapid colorimetric determination of acetylcho-linesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Eyer, P.; Worek, F.; Kiderlen, D.; Sinko, G.; Stuglin, A.; Simeon-Rudolf, V.; Reiner, E. Molar absorption coefficients for the reduced Ellman reagent: Reassessment. Anal. Biochem. 2003, 312, 224–227. [Google Scholar] [CrossRef]
- Carvey, P.M.; Hendey, B.; Monahan, A.J. The blood-brain barrier in neurodegenerative disease: A rhetorical perspective. J. Neurochem. 2009, 111, 291–314. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, Z. CNS Drug Design: Balancing Physicochemical Properties for Optimal Brain Exposure. J. Med. Chem. 2015, 58, 2584–2608. [Google Scholar] [CrossRef] [PubMed]
- Chemicalize, Calculation Module. 2018. Available online: https://chemicalize.com/developedbyChemAxon (accessed on 1 November 2020).
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRX 2005, 2, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Bosak, A.; Smilović, I.G.; Štimac, A.; Vinković, V.; Šinko, G.; Kovarik, Z. Peripheral site and acyl pocket define selective inhibition of mouse butyrylcholinesterase by two biscarbamates. Arch. Biochem. Biophys. 2013, 529, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Pistolozzi, M.; Liu, S.; Tan, W. Design, synthesis and biological evaluation of novel carbamates as potential inhibitors of acetylcholinesterase and butyrylcholinesterase. Bioorg. Med. Chem. 2020, 28, 115324. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, P.; Millard, C.B.; Harel, M.; Dvir, H.; Enz, A.; Sussman, J.L.; Silman, I. Kinetic and structural studies on the interaction of cholinesterases with the Anti-Alzheimer drug Rivastigmine. Biochemistry 2002, 41, 3555–3564. [Google Scholar] [CrossRef]
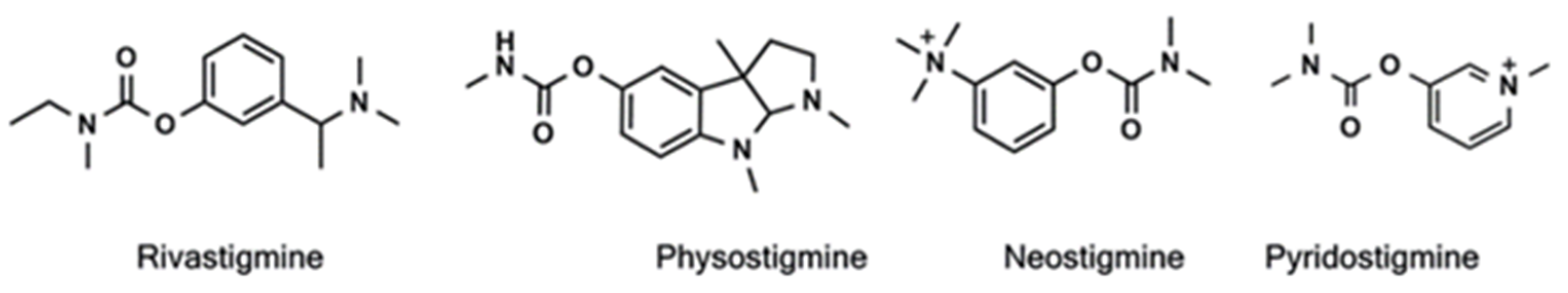
- Groner, E.; Ashani, Y.; Schorer-Apelbaum, D.; Sterling, J.; Herzig, Y.; Weinstock, M. The Kinetics of Inhibition of Human Acetylcholinesterase and Butyrylcholinesterase by Two Series of Novel Carbamates. Mol. Pharmacol. 2007, 71, 1610–1617. [Google Scholar] [CrossRef] [Green Version]
- Darvesh, S.; Darvesh, K.V.; McDonald, R.S.; Mataija, D.; Walsh, R.; Mothana, S.; Lockridge, O.; Martin, E. Carbamates with differential mechanism of inhibiton toward acetylcholinesterase and butyrylcholinesterase. J. Med. Chem. 2008, 51, 4200–4212. [Google Scholar] [CrossRef]
- Simeon, V.; Reiner, E. Comparasion between inhibiton of acetylcolinesterase an cholinesterase by some N-methyl- and N,N-dimethyl carbamates. Arch. Hig. Rad. 1973, 24, 199–206. [Google Scholar]
- Reiner, E.; Simeon-Rudolf, V. Cholinesterase: Substrate inhibition and substrate activation. Pflug. Arch. 2000, 440, 118–120. [Google Scholar] [CrossRef]
- Hrenar, T. moonee, Code for Manipulation and Analysis of Multi-and Univariate Data; Revision 0.6826; University of Zagreb Faculty of Science: Zagreb, Croatia, 2021. [Google Scholar]
