
Structural Analysis of the Simultaneous Activation and Inhibition of γ-Secretase Activity in the Development of Drugs for Alzheimer’s Disease





Abstract
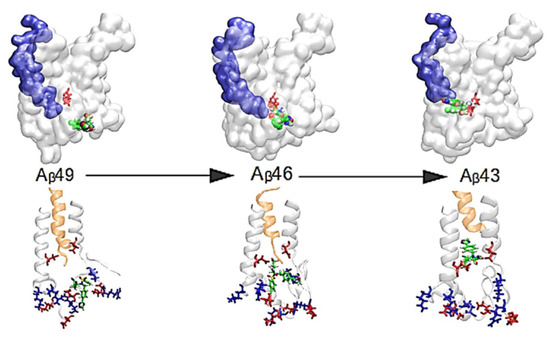
:
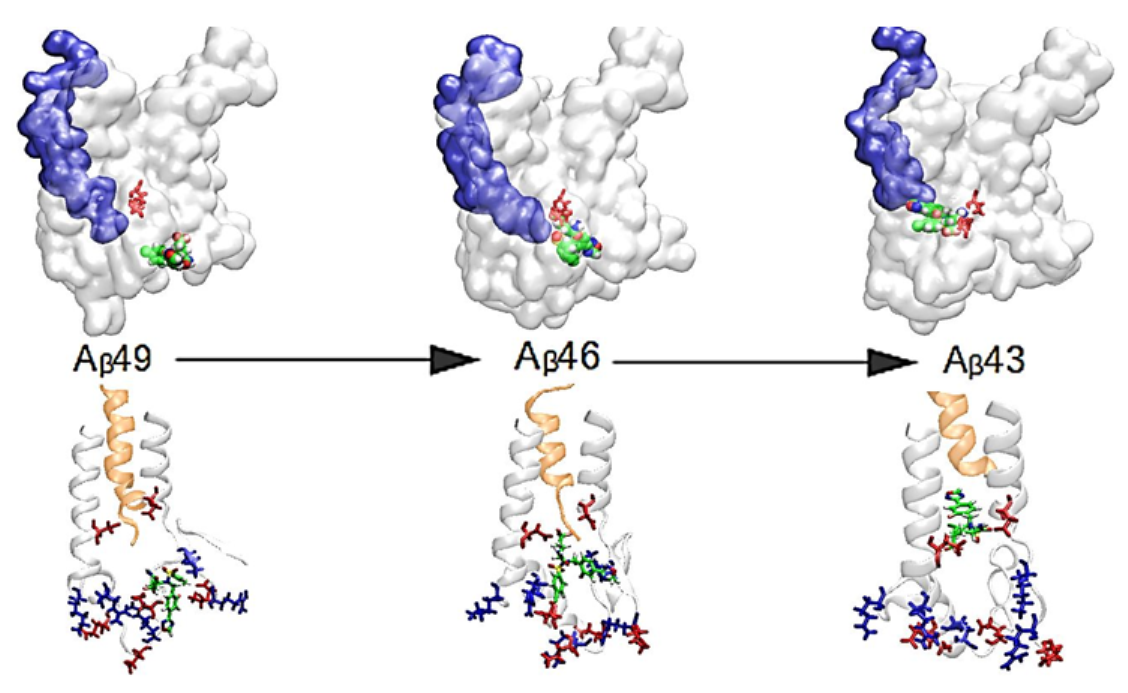{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
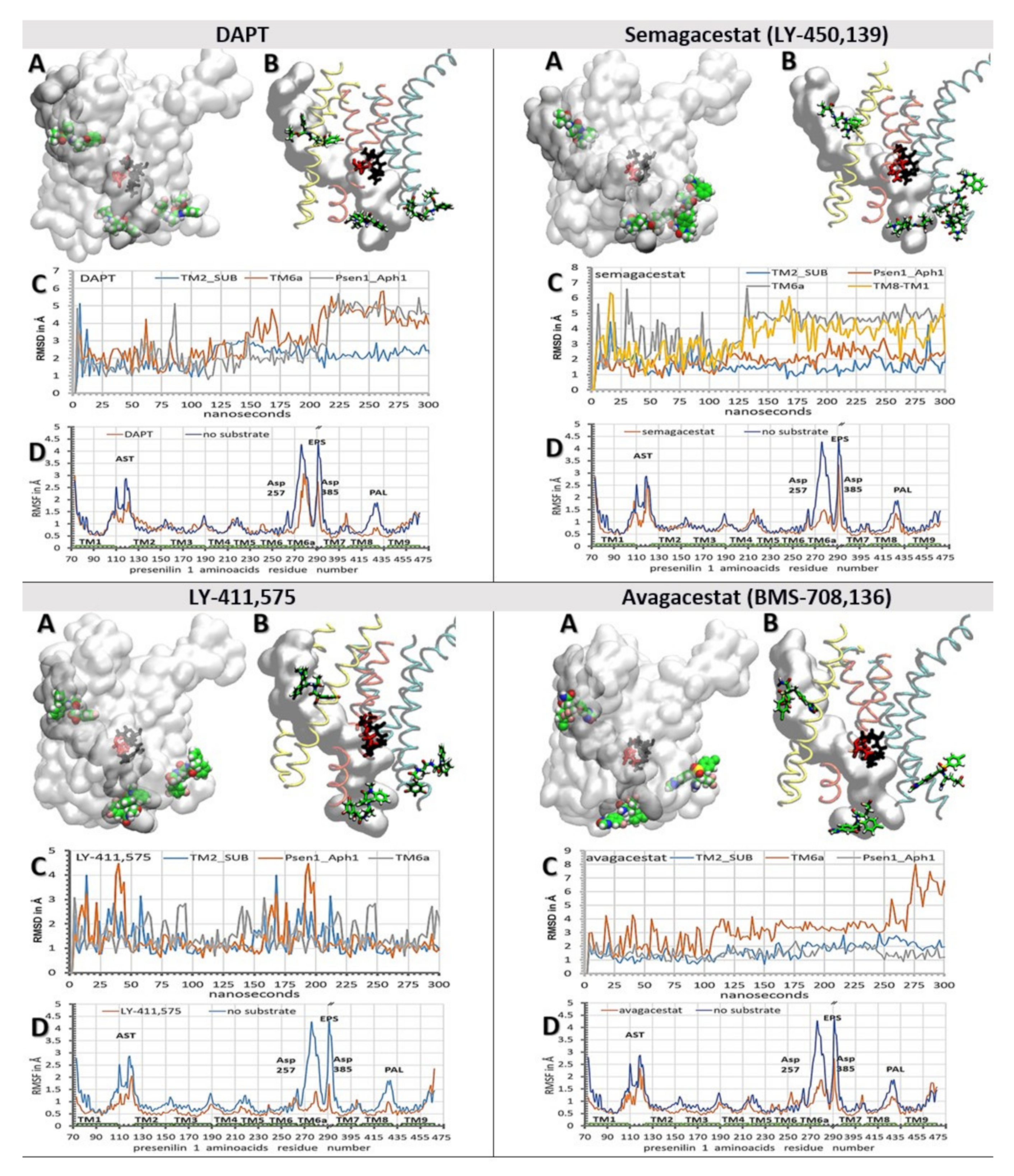{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Biphasic Activation–Inhibition Dose-Response Curves for DAPT, Semagacestat, Ly-411,575 and Avagacestat in Cultures of SH-SY5 Cells.