- Geladi, P.; Kowalski, B.R. Partial least-squares regression: A tutorial. Anal. Chim. Acta 1986, 185, 1–17. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | AChE | BChE | ki(BChE)/ki(AChE | ||||
|---|---|---|---|---|---|---|---|
| ki∙103 [M−1min−1] | Ka [μM] | kmax [min−1] | ki∙103 [M−1min−1] | Ka [μM] | kmax [min−1] | ||
| 1 | 2.6 ± 0.6 | 72 ± 15 | 0.19 ± 0.02 | 3.1 ± 0.7 | 60 ± 9 | 0.20 ± 0.02 | 1.3 |
| 2 | 5.5 ± 1.4 | 22 ± 5 | 0.12 ± 0.01 | 5.4 ± 1.5 | 18 ± 4 | 0.095 ± 0.007 | 0.98 |
| 3 | 3.4 ± 1.3 | 124 ± 63 | 0.34 ± 0.11 | 4.9 ± 0.7 | 32 ± 4 | 0.16 ± 0.01 | 1.4 |
| 4 | 5.9 ± 2.5 | 25 ± 10 | 0.15 ± 0.03 | 7.5 ± 1.9 | 15 ± 4 | 0.13 ± 0.01 | 1.3 |
| 5 | 5.5 ± 1.4 | 29 ± 7 | 0.16 ± 0.01 | 3.6 ± 0.8 | 80 ± 17 | 0.29 ± 0.03 | 0.65 |
| 6 | 2.9 ± 0.9 | 76 ± 19 | 0.22 ± 0.03 | 3.8 ± 0.9 | 41 ± 9 | 0.16 ± 0.01 | 1.3 |
| 7 | 3.5 ± 1.1 | 46 ± 13 | 0.16 ± 0.02 | 1.5 ± 0.0 | - | - | 0.42 |
| 8 | 15 ± 1 | - | - | 24 ± 6 | 7.9 ± 1.9 | 0.19 ± 0.02 | 1.6 |
| 9 | 6.8 ± 1.5 | 40 ± 8 | 0.27 ± 0.03 | 7.3 ± 2.1 | 32 ± 9 | 0.23 ± 0.03 | 1.0 |
| 10 | 3.2 ± 0.7 | 61 ± 13 | 0.20 ± 0.02 | 3.4 ± 2.0 | 46 ± 4 | 0.16 ± 0.05 | 1.0 |
| 11 | 1.0 ± 0.2 | - | - | 3.0 ± 0.9 | 65 ± 18 | 0.20 ± 0.03 | 3.0 |
| 12 | 3.7 ± 0.8 | 25 ± 5 | 0.09 ± 0.01 | 3.1 ± 1.1 | 103 ± 31 | 0.32 ± 0.05 | 0.83 |
| 13 | 3.2 ± 0.5 | 88 ± 13 | 0.28 ± 0.02 | 3.1 ± 1.0 | 89 ± 22 | 0.28 ± 0.04 | 0.98 |
| Rivastigmine [48] | 4.54 | 333 | 73 | ||||
| Physostigmine | 4900 ± 380 | 66 ± 27 | 0.32 ± 0.22 | 2800 ± 940 | 250 ± 24 | 0.61 ± 0.22 | 1.8 |
| Principal Component | Variance/% | Total/% |
|---|---|---|
| PC01 | 72.28 | 72.28 |
| PC02 | 2.79 | 75.07 |
| PC03 | 2.66 | 77.74 |
| PC04 | 2.60 | 80.34 |
| PC05 | 2.56 | 82.90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matošević, A.; Radman Kastelic, A.; Mikelić, A.; Zandona, A.; Katalinić, M.; Primožič, I.; Bosak, A.; Hrenar, T. Quinuclidine-Based Carbamates as Potential CNS Active Compounds. Pharmaceutics 2021, 13, 420. https://doi.org/10.3390/pharmaceutics13030420
Matošević A, Radman Kastelic A, Mikelić A, Zandona A, Katalinić M, Primožič I, Bosak A, Hrenar T. Quinuclidine-Based Carbamates as Potential CNS Active Compounds. Pharmaceutics. 2021; 13(3):420. https://doi.org/10.3390/pharmaceutics13030420
Chicago/Turabian StyleMatošević, Ana, Andreja Radman Kastelic, Ana Mikelić, Antonio Zandona, Maja Katalinić, Ines Primožič, Anita Bosak, and Tomica Hrenar. 2021. "Quinuclidine-Based Carbamates as Potential CNS Active Compounds" Pharmaceutics 13, no. 3: 420. https://doi.org/10.3390/pharmaceutics13030420
APA StyleMatošević, A., Radman Kastelic, A., Mikelić, A., Zandona, A., Katalinić, M., Primožič, I., Bosak, A., & Hrenar, T. (2021). Quinuclidine-Based Carbamates as Potential CNS Active Compounds. Pharmaceutics, 13(3), 420. https://doi.org/10.3390/pharmaceutics13030420