2.2. Multiscale Molecular Dynamics Studies of the Γ-Secretase Structure in Different Steps in the Catalytic Cycle
2.3. Initial Screening for Drug-Binding Sites Using Molecular Docking Studies
2.4. All-Atom Molecular Dynamics Studies Of Binding Interactions Between Biphasic Drugs and γ-Secretase in The Presence and Absence of A Substrate
2.5. All-Atom Molecular Dynamic Studies of γ-Secretase Structure With Aβ Catalytic Intermediates in Processive Catalysis
3. Discussion and Conclusions
3.1. Biphasic Drugs: The Activation Mechanism
3.2. Biphasic Drugs: The Inhibition Mechanism
3.3. Biphasic Drugs and FAD Utations
3.4. Concluding Remarks on Future Drug Development Strategies with γ-Secretase as the Target Enzyme
4. Materials and Methods
4.1. Chemicals
4.2. Secretion of Aβ 1-40 in Cultures of SH-SY5 Cells in the Presence of Increasing Concentrations of Drugs
4.3. Sandwich ELISA for Quantitative Detection of Aβ 1-40
4.4. Inhibitor Docking Studies
4.5. Residue Basedcoarse-Grained Molecular Dynamics Studies
4.6. All-Atom Molecular Dynamics Studies
4.7. Data Analysis and Presentation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Toyn, J.H.; Ahlijanian, M.K. Interpreting Alzheimer’s disease clinical trials in light of the effects on amyloid-β. Alzheimer Res. Ther. 2014, 6, 14. [Google Scholar] [CrossRef]
- Imbimbo, B.P.; Lucca, U.; Watling, M. Can anti-β-amyloid monoclonal antibodies work in autosomal dominant Alzheimer disease? Neurol. Genet. 2021, 7, e535. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Tomita, T. Structure-activity relationship of presenilin in γ-secretase-mediated intramembrane cleavage. Semin. Cell Dev. Biol. 2020, 105, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Sambamurti, K.; Greig, N.H.; Utsuki, T.; Barnwell, E.L.; Sharma, E.; Mazell, C.; Bhat, N.R.; Kindy, M.S.; Lahiri, D.K.; Pappolla, M.A. Targets for AD treatment: Conflicting messages from gamma-secretase inhibitors. J. Neurochem. 2011, 117, 359–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisby, B.; Jarrell, J.T.; Agar, M.E.; Cohen, D.S.; Rosin, E.R.; Cahill, C.M.; Rogers, J.T.; Huang, X. Alzheimer’s disease and its potential alternative therapeutics. J. Alzheimer Dis. Parkinsonism 2019, 9, 477. [Google Scholar]
- Tagami, S.; Yanagida, K.; Kodama, T.S.; Takami, M.; Mizuta, N.; Oyama, H.; Nishitomi, K.; Chiu, Y.W.; Okamoto, T.; Ikeuchi, T.; et al. Semagacestat is a pseudo-inhibitor of γ-secretase. Cell Rep. 2017, 21, 259–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbimbo, B.P.; Panza, F.; Frisardi, V.; Solfrizzi, V.; D’Onofrio, G.; Logroscino, G.; Seripa, D.; Pilotto, A. Therapeutic intervention for Alzheimer’s disease with gamma-secretase inhibitors: Still a viable option? Expert Opin. Investig. Drugs 2010, 20, 325–341. [Google Scholar] [CrossRef]
- Burton, C.R.; Meredith, J.E.; Barten, D.M.; Goldstein, M.E.; Krause, C.M.; Kieras, C.J.; Sisk, L.; Iben, L.G.; Polson, C.; Thompson, M.W.; et al. The amyloid-beta rise and gamma-secretase inhibitor potency depend on the level of substrate expression. J. Biol. Chem. 2008, 283, 22992–23003. [Google Scholar] [CrossRef] [Green Version]
- Tong, G.; Wang, J.S.; Sverdlov, O.; Huang, S.P.; Slemmon, R.; Croop, R.; Castaneda, L.; Gu, H.; Wong, O.; Li, H.; et al. Multicenter, randomized, double-blind, placebo-controlled, single-ascending dose study of the oral gamma-secretase inhibitor BMS-708163 (Avagacestat): Tolerability profile, pharmacokinetic parameters, and pharmacodynamic markers. Clin. Ther. 2012, 34, 654–667. [Google Scholar] [CrossRef]
- Svedružić, Z.M.; Popovic, K.; Sendula-Jengic, V. Modulators of gamma-secretase activity can facilitate the toxic side-effects and pathogenesis of Alzheimer’s disease. PLoS ONE 2013, 8, e50759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, R. Are improper kinetic models hampering drug development? PeerJ 2014, 2, e649. [Google Scholar] [CrossRef] [Green Version]
- Yagishita, S.; Morishima-Kawashima, M.; Tanimura, Y.; Ishiura, S.; Ihara, Y. DAPT-induced intracellular accumulations of longer amyloid beta-proteins: Further implications for the mechanism of intramembrane cleavage by gamma-secretase. Biochemistry 2006, 45, 3952–3960. [Google Scholar] [CrossRef]
- Gillman, K.W.; Starrett, J.E., Jr.; Parker, M.F.; Xie, K.; Bronson, J.J.; Marcin, L.R.; McElhone, K.E.; Bergstrom, C.P.; Mate, R.A.; Williams, R.; et al. Discovery and evaluation of BMS-708163, a potent, selective and orally bioavailable γ-secretase inhibitor. ACS Med. Chem. Lett. 2010, 1, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Jämsä, A.; Belda, O.; Edlund, M.; Lindström, E. BACE-1 inhibition prevents the γ-secretase inhibitor evoked Aβ rise in human neuroblastoma SH-SY5Y cells. J. Biomed. Sci. 2011, 18, 76. [Google Scholar] [CrossRef] [Green Version]
- Mitani, Y.; Yarimizu, J.; Saita, K.; Uchino, H.; Akashiba, H.; Shitaka, Y.; Ni, K.; Matsuoka, N. Differential effects between gamma-secretase inhibitors and modulators on cognitive function in amyloid precursor protein-transgenic and nontransgenic mice. J. Neurosci. 2012, 32, 2037–2050. [Google Scholar] [CrossRef] [Green Version]
- Svedružić, Ž.M.; Popović, K.; Šendula-Jengić, V. Decrease in catalytic capacity of γ-secretase can facilitate pathogenesis in sporadic and Familial Alzheimer’s disease. Mol. Cell. Neurosci. 2015, 67, 55–65. [Google Scholar] [CrossRef]
- Fersht, A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding (Hardcover), 1st ed.; Freeman, W.H., Ed.; Macmillan: New York, NY, USA, 1998; p. 650. [Google Scholar]
- Tipton, K.F. (Ed.) Enzyme Assays, 2nd ed.; Oxford University Press: Oxford, UK, 2002; p. 282. [Google Scholar]
- Svedružić, Ž.M.; Odorčić, I.; Chang, C.H.; Svedružić, D. Substrate channeling via a transient protein-protein complex: The case of D-glyceraldehyde-3-phosphate dehydrogenase and L-lactate dehydrogenase. Sci. Rep. 2020, 10, 10404. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, D.K.; Bernhard, S.A. Biophysical chemistry of metabolic reaction sequences in concentrated enzyme solution and in the cell. Annu. Rev. Biophys. Biophys. Chem. 1987, 16, 175–204. [Google Scholar] [CrossRef] [PubMed]
- Svedružić, Z.M.; Popovic, K.; Smoljan, I.; Sendula-Jengic, V. Modulation of gamma-secretase activity by multiple enzyme-substrate interactions: Implications in pathogenesis of Alzheimer’s disease. PLoS ONE 2012, 7, e32293. [Google Scholar] [CrossRef] [PubMed]
- Kakuda, N.; Funamoto, S.; Yagishita, S.; Takami, M.; Osawa, S.; Dohmae, N.; Ihara, Y. Equimolar production of amyloid beta-protein and amyloid precursor protein intracellular domain from beta-carboxyl-terminal fragment by gamma-secretase. J. Biol. Chem. 2006, 281, 14776–14786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motulsky, H.; Christopoulos, A. Fitting Models to Biological Data Using Linear and Nonlinear Regression: A Practical Guide to Curve Fitting, 1st ed.; Oxford University Press: New York, NY, USA, 2004; p. 352. [Google Scholar]
- Hochard, A.; Oumata, N.; Bettayeb, K.; Gloulou, O.; Fant, X.; Durieu, E.; Buron, N.; Porceddu, M.; Borgne-Sanchez, A.; Galons, H.; et al. Aftins increase amyloid-beta42, lower amyloid-beta38, and do not alter amyloid-beta40 extracellular production in vitro: Toward a chemical model of Alzheimer’s disease? J. Alzheimer Dis. 2013, 35, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Chavez-Gutierrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of gamma-secretase dysfunction in familial Alzheimer disease. EMBO J. 2012, 31, 2261–2274. [Google Scholar] [CrossRef]
- Yagishita, S.; Morishima-Kawashima, M.; Ishiura, S.; Ihara, Y. Abeta46 is processed to Abeta40 and Abeta43, but not to Abeta42, in the low density membrane domains. J. Biol. Chem. 2008, 283, 733–738. [Google Scholar] [CrossRef] [Green Version]
- Coric, V.; van Dyck, C.H.; Salloway, S.; Andreasen, N.; Brody, M.; Richter, R.W.; Soininen, H.; Thein, S.; Shiovitz, T.; Pilcher, G.; et al. Safety and tolerability of the gamma-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch. Neurol. 2012, 69, 1430–1440. [Google Scholar] [CrossRef] [Green Version]
- Tamayev, R.; D’Adamio, L. Inhibition of gamma-secretase worsens memory deficits in a genetically congruous mouse model of Danish dementia. Mol. Neurodegener. 2012, 7, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef]
- Henley, D.B.; May, P.C.; Dean, R.A.; Siemers, E.R. Development of semagacestat (LY450139), a functional gamma-secretase inhibitor, for the treatment of Alzheimer’s disease. Expert Opin. Pharmacother. 2009, 10, 1657–1664. [Google Scholar] [CrossRef]
- Yin, Y.I.; Bassit, B.; Zhu, L.; Yang, X.; Wang, C.; Li, Y.M. γ-Secretase substrate concentration modulates the Abeta42/Abeta40 ratio: Implications for Alzheimer’s disease. J. Biol. Chem. 2007, 282, 23639–23644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.C.; Rajendra, E.; Yang, G.; Shi, Y.; Scheres, S.H. Sampling the conformational space of the catalytic subunit of human γ-secretase. Elife 2015, 4. [Google Scholar] [CrossRef]
- Barnwell, E.; Padmaraju, V.; Baranello, R.; Pacheco-Quinto, J.; Crosson, C.; Ablonczy, Z.; Eckman, E.; Eckman, C.B.; Ramakrishnan, V.; Greig, N.H.; et al. Evidence of a novel mechanism for partial gamma-secretase inhibition induced paradoxical increase in secreted amyloid beta protein. PLoS ONE 2014, 9, e91531. [Google Scholar] [CrossRef] [Green Version]
- Morohashi, Y.; Kan, T.; Tominari, Y.; Fuwa, H.; Okamura, Y.; Watanabe, N.; Sato, C.; Natsugari, H.; Fukuyama, T.; Iwatsubo, T.; et al. C-terminal fragment of presenilin is the molecular target of a dipeptidic gamma-secretase-specific inhibitor DAPT (N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester). J. Biol. Chem. 2006, 281, 14670–14676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanz, T.A.; Hosley, J.D.; Adams, W.J.; Merchant, K.M. Studies of Abeta pharmacodynamics in the brain, cerebrospinal fluid, and plasma in young (plaque-free) Tg2576 mice using the gamma-secretase inhibitor N2-[(2S)-2-(3,5-difluorophenyl)-2-hydroxyethanoyl]-N1-[(7S)-5-methyl-6-oxo-6,7-di hydro-5H-dibenzo[b,d]azepin-7-yl]-L-alaninamide (LY-411575). J. Pharmacol. Exp. Ther. 2004, 309, 49–55. [Google Scholar]
- Arnarez, C.; Uusitalo, J.J.; Masman, M.F.; Ingólfsson, H.I.; de Jong, D.H.; Melo, M.N.; Periole, X.; de Vries, A.H.; Marrink, S.J. Dry Martini, a coarse-grained force field for lipid membrane simulations with implicit solvent. J. Chem. Theory Comput. 2015, 11, 260–275. [Google Scholar] [CrossRef]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the amyloid precursor protein by human γ-secretase. Science 2019, 363. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.C.; Yan, C.; Yang, G.; Lu, P.; Ma, D.; Sun, L.; Zhou, R.; Scheres, S.H.W.; Shi, Y. An atomic structure of human γ-secretase. Nature 2015, 525, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Grant, B.J.; Skjærven, L.; Yao, X.Q. Comparative protein structure analysis with Bio3D-web. Methods Mol. Biol. 2020, 2112, 15–28. [Google Scholar] [CrossRef]
- Skjærven, L.; Yao, X.Q.; Scarabelli, G.; Grant, B.J. Integrating protein structural dynamics and evolutionary analysis with Bio3D. BMC Bioinform. 2014, 15, 399. [Google Scholar] [CrossRef] [Green Version]
- Sato, C.; Takagi, S.; Tomita, T.; Iwatsubo, T. The C-terminal PAL motif and transmembrane domain 9 of presenilin 1 are involved in the formation of the catalytic pore of the gamma-secretase. J. Neurosci. 2008, 28, 6264–6271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, S.; Tominaga, A.; Sato, C.; Tomita, T.; Iwatsubo, T. Participation of transmembrane domain 1 of presenilin 1 in the catalytic pore structure of the gamma-secretase. J. Neurosci. 2011, 30, 15943–15950. [Google Scholar] [CrossRef]
- Gertsik, N.; Am Ende, C.W.; Geoghegan, K.F.; Nguyen, C.; Mukherjee, P.; Mente, S.; Seneviratne, U.; Johnson, D.S.; Li, Y.M. Mapping the binding site of BMS-708163 on γ-secretase with cleavable photoprobes. Cell Chem. Biol. 2017, 24, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Pozdnyakov, N.; Murrey, H.E.; Crump, C.J.; Pettersson, M.; Ballard, T.E.; Am Ende, C.W.; Ahn, K.; Li, Y.M.; Bales, K.R.; Johnson, D.S. γ-Secretase modulator (GSM) photoaffinity probes reveal distinct allosteric binding sites on presenilin. J. Biol. Chem. 2013, 288, 9710–9720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebke, A.; Luebbers, T.; Fukumori, A.; Shirotani, K.; Haass, C.; Baumann, K.; Steiner, H. Novel gamma-secretase enzyme modulators directly target presenilin protein. J. Biol. Chem. 2011, 286, 37181–37186. [Google Scholar] [CrossRef] [Green Version]
- Tian, G.; Ghanekar, S.V.; Aharony, D.; Shenvi, A.B.; Jacobs, R.T.; Liu, X.; Greenberg, B.D. The mechanism of gamma-secretase: Multiple inhibitor binding sites for transition state analogs and small molecule inhibitors. J. Biol. Chem. 2003, 278, 28968–28975. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Carmona, S.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Zhou, R.; Guo, X.; Yan, C.; Lei, J.; Shi, Y. Structural basis of γ-secretase inhibition and modulation by small molecule drugs. Cell 2021, 184, 521–533.e14. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Beel, A.J.; Barrett, P.; Schnier, P.D.; Hitchcock, S.A.; Bagal, D.; Sanders, C.R.; Jordan, J.B. Nonspecificity of binding of gamma-secretase modulators to the amyloid precursor protein. Biochemistry 2009, 48, 11837–11839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serneels, L.; Van Biervliet, J.; Craessaerts, K.; Dejaegere, T.; Horre, K.; Van Houtvin, T.; Esselmann, H.; Paul, S.; Schafer, M.K.; Berezovska, O.; et al. γ-Secretase heterogeneity in the Aph1 subunit: Relevance for Alzheimer’s disease. Science 2009, 324, 639–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedi, R.K.; Patel, C.; Mishra, V.; Xiao, H.; Yada, R.Y.; Bhaumik, P. Understanding the structural basis of substrate recognition by Plasmodium falciparum plasmepsin V to aid in the design of potent inhibitors. Sci. Rep. 2016, 6, 31420. [Google Scholar] [CrossRef] [Green Version]
- Mahanti, M.; Bhakat, S.; Nilsson, U.J.; Söderhjelm, P. Flap dynamics in aspartic proteases: A computational perspective. Chem. Biol. Drug Des. 2016, 88, 159–177. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T. Secretase inhibitors and modulators for Alzheimer’s disease treatment. Expert Rev. Neurother. 2009, 9, 661–679. [Google Scholar] [CrossRef]
- Potter, R.; Patterson, B.W.; Elbert, D.L.; Ovod, V.; Kasten, T.; Sigurdson, W.; Mawuenyega, K.; Blazey, T.; Goate, A.; Chott, R.; et al. Increased in vivo amyloid-beta42 production, exchange, and loss in presenilin mutation carriers. Sci. Transl. Med. 2013, 5, 189ra177. [Google Scholar] [CrossRef] [Green Version]
- Kumar-Singh, S.; Theuns, J.; Van Broeck, B.; Pirici, D.; Vennekens, K.; Corsmit, E.; Cruts, M.; Dermaut, B.; Wang, R.; Van Broeckhoven, C. Mean age-of-onset of familial Alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum. Mutat. 2006, 27, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Koch, P.; Tamboli, I.Y.; Mertens, J.; Wunderlich, P.; Ladewig, J.; Stuber, K.; Esselmann, H.; Wiltfang, J.; Brustle, O.; Walter, J. Presenilin-1 L166P mutant human pluripotent stem cell-derived neurons exhibit partial loss of gamma-secretase activity in endogenous amyloid-beta generation. Am. J. Pathol. 2012, 180, 2404–2416. [Google Scholar] [CrossRef]
- Fukumoto, H.; Rosene, D.L.; Moss, M.B.; Raju, S.; Hyman, B.T.; Irizarry, M.C. Beta-secretase activity increases with aging in human, monkey, and mouse brain. Am. J. Pathol. 2004, 164, 719–725. [Google Scholar] [CrossRef]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef]
- Theuns, J.; Remacle, J.; Killick, R.; Corsmit, E.; Vennekens, K.; Huylebroeck, D.; Cruts, M.; Van Broeckhoven, C. Alzheimer-associated C allele of the promoter polymorphism -22C>T causes a critical neuron-specific decrease of presenilin 1 expression. Hum. Mol. Genet. 2003, 12, 869–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovelet-Lecrux, A.; Hannequin, D.; Raux, G.; Le Meur, N.; Laquerriere, A.; Vital, A.; Dumanchin, C.; Feuillette, S.; Brice, A.; Vercelletto, M.; et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 2006, 38, 24–26. [Google Scholar] [CrossRef]
- Guyant-Marechal, L.; Rovelet-Lecrux, A.; Goumidi, L.; Cousin, E.; Hannequin, D.; Raux, G.; Penet, C.; Ricard, S.; Mace, S.; Amouyel, P.; et al. Variations in the APP gene promoter region and risk of Alzheimer disease. Neurology 2007, 68, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Lindholm, K.; Yang, L.B.; Yue, X.; Citron, M.; Yan, R.; Beach, T.; Sue, L.; Sabbagh, M.; Cai, H.; et al. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2004, 101, 3632–3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schor, N.F. What the halted phase III gamma-secretase inhibitor trial may (or may not) be telling us. Ann. Neurol. 2011, 69, 237–239. [Google Scholar] [CrossRef]
- Miletić, V.; Odorčić, I.; Nikolić, P.; Svedružić, Ž.M. In silico design of the first DNA-independent mechanism-based inhibitor of mammalian DNA methyltransferase Dnmt1. PLoS ONE 2017, 12, e0174410. [Google Scholar] [CrossRef]
- Qi, Y.; Ingolfsson, H.I.; Cheng, X.; Lee, J.; Marrink, S.J.; Im, W. CHARMM-GUI martini maker for coarse-grained simulations with the martini force field. J. Chem. Theory Comput. 2015, 11, 4486–4494. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Audagnotto, M.; Kengo Lorkowski, A.; Dal Peraro, M. Recruitment of the amyloid precursor protein by γ-secretase at the synaptic plasma membrane. Biochem. Biophys. Res. Commun. 2018, 498, 334–341. [Google Scholar] [CrossRef]
- Krzemińska, A.; Moliner, V.; Świderek, K. Dynamic and electrostatic effects on the reaction catalyzed by HIV-1 protease. J. Am. Chem. Soc. 2016, 138, 16283–16298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis vesrion. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Patel, D.S.; Ståhle, J.; Park, S.J.; Kern, N.R.; Kim, S.; Lee, J.; Cheng, X.; Valvano, M.A.; Holst, O.; et al. CHARMM-GUI membrane builder for complex biological membrane simulations with glycolipids and lipoglycans. J. Chem. Theory Comput. 2019, 15, 775–786. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.M.; Lai, M.T.; Xu, M.; Huang, Q.; DiMuzio-Mower, J.; Sardana, M.K.; Shi, X.P.; Yin, K.C.; Shafer, J.A.; Gardell, S.J. Presenilin 1 is linked with gamma-secretase activity in the detergent solubilized state. Proc. Natl. Acad. Sci. USA 2000, 97, 6138–6143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, A.W.S.; Vranken, W.F. ACPYPE-Antechamber python parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Lee, J.; Jo, S.; Brooks, C.L., 3rd; Lee, H.S.; Im, W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Svedružić, Ž.M.; Vrbnjak, K.; Martinović, M.; Miletić, V. Structural Analysis of the Simultaneous Activation and Inhibition of γ-Secretase Activity in the Development of Drugs for Alzheimer’s Disease. Pharmaceutics 2021, 13, 514. https://doi.org/10.3390/pharmaceutics13040514
Svedružić ŽM, Vrbnjak K, Martinović M, Miletić V. Structural Analysis of the Simultaneous Activation and Inhibition of γ-Secretase Activity in the Development of Drugs for Alzheimer’s Disease. Pharmaceutics. 2021; 13(4):514. https://doi.org/10.3390/pharmaceutics13040514
Chicago/Turabian StyleSvedružić, Željko M., Katarina Vrbnjak, Manuel Martinović, and Vedran Miletić. 2021. "Structural Analysis of the Simultaneous Activation and Inhibition of γ-Secretase Activity in the Development of Drugs for Alzheimer’s Disease" Pharmaceutics 13, no. 4: 514. https://doi.org/10.3390/pharmaceutics13040514
APA StyleSvedružić, Ž. M., Vrbnjak, K., Martinović, M., & Miletić, V. (2021). Structural Analysis of the Simultaneous Activation and Inhibition of γ-Secretase Activity in the Development of Drugs for Alzheimer’s Disease. Pharmaceutics, 13(4), 514. https://doi.org/10.3390/pharmaceutics13040514